

Abstract
The paraventricular nucleus of hypothalamus plays important roles in the regulation of energy balance and fetal growth. However, the molecular mechanisms underlying its formation and function have not been clearly elucidated. Various mutations in the human COUP-TFII gene, which encodes a nuclear receptor, result in growth retardation, congenital diaphragmatic hernia and congenital heart defects. Here, we show that COUP-TFII gene is expressed in the developing hypothalamus in mouse. The ventral forebrain-specific RXCre/+; COUP-TFIIF/F mutant mice display growth retardation. The development of the paraventricular nucleus of hypothalamus is compromised in the COUP-TFII mutant mainly because of increased apoptosis and mis-migration of the Brn2+ neurons. Moreover, hypoplastic anterior pituitary with blood cell clusters and shrunken posterior pituitary lacking AVP/OT neuron innervations are observed in the mutant, indicating the failure of formation of the hypothalamic-pituitary axis. Mechanistic studies show that the expression of Bdnf and Nrp1 genes is reduced in the mutant embryo, and that Bdnf is a direct downstream target of the COUP-TFII protein. Thus, our findings provide a novel functional validation that COUP-TFII gene promotes the expression of Bdnf and Nrp1 genes to ensure the appropriate morphogenesis of the hypothalamic-pituitary axis, especially the paraventricular nucleus of hypothalamus, and to prevent growth retardation.
Introduction
The central nervous system, especially the hypothalamus, plays pivotal roles in the regulation of energy balance1–3. Several types of neurons in various murine hypothalamic nuclei have been identified to regulate food intake and energy expenditure, including AgRP and POMC neurons in the arcuate nucleus of hypothalamus4–6, oxytocin (OT) and melanocortin-4 receptor neurons in the paraventricular nucleus of hypothalamus (PVH)7–10, and Orexin and MCH neurons in the lateral hypothalamic area (LHA)11–13. Nevertheless, so far how these hypothalamic nuclei and molecularly defined neurons are generated remains largely unknown.
The PVH nucleus, which is located dorsally on either side of the third ventricle, participates in not only the regulation of energy balance but also the formation of the hypothalamic-pituitary (HP) axis1,14. There are two main groups of secretory neurons in the PVH nucleus: magnocellular neurons synthesizing the peptide hormones arginine vasopressin (AVP) or OT and parvocellular neurons secreting corticotropin-releasing hormone (CRH) or thyrotropin-releasing hormone (TRH)15,16. The magnocellular neurons in the PVH nucleus and the supraoptic nucleus (SON) project to the posterior pituitary, where AVP and OT are released. Several genes including Brn2, Sim1, Otp and Arnt2 play important roles in the development of the PVH nucleus and posterior pituitary17–24. Growth retardation is observed in Brn2−/−, Otp−/− and Arnt2−/− mouse mainly because of the failure of formation of the HP axis17,18,23,24. Nonetheless, the molecular mechanism responsible for the appropriate formation of the HP axis, especially regarding the PVH nucleus, has not been fully understood.
Various rare deletions in chromosome 15q26 have been identified in patients with pre- and postnatal growth retardation, congenital heart defects (CHD), congenital diaphragmatic hernia (CDH), and high mortality25–31. Chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) gene (also known as Arp-1, Nr2f2, according to the Nuclear Receptors Nomenclature Committee 1999), mapped to 15q26.2, belongs to the steroid nuclear receptor superfamily32. Various copy number variants (CNVs) of the COUP-TFII gene have been identified in patients with growth restriction, CHD and CDH25,27–30,33. Especially, 11 out of 15 patients with COUP-TFII deletion have CDH27,28,34. One recent clinical study further demonstrated that several de novo single-nucleotide variants of COUP-TFII gene are responsible for the development of CHD35. COUP-TFII homozygous null mutant mouse is early embryonic lethal because of the failure of angiogenesis and heart development36. Conditional COUP-TFII homozygous mutant mouse with Nkx3.2Cre generates Bochdalek-type CDH37. The findings in mouse studies support the association of COUP-TFII with CHD and CDH. Consistent with the haploinsufficiency of COUP-TFII in patients, COUP-TFII gene heterozygous null mutant displays growth deficit and poor postnatal viability36. Nevertheless, so far how mutations of COUP-TFII gene cause growth failure remains unknown.
Here, we found that COUP-TFII is expressed in the developing hypothalamus of mouse. Ventral forebrain-specific RXCre/+; COUP-TFIIF/F homozygous mutant mice generate postnatal growth retardation and poor viability. Hypocellular PVH and SON nuclei are observed in the mutants, which may be caused by increased apoptosis and mis-migration of the Brn2+ neurons. In addition, hypoplastic anterior pituitary with blood cell clusters and shrunken posterior pituitary lacking AVP/OT neuron projection are observed in the mutant. Furthermore, the expression of Bdnf, Nrp1 and Avp genes is reduced in the ventral forebrain of the mutant, and Bdnf is a direct downstream target of the COUP-TFII protein.
Results
COUP-TFII gene is expressed in the developing hypothalamus of mouse
Since patients with various 15q26 deletions including the COUP-TFII gene display growth retardation and COUP-TFII gene heterozygous mutant mice generate developmental delay and poor postnatal viability36, we asked whether COUP-TFII in the hypothalamus contributes to the regulation of growth deficit. To answer this question, we first assessed the expression of COUP-TFII in mouse embryonic forebrain by immunofluorescence assays. At E10.5, the expression of COUP-TFII was detected in the ventricular zone of the hypothalamus, where reside neuronal progenitor cells (NPCs) (Fig. 1A,B). The expression of COUP-TFII was confined to the hypothalamic region at E12.5 (Fig. 1C,D) and E14.5 (Fig. 1F,G). Thus, COUP-TFII is expressed in the NPCs and early differentiating neurons of the mouse hypothalamus.
Figure 1.
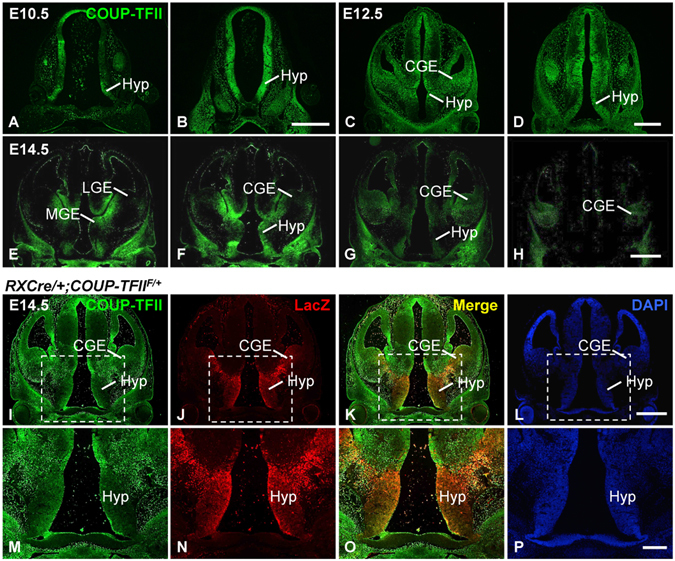
Expression of COUP-TFII in the developing hypothalamus and the RXCre recombinase activity in the hypothalamus. The expression of COUP-TFII protein is detected in neuronal progenitor cells at the ventricular zone of the hypothalamus at E10.5 (A,B), in the hypothalamic region at E12.5 (C,D) and E14.5 (E–H). (I–P) COUP-TFII (green) and LacZ (red) are colocalized at the hypothalamus of the RXCre/+; COUP-TFIIF/+ heterozygous mutant at E14.5. (M–P) Inserts in (I–L). CGE, caudal ganglionic eminence; Hyp, Hypothalamus; LGE, lateral ganglionic eminence; MGE, medial ganglionic eminence. Two or three embryos were analyzed at each stage. Scale bars, (A–D,E–H,I–L) 500 μm; (M–P) 200 μm.
It has been shown that the activity of RXCre recombinase is detected in the mouse embryonic ventral forebrain including the eye and the hypothalamus38, and LacZ expression can be used as an indicator for the deletion of COUP-TFII gene39. We performed double immunofluorescence staining assays with antibodies against COUP-TFII and LacZ on coronal sections of a RXCre/+; COUP-TFIIF/+ heterozygous mutant embryo at E14.5. COUP-TFII was expressed at the hypothalamus (Fig. 1I,M). The expression of LacZ was also detected at the hypothalamus and the caudal ganglionic eminence (Fig. 1J,N). Merged images revealed that the green COUP-TFII signals and the red LacZ signals were highly colocalized at the hypothalamus (Fig. 1K,O), suggesting that RXCre recombinase can efficiently excise the COUP-TFII gene in the embryonic hypothalamus.
RXCre/+; COUP-TFIIF/F mutant mice display growth retardation and compromised PVH nucleus
RXCre mouse was used to generate the RXCre/+; COUP-TFIIF/F conditional homozygous mutant mouse, referred to as COUP-TFII mutant or mutant hereafter. At birth, all the pups in the same litter were similar. Nevertheless, the mutant pups were smaller than their control littermates at postnatal day 3 (P3) and day 4 (P4) (data not shown). Some mutant mice did not survive between P18 and P26, and some survived to adulthood. No obvious differences were observed among COUP-TFIIF/+, COUP-TFIIF/F and RXCre/+; COUP-TFIIF/+ mice; therefore, they were used as the control in the study. The body weight of the mutant mice was only approximately half that of the control mice at wean (Fig. 2A). Clearly, RXCre/+; COUP-TFIIF/F homozygous mutant mice displayed growth retardation.
Figure 2.
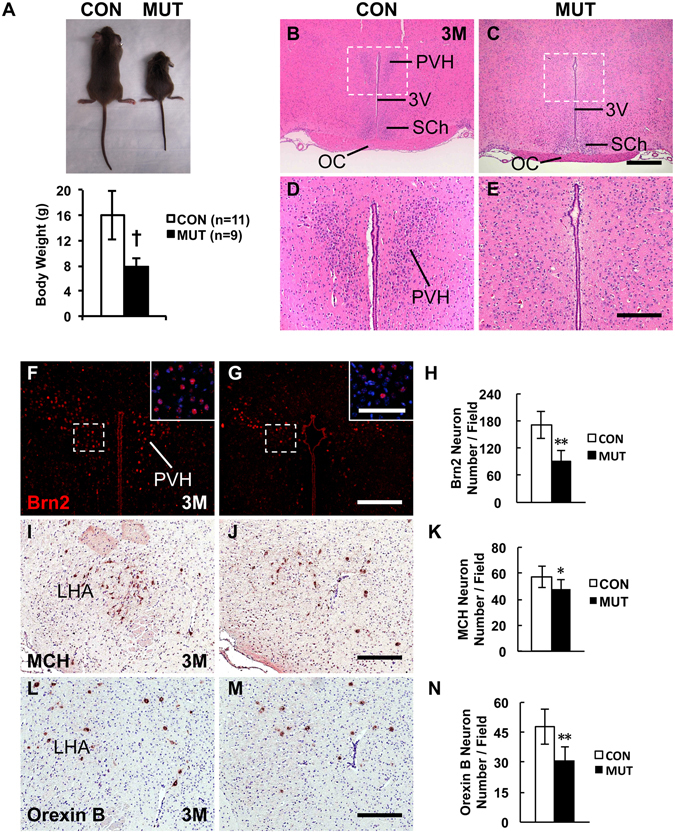
Adult COUP-TFII gene mutant mice with RXCre display growth retardation and compromised PVH nucleus. (A) Body weight of the RXCre/+; COUP-TFIIF/F male mutant mice (n = 6) is approximately half of that of the control (n = 8) at wean. The H&E staining data reveal that compared with the control (B,D), the PVH nucleus is barely detectable in the mutant at 3 M (C,E). (D,E) Inserts in (B,C). Compared with the control (F), there are very few Brn2+ PVH neurons in the mutant at 3 M (G and insert), and the reduction is significant (H and insert). Compared with the control (I), there are fewer MCH neurons in the mutant LHA region at 3 M (J), and the reduction is significant (K). Compared with the control (L), there are fewer Orexin B neurons in the mutant LHA region at 3 M (M), and the reduction is significant (N). 3 V, third ventricle; LHA, lateral hypothalamic area; OC, optic chiasm; PVH, paraventricular nucleus of hypothalamus; SCh, suprachiasmatic nucleus. The quantitative data in (H,K and N) were generated from the analysis of three pairs of control and mutant mice. The data indicate the mean ± SD. Student’s t-test, *P < 0.05; **P < 0.01; †P < 0.001. Scale bars, (B,C) 500 μm; (D,E,F,G) 200 μm; inserts of (F,G) 100 μm; (I,J,L,M) 100 μm.
To investigate the cause of growth restriction, we performed H&E staining on coronal sections from the control and the mutant mouse at 3 weeks (3 W) and 3 months (3 M) after birth. The same phenotypes were observed at both stages, and images generated from mice at 3 M were shown. Compared with the control (Fig. 2B,D), the PVH nucleus was barely observed in the mutant (Fig. 2C,E). Brn2 gene is specifically expressed in the PVH neurons17,18. Immunofluorescence staining showed that compared with the control (Fig. 2F and insert), there were much fewer Brn2+ PVH neurons in the mutant (Fig. 2G and insert). Quantitative analysis from three pairs of mice showed that the reduction of Brn2+ PVH neurons in the mutant was significant (Fig. 2H). Overall, therefore, the development of the PVH nucleus is morphologically and molecularly compromised in the COUP-TFII mutant mouse.
MCH neurons and Orexin B neurons in the LHA promote food intake, and MCH mutant or Orexin B neuron-ablating mouse displays hypophagia11–13. Compared with the control (Fig. 2I), the number of MCH neurons was significantly reduced in the mutant LHA (Fig. 2J,K), as was the number of Orexin B neurons in the mutant (Fig. 2L–N). POMC and NPY neurons in the arcuate nucleus also participate in the regulation of energy balance4–6. The expression of POMC and NPY in the arcuate nucleus was comparable between the control and the mutant mouse (data not shown). GHRH neurons in the arcuate nucleus control body growth through regulating growth hormone pathway40–42. Compared with the control (Fig. S1A,C), there were much few GHRH neurons in the arcuate nucleus in the mutant at 1 M (Fig. S1B,D), and the reduction was significant (Fig. S1E). Similar as the RXCre/+; COUP-TFIIF/+ control mouse (Fig. S1F,H), the expression of LacZ was barely detected in the arcuate nucleus in the RXCre/+; COUP-TFIIF/F mutant mouse (Fig. S1G,I), indicating COUP-TFII gene is not deleted in the mutant arcuate nucleus. The data above suggest that the reduced MCH, Orexin B and GHRH neurons in the hypothalamic regions may contribute to growth defect in the COUP-TFII mutant.
The expression of COUP-TFII in the ventromedial nucleus of hypothalamus (VMH) of mouse is related to hypoglycemia-associated autonomic failure43. Consistent with a previous report43, the expression of COUP-TFII was readily detected in the majority of SF1+ VMH neurons at 1 M (Fig. 3A–D). Double immunostaining with antibodies against COUP-TFII and LacZ was conducted in the RXCre/+; COUP-TFIIF/+ heterozygous and RXCre/+; COUP-TFIIF/F homozygous mice at 3 M. The expression of both COUP-TFII and LacZ in the VMH nucleus was similar between the control and the mutant mouse (Fig. 3E–L). It seems that RXCre recombinase does not target the VMH neurons, and the development of the VMH nucleus is normal in the COUP-TFII mutant mouse. Thus, in the mutant hypothalamus, the differentiation of Brn2+ neurons in the PVH nucleus, MCH and Orexin B neurons in the LHA was affected, but not POMC and NPY neurons in the arcuate nucleus and COUP-TFII+ neurons in the VMH nucleus.
Figure 3.
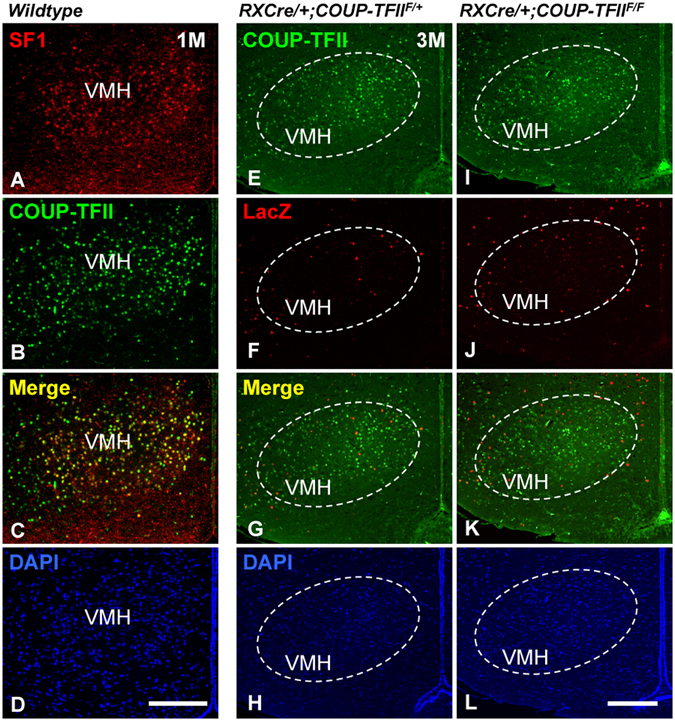
Development of the VMH nucleus is normal in the adult COUP-TFII mutant. (A–D) The expression of COUP-TFII (green) is readily detected in the majority of SF1+ VMH neurons (red) at 1 M. (E–L) The expression of COUP-TFII (green) in the VMH nucleus was not altered between the control and the mutant at 3 M; additionally, there were few LacZ signals (red) in the VMH nucleus of the control and the mutant. VMH, ventromedial nucleus of hypothalamus. Scale bars, (A–D,E–L) 200 μm.
COUP-TFII is expressed in Brn2+ early differentiating PVH neurons
Hypocellular PVH nucleus is the most obvious defect observed in the COUP-TFII mutant (Fig. 2B–H). It has been reported that Brn2 gene plays a pivotal role in the differentiation of the PVH neuron17,18. We performed double immunofluorescence staining with antibodies against COUP-TFII and Brn2 on coronal sections containing the PVH nucleus at E14.5, E17.5, P1 and 6 M. The expression of Brn2 was detected at the prospective PVH nucleus at E14.5 (Fig. 4A), in the late differentiating PVH neuron at E17.5 (Fig. 4E) and at P1 (Fig. 4I), and in the mature PVH neurons at 6 M (Fig. 4M). The expression of COUP-TFII was broadly detected in the hypothalamic regions, and was colocalized with Brn2 in the early differentiating PVH neurons at E14.5 (Fig. 4B–D). At E17.5, the expression of COUP-TFII was sharply decreased in the Brn2+ differentiating PVH neurons, but remained high in the neurons localized dorsally to the PVH nucleus (Fig. 4F–H). The expression of COUP-TFII was barely detectable in the Brn2+ late differentiating PVH neurons at P1 (Fig. 4J–L) and the Brn2+ mature PVH neurons at 6 M (Fig. 4N–P). These data show that COUP-TFII gene is expressed in the early differentiating PVH neurons, but not in the late differentiating and the mature PVH neurons.
Figure 4.
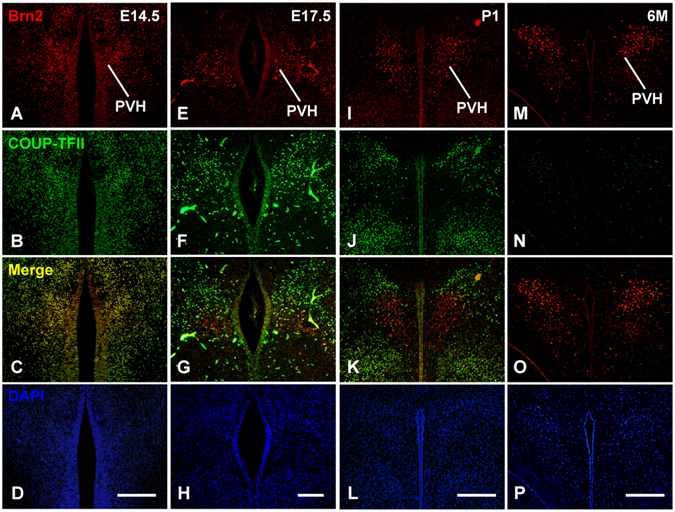
COUP-TFII is co-expressed with Brn2 in the early differentiating PVH neurons, but not in the late differentiating PVH neurons and the mature PVH neurons. (A–D) Colocalization of Brn2 (red) and COUP-TFII (green) in the early differentiating PVH neurons at E14.5. (E–H) Expression of Brn2 and COUP-TFII in the differentiating PVH neurons at E17.5. (I–L) Expression of Brn2 and COUP-TFII in the late differentiating PVH neurons at P1. (M–P) Expression of Brn2 and COUP-TFII in the mature PVH neurons at 6 M. PVH, paraventricular nucleus of hypothalamus. Scale bars, (A–D,I–L,M–P) 200 μm; (E–H) 100 μm.
Reduced Brn2+ early differentiating PVH neurons, hypocellular SON nucleus, and increased apoptosis in the COUP-TFII mutant embryo
Next, we asked whether the development of the PVH nucleus is normal at embryonic stages. As shown in Fig. 5Aa,c,e, there were many Brn2+ neurons at the PVH region along the rostro-caudal axis in the control at E15.5. In contrast, there were very few Brn2+ neurons at the prospected mutant PVH nucleus (Fig. 5Ab,d,f). Quantitative data from three pairs of animals at the same stage revealed that compared with the control, the reduction of the Brn2+ early differentiating PVH neurons was significant in the mutant (Fig. 5Ag).
Figure 5.
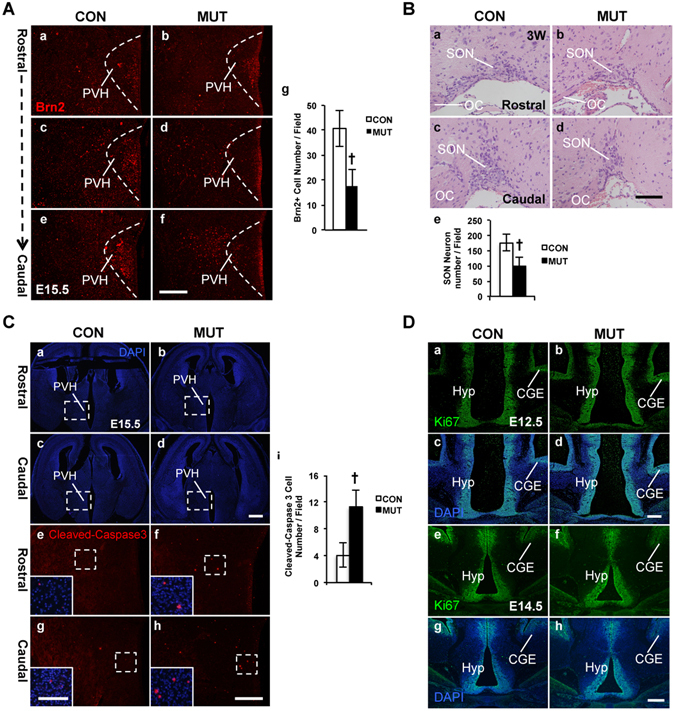
Reduction of the Brn2+ early differentiating PVH neurons, hypocellular SON nucleus, and increased apoptosis in the COUP-TFII mutant. (A) Brn2+ early differentiating PVH neurons are reduced in the mutant embryo. Compared with the control (Aa–c), there are much fewer Brn2+ early differentiating PVH neurons along the rostro-caudal axis in the mutant at E15.5 (Ad–f), and the reduction is significant (Ag). The data are collected from three pairs of control and mutant embryos. (B) Development of the SON nucleus is abnormal in the adult mutant mouse. The H&E staining data show that compared with the control (Ba,b), there are much fewer SON neurons in the COUP-TFII mutant (Bc,d). The reduction of magnocellular SON neurons is significant in the mutant (Be). (Ca–h) Increased apoptosis is detected in the COUP-TFII mutant embryo. DAPI staining images of coronal sections with PVH region in the control (Ca,c) and the mutant (Cb,d) at E15.5. Compared with the control (Ce,g and inserts), there are more cleaved-Caspase-3 signals in the mutant PVH region at E15.5 (Cf,h and inserts), and the increase of the cleaved-Caspase-3 signals is significant (Ci). (Da–h) Proliferation is not affected in the COUP-TFII mutant. The expression of Ki67 is comparable between the control (Da,c) and mutant (Db,d) at E12.5. Expression of Ki67 is comparable between the control (De,g) and mutant (Df,h) at E14.5. CGE, caudal ganglionic eminence; OC, optic chiasm; PVH, paraventricular nucleus of hypothalamus; SON, supraoptic nucleus. At least three pairs of the control and the mutant mice were used in each study. The data indicate the mean ± SD. Student’s t-test, †P < 0.001. Scale bars, (Aa–f) 200 μm; (Ba–d) 100 μm; (Ca–d) 500 μm; (Ce–h,Da–h) 100 μm; inserts of (Ce–h) 50 μm.
Interestingly, many Brn2+ neurons were localized laterally to the prospected caudal PVH nucleus in the mutant (Fig. 5Ad,f). Calbindin is another marker of the SON and PVH neurons17,44. Similar to the Brn2+ neurons, compared with the control (Fig. S2A,C,E,G), there were more mis-migrating Calbindin+ neurons beside the caudal PVH nucleus in the mutant (Fig. S2B,D,F,H). The SON nucleus lies lateral to optic tracts and dorsal to pial surface of the brain. Neurons in the PVH nucleus and the SON nucleus originate from the same group of NPCs at E10.545. During development, the PVH neurons are differentiated locally, while the SON neurons migrate ventro-laterally to their final destination45. Probably, those mis-located Brn2+ or Calbindin+ neurons are related to the SON neurons. As expected, H&E staining results revealed that compared with the control (Fig. 5Ba,c), the number of magnocellular SON neurons in the mutant was reduced (Fig. 5Bb,d), and the reduction was significant (Fig. 5Be). Thus, the formation of the SON nucleus is also affected in the COUP-TFII mutant.
There are several possibilities for the loss of the Brn2+ PVH neurons in the COUP-TFII mutant embryos, such as apoptosis and proliferation defect. To investigate the mechanism for the reduction of the Brn2+ PVH neurons, we performed immunostaining assay to examine the expression of cleaved-Caspase-3, an apoptotic marker. Compared with the control at E15.5 (Fig. 5Ca,c,e,g, and inserts), there were more cleaved-Caspase-3 signals in the mutant PVH region (Fig. 5Cb,d,f,h, and inserts). Quantitative assays with samples from 3 pairs of animals at E15.5 confirmed that there were significantly more apoptotic cells in the mutant PVH than in the control (Fig. 5Ci). In addition, there were also more cleaved-Caspase-3 signals in the hypothalamus of the mutant than the control at E13.5 (Fig. S3A–F), indicating that abnormal apoptosis occurs at earlier embryonic stages. Next, we assessed the expression of Ki67, a proliferation marker, at the PVH region at E12.5 and at E14.5. The expression of Ki67 was comparable between the control (Fig. 5Da,c,e,g) and the mutant (Fig. 5Db,d,f,h) at both stages, suggesting that proliferation was not altered in the mutant. Thus, our data show that increased apoptosis is one possible mechanism that leads to the reduction of the Brn2+ PVH neurons in the COUP-TFII mutant.
Bdnf gene is a direct downstream target of COUP-TFII
To investigate molecular mechanism responsible for the defects in the COUP-TFII mutant, real-time quantitative PCR (qPCR) assays were performed with total RNAs prepared from ventral forebrains of the control and the mutant at E14.5. As shown in Fig. 6A, the expression of COUP-TFII transcripts was reduced by approximately 50% in the mutant compared with the control. The expression of the COUP-TFI gene was not altered. Brn2, Sim1, Otp and Arnt2 genes are essential for the development of the PVH nucleus17–23. Compared with the control, the expression of these genes was slightly reduced in the mutant mouse at E14.5, but the difference was not significant (Fig. 6A), indicating that the function of COUP-TFII in the development of the PVH nucleus may be independent of these known regulatory genes.
Figure 6.
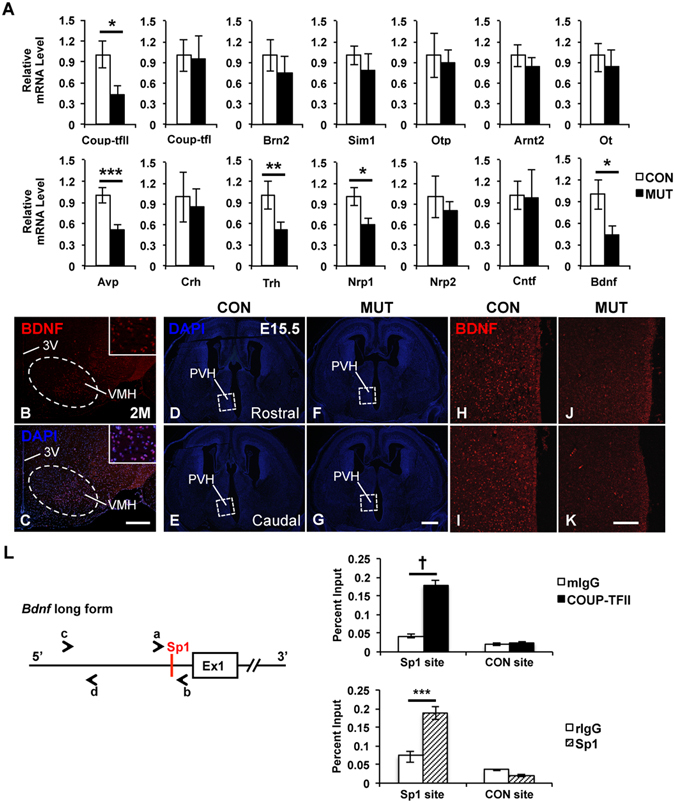
Expression of Bdnf and Nrp1 is reduced in the COUP-TFII mutant embryo, and Bdnf is a downstream target of COUP-TFII. (A) Real-time quantitative PCR data with samples from the ventral forebrain of the control (n = 4) and mutant (n = 3) embryos at E14.5. The expression of COUP-TFII, Avp, Bdnf, Nrp1 and Trh transcripts is significantly reduced in the mutant. A BDNF-specific antibody detects the expression of BDNF in the cytoplasm of neurons at the ventro-lateral region of the mouse VMH nucleus at 2 M (B,C). DAPI staining images of coronal sections with the PVH region in the control (D,E) and the mutant (F,G) at E15.5. Compared with the control (H,I), the expression of BDNF protein is noticeably reduced in the prospected mutant hypothalamus at E15.5 (J–K). L, An evolutionarily conserved Sp1 binding site is identified at the promoter region of the long form of the Bdnf gene. Primers a/b and c/d were used to amplify the Sp1 locus and a 2 kb up-stream non-Sp1 control locus, respectively. In chromatin immunoprecipitation assays, the binding of both COUP-TFII and Sp1 protein is enriched at the conserved Sp1 site but not at the negative control site. 3 V, third ventricle; PVH, paraventricular nucleus of hypothalamus; VMH, ventromedial nucleus of hypothalamus. Three independent assays were performed in each real-time quantitative PCR experiment. The data indicate the mean ± SD. Student’s t-test, *P < 0.05; **P < 0.01; ***P < 0.005; †P < 0.001. Scale bar, (B,C) 300 μm; (D–G) 500 μm; (H–K) 100 μm.
There are several major secretory neurons in the PVH nucleus including the AVP, OT, CRH and TRH neurons15,16. Interestingly, the expression of Avp and Trh genes but not Ot and Crh genes was significantly reduced in the mutant at E14.5 (Fig. 6A), suggesting that COUP-TFII gene may be specifically required for the early differentiation of the AVP and TRH neurons. Nrp1 and Nrp2 genes, which are involved in cell migration and axon guidance, are regulated directly by COUP-TFII protein39. The expression of both Nrp1 and Nrp2 genes is reduced in the ventral forebrain of RXCre/+; COUP-TFIIF/F mutant mouse at E12.539. The expression of Nrp1 but not Nrp2 was decreased significantly in the mutant at E14.5 (Fig. 6A), suggesting that Nrp1 might specifically mediate the migration of the Brn2+ neurons in the hypothalamus.
Neurotrophin factors, such as NGF, BDNF, NT3 and CNTF, are essential for the survival and differentiation of neurons46–48. Since the increased apoptosis was detected in the mutant hypothalamus (Figs 5C and S3), the expression of neurotrophin factors was assessed by qPCR assays. The expression of the Ngf and Nt3 genes was undetectable in both the control and the mutant (data not shown). The expression of Cntf transcripts was not altered in the mutant, whereas the expression of Bdnf was significant lower in the mutant than the control (Fig. 6A). Bdnf transcripts are preferentially expressed at the ventro-lateral region of the VMH nucleus49. A specific BDNF antibody could detect the expression of BDNF protein in the cytoplasm of neurons at the ventro-lateral VMH nucleus in the adult mouse at 2 M (Fig. 6B,C). The expression of BDNF was readily detected in the rostral and the caudal part of the control hypothalamic areas including the PVH nucleus at E15.5 (Fig. 6D,E,H,I); in contrast, its expression was barely detectable in the mutant (Fig. 6F,G,J,K). Thus, the expression of Bdnf was reduced in the COUP-TFII mutant at both transcriptional and translational levels.
To determine the direct downstream targets of COUP-TFII among the Avp, Bdnf, Nrp1 and Trh genes, chromatin immuoprecipitation (ChIP) assay was performed with chromatin prepared from the hypothalamus of mouse embryos at E14.5. COUP-TFII positively regulates the expression of target genes through the Sp1 site by tethering to Sp1 protein32,50,51. COUP-TFII may promote the expression of Nrp1 gene in the ventral forebrain through a conserved Sp1 site in intron 1239. Our ChIP-qPCR assays showed that both COUP-TFII and Sp1 were recruited at the conserved Sp1 site of Nrp1 gene, but not at the control non-Sp1 site at 3′ UTR (Fig. S4). Next, an Sp1 site, which is evolutionarily conserved among human, chimpanzee, mouse, cow and opossum, was identified at the promoter region of the long form of the Bdnf gene (Fig. 6L). The binding of both COUP-TFII and Sp1 was enriched at the conserved Sp1 site of Bdnf gene, but not at a negative non-Sp1 control site, which was located 2 kb upstream (Fig. 6L). Unfortunately, no such evolutionarily conserved Sp1 site was identified in the genomic locus of either Avp or Trh. The findings above suggest that the Bdnf gene is a direct downstream target of COUP-TFII, and the reduced expression of Bdnf is a possible cause for the increased apoptosis in the COUP-TFII mutant.
The development of pituitary is compromised in the COUP-TFII mutant
In Brn2−/− null mutant mouse, the absence of the PVH and SON nuclei leads to the loss of the posterior pituitary, failure of formation of the HP axis, and growth retardation17,18. The pituitary gland consists of anterior (A), intermediate (I) and posterior (P) pituitary lobes (Fig. 7A). H&E staining results revealed that compared with the control (Fig. 7Aa,c,e,g,i,k), the posterior pituitary was noticeably smaller in the COUP-TFII mutant at P0 (Fig. 7Ab,d,f,h,j,l). Moreover, images at higher magnification revealed that there were a few isolated blood cells in the anterior pituitary of the control (Fig. 7Am,o,q); nevertheless, many blood cell clusters, indicated by white arrowheads, were observed in the anterior pituitary of the mutant (Fig. 7n,p,r), indicating a hypoplastic anterior pituitary. Most likely, the defective anterior and posterior pituitary is a cause of growth restriction of the COUP-TFII mutant.
Figure 7.
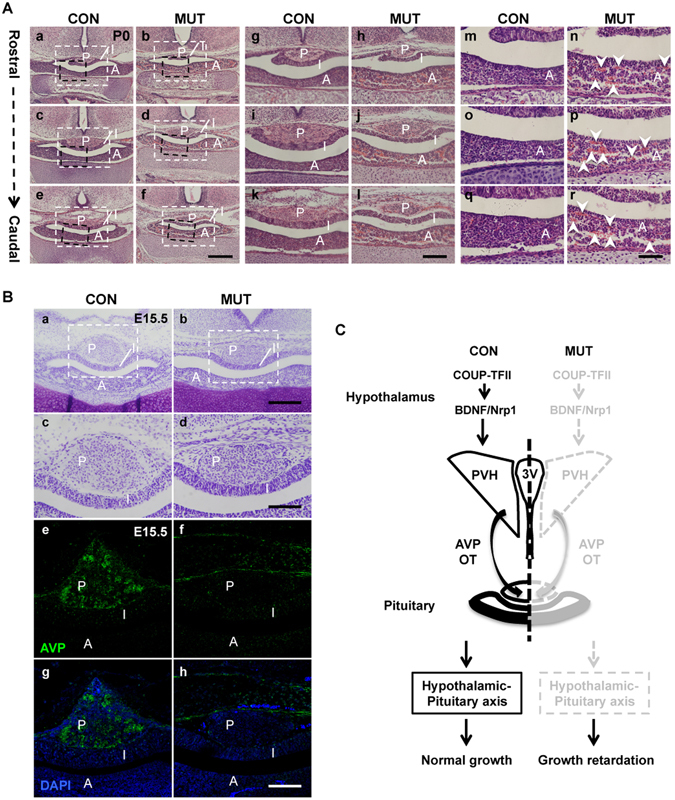
COUP-TFII ensures the appropriate morphogenesis and function of the hypothalamic-pituitary axis, especially the PVH nucleus, to prevent growth retardation. (A), The development of both posterior and anterior pituitary is abnormal in the newborn mutant. (Ag–i) White inserts in (Aa–f). (Am–r) Black inserts in (Aa–f). H&E staining showed that compared with the control (Aa,c,e,g,i,k), the posterior pituitary is shrunken along the rostro-caudal axis in the mutant at P0 (Ab,d,f,h,j,l). There are a few isolated blood cells in the anterior pituitary of the control (Am,o,q); however, many blood cell clusters, indicated by white arrow-heads, are observed in the mutant anterior pituitary (An,p,r). (B) The projection of AVP neuron to the posterior pituitary is abnormal in the mutant embryo at E15.5. Nissl staining images of the pituitary in the control (Ba,c) and the mutant at E15.5 (Bb,d). (Bc,d) Inserts in (Ba,b). The expression of AVP is readily detected in the control posterior pituitary at E15.5 (Be,g), but not in the mutant posterior pituitary (Bf,h). (C) A working model: during early development, COUP-TFII may activate or maintain the expression of Bdnf and Nrp1 genes to ensure the appropriate morphogenesis and function of the hypothalamic-pituitary axis, especially the PVH nucleus, and to prevent the growth failure. 3 V, third ventricle; A, anterior pituitary lobe; I, intermediate pituitary lobe; P, posterior pituitary lobe; PVH, paraventricular nucleus of hypothalamus. Scale bar, (Aa–f) 400 μm; (Ag–l) 200 μm; (Am–r) 100 μm; (Ba,b) 200 μm; (Bc–h) 100 μm.
Axon projections of AVP or OT magnocellular neurons target the posterior pituitary between E15.5 and E16.552. The expression of AVP was readily detected in the control at E15.5 (Fig. 7Be,g); however, its expression was absent from the posterior pituitary of the mutant (Fig. 7Bf,h). In addition, compared with the control (Fig. S5A,C,E,G), the expression of both AVP and OT was barely detected in the mutant posterior pituitary at P0 (Fig. S5B,D,F,H). In the COUP-TFII mutant mouse, hypocellular PVH/SON nuclei, hypoplastic anterior pituitary, and shrunken posterior pituitary lead to the failure of formation of the HP axis, which could be a main reason for growth retardation of these mice.
Discussion
In the present study, we have observed that COUP-TFII is expressed in the developing embryonic hypothalamus. Similar to the phenotype of hemizygous deletion of COUP-TFII in both human and mouse, RXCre/+; COUP-TFIIF/F mutant mice display noticeable growth restriction and poor postnatal viability. The development of the PVH and SON nuclei is affected in the COUP-TFII mutant mouse, with defects in both the anterior and the posterior pituitary. Moreover, increased apoptosis and mis-migration of the Brn2+ neurons are observed in the mutant. Mechanistic studies reveal that COUP-TFII may maintain or activate the expression of Bdnf and Nrp1 genes to ensure the appropriate morphogenesis and function of the HP axis, especially the PVH nucleus.
All patients with various 15q26 deletions generate growth retardation, associated with CDH and/or CHD25–31. The evidence from both clinical and mouse studies suggests that among the genes at the 15q26 locus, the loss of IGF1R at 15q26.3 is responsible for pre- and postnatal growth restriction31,53–55. Interestingly, various CNVs of the COUP-TFII gene were also identified in patients with different 15q26 deletions25,27–30,33. Moreover, the loss of an allele of the COUP-TFII gene leads to growth retardation and poor postnatal viability in pure-bred 129 Sv, C57BL/6 or ICR mouse and in 129 Sv/C57BL/6 mixture mouse36, indicating complete penetrance of the defect. Here, we find that the RXCre/+; COUP-TFIIF/F mutant mice are half the size of the control mice at wean (Fig. 2), suggesting that the RXCre/+; COUP-TFIIF/F mutant mouse phenocopies the COUP-TFII heterozygous null mouse. Therefore, other than IGF1R, haploinsufficiency of COUP-TFII is a possible cause for growth deficit in patients with 15q26 deletions.
The HP axis is an important neuroendocrine system coordinating the periphery and brain signals required for physiologic homeostasis and survival16,56. However, the detailed molecular and cellular mechanism responsible for the formation of the HP axis has not been fully clarified. Because the COUP-TFII gene is not expressed in the pituitary gland57, we mainly focused on its function in the hypothalamus. COUP-TFII is expressed in the hypothalamic NPCs and early differentiating PVH neurons, but not in the late differentiating and the mature PVH neurons (Figs 1 and 4), suggesting that COUP-TFII may play crucial roles in the early development of the hypothalamus. Indeed, the morphogenesis of the PVH nucleus is compromised in the COUP-TFII mutant with hypocellular SON nucleus, hypoplastic anterior pituitary with blood cell clusters, and shrunken posterior pituitary lacking AVP/OT projections (Figs 2, 5 and 7 and S5). The COUP-TFII mutant mouse phenocopies Brn2−/−, Sim1−/− and Arnt2−/− mouse in terms of growth restriction and compromised posterior pituitary17–23. However, the development of the anterior pituitary is not affected in the Brn2−/−, Sim1−/− and Arnt2−/− mouse17–23. In contrast, hypoplastic anterior pituitary is detected in the COUP-TFII mutant (Fig. 7), suggesting that the failure of formation of the HP axis, especially a defective anterior pituitary, is a possible cause for growth failure of the COUP-TFII mutant. The hypoplastic anterior pituitary could be caused by the reduction of GHRH neurons in the arcuate nucleus in the mutant (Fig. S1). The expression of Brn2, Sim1, Otp and Arnt2 transcripts is comparable between the control and the mutant (Fig. 6). The development of PVH NPCs remains normal in either Brn2−/− or Sim1−/− null mutant mouse at E15.517–19. Nonetheless, the Brn2+ early differentiating PVH neurons are noticeably reduced in the COUP-TFII mutant at E15.5 (Fig. 5). Therefore, distinct from Brn2 and Sim1, which participate in the terminal differentiation of PVH neurons, COUP-TFII is a novel key regulatory gene mediating the early morphogenesis of the HP axis, especially the PVH nucleus.
A hypocellular PVH nucleus is one of the most significant phenotypes in the COUP-TFII mutant (Fig. 2), which may be caused by several possibilities including abnormal apoptosis, lower proliferation, mis-migration or inappropriate differentiation. The Ki67 staining data show that proliferation is not altered in the mutant (Fig. 6). Both the PVH and SON neurons originate from the same population of NPCs at E10.545. A few Brn2+ or Calbindin+ neurons are mis-located laterally to the prospective mutant PVH nucleus (Figs 5 and S2), indicating that mis-migration may contribute to the hypocelluar PVH and SON nuclei in the mutant. As the direct targets of COUP-TFII protein, the Nrp1 and Nrp2 genes mediate the migration of Pax6+ neurons from the caudal ganglionic eminence to the basal medial amygdala nucleus39. Consistently, the expression of Nrp1 transcripts is reduced in the mutant at E14.5 (Fig. 6), suggesting that Nrp1 may specifically participate in the regulation of migration of the Brn2+ neuron in the hypothalamus. Furthermore, the number of cleaved-Caspase-3+ apoptotic cells is significantly increased in the mutant (Fig. 5). Neurotrophins, such as NGF and BDNF, are essential for the survival and growth of neurons during development46,47,58. Intriguingly, Bdnf−/− mouse displays postnatal growth retardation59; however, the cause of growth restriction in Bdnf−/− mouse is not clear. In the COUP-TFII mutant embryo, the expression of Bdnf is reduced at both the transcriptional and translational levels (Fig. 6). ChIP assay in vivo data demonstrate further that the Bdnf gene is a direct downstream target of COUP-TFII (Fig. 6). RXCre recombinase deletes the COUP-TFII gene in the ventral forebrain including the hypothalamus and caudal ganglionic eminence (Fig. 1)39. The reduced expression of Bdnf protein is also observed in the ventral forebrain regions of the mutant (Fig. 6), which may explain the reduction of the Brn2+ PVH neurons, as well as MCH neurons and Orexin B neurons in the LHA in the adult mutant. Clearly, both abnormal apoptosis and mis-migration are responsible for the reduction of the Brn2+ neurons. Nonetheless, we cannot exclude the possibility that COUP-TFII gene may also regulate the differentiation of some specific subtypes of PVH neurons, since the expression of Avp and Trh transcripts is specifically reduced in the mutant (Fig. 6).
In summary, our data provide a functional validation that CNVs of the COUP-TFII gene are possible causes for growth retardation. In the control mouse embryonic hypothalamus, COUP-TFII is required to activate or maintain the expression of Bdnf and Nrp1, which ensure appropriate morphogenesis and function of the HP axis, especially the PVH nucleus, to coordinate the normal growth. In the mutant, the loss of COUP-TFII results in reduced expression of Bdnf, Nrp1 and Avp, which may cause apoptosis and mis-migration of the Brn2+ neurons, hypocelluar PVH nucleus, hypoplastic anterior pituitary with blood cell clusters and shrunken posterior pituitary lacking AVP/OT neural innervations. The defective formation of the HP axis, especially the anterior pituitary, leads to growth deficit (Fig. 7C).
Our findings together with other clinical and mouse studies, reveal that COUP-TFII gene mutations are strongly associated with growth retardation, CHD, CDH, congenital coloboma, and postnatal viability27,28,34–37,51,60,61. Furthermore, enhanced expression of COUP-TFII is correlated with dilated cardiomyopathy62 as well as the recurrence and progression of prostate cancer63. Thus, COUP-TFII mutations should be included in the differential diagnosis of birth defects, CHD, CDH, ocular defects and cancer. Our study will benefit the prediction, prevention and treatment of human diseases.
Methods
Ethics statement
All animal experiments were carried out following protocols approved by the Animal Ethics Committee of the Shanghai Institute of Biochemistry and Cell Biology. We confirm that all methods were performed in accordance with the relevant guidelines and regulations.
Animals
COUP-TFII-floxed mice and RXCre mice used in the study were of the C57B6/129 mixed background. Male mice were used in body weight study. Noon of the day of vaginal plugs was designated as embryonic day 0.5 (E0.5).
Hematoxylin and eosin (H&E) staining, Nissl staining, immunofluorescent staining, and immunohistochemical staining
For H&E staining, paraffin sections on slides were dewaxed in xylene for 3 min for three times, and rehydrated in 100% ethanol three times, 95% and 70% ethanol once, with 1 min each. The slides were rinsed in the distilled water, and were stained in hematoxylin solution for 30 sec. Then the slides were washed in running tap water for 2 min. The slides were immersed in acid alcohol for decolorizing, and then rinsed in distilled water. The slides were immersed in Lithium Carbonate solution, and also washed in distilled water. The slides were counterstained in eosin solution for 10 sec. And then, the slides were dehydrated with 95% ethanol once, and 100% ethanol three times with 1 min each. The slides were cleared in xylene twice with 2 min each. The slides were mounted in fume hood. Nissl staining was conducted with 0.1% Cresyl Violet for 10 minutes.
For immunofluorescence staining, the slides were dewaxed and rehydrated the same as H&E staining. Then the slides were washed in distilled water and 1X phosphate buffer solution (PBS) with 1 min each. The slides were treated with boiling 1X antigen retrieval solution (DAKO) for 15 min. After cooling down to room temperature (RT), the slides were rinsed with 1XPBS for 10 min for 3 times. The slides were treated with 3% H2O2 in 1XPBS for 30 min, and then washed with 1XPBS for 10 min for 3 times. The sections were blocked with buffer containing 1% BSA, 5% Serum in 1XPBS for 1 h at RT, and then incubated with primary antibody in hybridization buffer overnight at 4 °C. After being washed with 1XPBS for 10 min for 3 times, sections were incubated with secondary antibody for 1 h at RT. The sections were washed and counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). After wash, the slides were mounted with mounting medium. In case, TSA kit (Invitrogen) was used. The samples were treated the same as the regular immunofluorescence staining till the completion of the primary antibody incubation. And then the processes were carried out with TSA kit by following the manufactory’s protocol.
For immunohistochemical staining, the sections were treated the same as the immunofluorescence staining till the incubation with the second antibody. The slides were incubated with biotinylated secondary antibody for 1 h at RT. After being washed in 1XPBS for 5 min for 3 times, the slides were incubated with ABC Reagent (Vectorlabs) for 30 min. After being washed in 1XPBS for 5 min for 3 times, the slides were incubated with fresh prepared DAB substrate solution (Vectorlabs). The reaction was terminated till desire signals observed. The slides were counterstained with hematoxylin solution, and then dehydrated, cleared and mounted as described in the H&E staining.
The following primary antibodies were used in the study: rabbit anti-AVP (1:4000, PenisulaLab), rabbit anti-BDNF (1:100, Santa Cruz), rabbit anti-Brn2 (1:400, Santa Cruz), rabbit anti-cleaved-Caspase3 (1:400, Cell Signaling), mouse anti-COUP-TFII (1:500, R&D), goat anti-β-galactosidase (LacZ) (1:400, Biogenesis), rabbit anti-Ki67 (1:400, BD Biosciences), rabbit anti-MCH (1:800, Phoenix Pharm), rabbit anti-Orexin B (1:800, PenisulaLab). The following secondary antibodies were used in the study: donkey anti-mouse IgG biotin-conjugated (1:400, JacksonImmuno); donkey anti-rabbit IgG biotin-conjugated (1:400, Jacksonimmuno); donkey anti-goat IgG biotin-conjugated (1:400, JacksonImmuno); donkey anti-rabbit IgG Alexa-594 (1:400, Invitrogen); donkey anti-goat IgG Alexa-594 (1:400, Invitrogen).
RNA isolation and quantitative real-time PCR
Total RNAs were prepared from the ventral forebrain of the control and the mutant embryos at E14.5 respectively with TRIzol Reagent (Invitrogen) by following the manufactory’s protocol. Transverse-transcription PCR and quantitative real-time PCR assays were carried out as described previously39,60. The universal probe library (Roche) was used in the study. A student’s t-test was used to compare the means of the relative mRNA levels between the control group and the mutant group. Primer sequences and probes are, Avp-f, 5′-ctacgctctccgcttgtttc-3′, Avp-r, 5′-gggcagttctggaagtagca-3′, Probe #40; Arnt2-f, 5′-aaacgcataccccagtcttg-3′, Arnt2-r, 5′-cgccactctgtccactctc-3′, Probe #109; Bdnf-f, 5′-agtctccaggacagcaaagc-3′, Bdnf-r, 5′-tgcaaccgaagtatgaaataacc-3′, Probe #31; Brn2-f, 5′-catcagtggaactagatggacct-3′, Brn2-r, 5′-cttttctgaaggtcccaggtt-3′, Probe #53; Cntf-f, 5′-gacctgactgctcttatggaatct-3′, Cntf-r, 5′-gcctggaggttctcttgga-3′, Probe #13; Coup-tfI-f, 5′-caaagccatcgtgctattca-3′, Coup-tfI-r, 5′-cctgcaggctttcgatgt-3′, Probe #89; Coup-tfII-f, 5′-cctcaaagtgggcatgagac-3′, Coup-tfII-r, 5′-tgggtaggctgggtaggag-3′, Probe #36; Crh-f, 5′-gaggcatcctgagagaagtcc-3′, Crh-r, 5′-tgttaggggcgctctcttc-3′, Probe #34; Nrp1-f, 5′-ccacacacagtgggcttg-3′, Nrp1-r, 5′-ggtccagctgtaggtgcttc-3′, Probe #26; Nrp2-f, 5′-ggcttctccgcacgttacta-3′, Nrp2-r, 5′-aaagggacattgcactgaaaa-3′, Probe #92; Ot-f, 5′-cacctacagcggatctcagac-3′, Ot-r, 5′-cgaggtcagagccagtaagc-3′, Probe #27; Otp-f, 5′-ccagcacagctcaacgaa-3′, Otp-r,5′-tgaagatgtcggggtagtga-3′, Probe #55; Sim1-f, 5′-actcggctctcatctactcca-3′, Sim1-r, 5′-tgaaatgtacatgatcttcccatc-3′, Probe #49; Trh-f, 5′-tgcagagtctccaccttgc-3′, Trh-r, 5′-ggggataccagttagcacga-3′, Probe #21.
Chromatin immunoprecipitation (ChIP) and real-time quantitative PCR (qPCR)
Chromatins were prepared from the hypothalamus of mouse embryos at E14.5. ChIP assays were carried out with an EZ ChIP Chromatin Immunoprecipitation Kit (Millipore) by following the manufactory’s protocol. The following qPCR assays were performed as described previously39. Mouse anti-COUP-TFII antibody, rabbit anti-Sp1 antibody (Millipore), normal mouse IgG and normal rabbit IgG were used in the study. Primer sequences are, a, 5′-aagccgcaaagaaggtaagc-3′; b, 5′-ctccacttcctcctctccac-3′; c, 5′-ctgtaccccaagacctctga-3′; d, 5′-ccatccatgaattagccagca-3′.
Electronic supplementary material
Acknowledgements
This work was supported by National Natural Science Foundation of China [81360124, 31671508] to K.T.; and in part by the “Strategic Priority Research Program” of the Chinese Academy of Sciences [Grant No. XDA01010201]; and National Key Basic Research and Development Program of China [2014CB964804, 2015CB964500]; and National Natural Science Foundation of China [31430058, 31571513, 31630043, 91519314, 31661143042] to N.J.
Author Contributions
S.F., C.X. and T.S. designed and conducted the experiments. S.F., N.J. and K.T. organized and wrote the manuscript. S.F., C.X., T.S., Y.Q., J.C., and R.W. collected and analyzed the data. J.Y.L., Z.L., X.Y., and S.A. assisted in conducting the experiments and analyzed the data. J.S.L., N.J. and K.T. conceived the project and approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Su Feng, Can Xing and Tingyu Shen contributed equally to this work.
A correction to this article is available online at https://doi.org/10.1038/s41598-018-25537-y.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05682-6
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
5/11/2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
Contributor Information
Naihe Jing, Email: njing@sibcb.ac.cn.
Ke Tang, Email: ktang.sc@gmail.com.
References
- 1.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 2.Morton GJ, Meek TH, Schwartz MW. Neurobiology of food intake in health and disease. Nature reviews. Neuroscience. 2014;15:367–378. doi: 10.1038/nrn3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gautron L, Elmquist JK, Williams KW. Neural control of energy balance: translating circuits to therapies. Cell. 2015;161:133–145. doi: 10.1016/j.cell.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nature medicine. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- 5.Ollmann MM, et al. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 6.Graham M, Shutter JR, Sarmiento U, Sarosi I, Stark KL. Overexpression of Agrt leads to obesity in transgenic mice. Nature genetics. 1997;17:273–274. doi: 10.1038/ng1197-273. [DOI] [PubMed] [Google Scholar]
- 7.Huszar D, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/S0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 8.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 9.Balthasar N, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 10.Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. Nature. 2012;488:172–177. doi: 10.1038/nature11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- 12.Sakurai, T. et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell92, 1 page following 696 (1998). [DOI] [PubMed]
- 13.Hara J, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/S0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- 14.Scott LV, Dinan TG. Vasopressin and the regulation of hypothalamic-pituitary-adrenal axis function: implications for the pathophysiology of depression. Life sciences. 1998;62:1985–1998. doi: 10.1016/S0024-3205(98)00027-7. [DOI] [PubMed] [Google Scholar]
- 15.Cunningham ET, Jr., Sawchenko PE. Reflex control of magnocellular vasopressin and oxytocin secretion. Trends in neurosciences. 1991;14:406–411. doi: 10.1016/0166-2236(91)90032-P. [DOI] [PubMed] [Google Scholar]
- 16.Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annual review of neuroscience. 1983;6:269–324. doi: 10.1146/annurev.ne.06.030183.001413. [DOI] [PubMed] [Google Scholar]
- 17.Nakai S, et al. The POU domain transcription factor Brn-2 is required for the determination of specific neuronal lineages in the hypothalamus of the mouse. Genes & development. 1995;9:3109–3121. doi: 10.1101/gad.9.24.3109. [DOI] [PubMed] [Google Scholar]
- 18.Schonemann MD, et al. Development and survival of the endocrine hypothalamus and posterior pituitary gland requires the neuronal POU domain factor Brn-2. Genes & development. 1995;9:3122–3135. doi: 10.1101/gad.9.24.3122. [DOI] [PubMed] [Google Scholar]
- 19.Michaud JL, Rosenquist T, May NR, Fan CM. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes & development. 1998;12:3264–3275. doi: 10.1101/gad.12.20.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Acampora D, et al. Progressive impairment of developing neuroendocrine cell lineages in the hypothalamus of mice lacking the Orthopedia gene. Genes & development. 1999;13:2787–2800. doi: 10.1101/gad.13.21.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosoya T, et al. Defective development of secretory neurones in the hypothalamus of Arnt2-knockout mice. Genes to cells: devoted to molecular & cellular mechanisms. 2001;6:361–374. doi: 10.1046/j.1365-2443.2001.00421.x. [DOI] [PubMed] [Google Scholar]
- 22.Michaud JL, DeRossi C, May NR, Holdener BC, Fan CM. ARNT2 acts as the dimerization partner of SIM1 for the development of the hypothalamus. Mechanisms of development. 2000;90:253–261. doi: 10.1016/S0925-4773(99)00328-7. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Lufkin T. The murine Otp homeobox gene plays an essential role in the specification of neuronal cell lineages in the developing hypothalamus. Developmental biology. 2000;227:432–449. doi: 10.1006/dbio.2000.9902. [DOI] [PubMed] [Google Scholar]
- 24.Keith B, Adelman DM, Simon MC. Targeted mutation of the murine arylhydrocarbon receptor nuclear translocator 2 (Arnt2) gene reveals partial redundancy with Arnt. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6692–6697. doi: 10.1073/pnas.121494298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura E, et al. 5.78 Mb terminal deletion of chromosome 15q in a girl, evaluation of NR2F2 as candidate gene for congenital heart defects. European journal of medical genetics. 2011;54:354–356. doi: 10.1016/j.ejmg.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Rudaks LI, Nicholl JK, Bratkovic D, Barnett CP. Short stature due to 15q26 microdeletion involving IGF1R: report of an additional case and review of the literature. American journal of medical genetics. Part A. 2011;155A:3139–3143. doi: 10.1002/ajmg.a.34310. [DOI] [PubMed] [Google Scholar]
- 27.Slavotinek AM, et al. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1-15q26.2. European journal of human genetics: EJHG. 2006;14:999–1008. doi: 10.1038/sj.ejhg.5201652. [DOI] [PubMed] [Google Scholar]
- 28.Klaassens M, et al. Congenital diaphragmatic hernia and chromosome 15q26: determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. American journal of human genetics. 2005;76:877–882. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biggio JR, Jr., Descartes MD, Carroll AJ, Holt RL. Congenital diaphragmatic hernia: is 15q26.1-26.2 a candidate locus? American journal of medical genetics. Part A. 2004;126A:183–185. doi: 10.1002/ajmg.a.20464. [DOI] [PubMed] [Google Scholar]
- 30.Arrington CB, et al. A family-based paradigm to identify candidate chromosomal regions for isolated congenital diaphragmatic hernia. American journal of medical genetics. Part A. 2012;158A:3137–3147. doi: 10.1002/ajmg.a.35664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roback EW, et al. An infant with deletion of the distal long arm of chromosome 15 (q26.1—qter) and loss of insulin-like growth factor 1 receptor gene. American journal of medical genetics. 1991;38:74–79. doi: 10.1002/ajmg.1320380117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai SY, Tsai MJ. Chick ovalbumin upstream promoter-transcription factors (COUP-TFs): coming of age. Endocrine reviews. 1997;18:229–240. doi: 10.1210/edrv.18.2.0294. [DOI] [PubMed] [Google Scholar]
- 33.High FA, et al. De novo frameshift mutation in COUP-TFII (NR2F2) in human congenital diaphragmatic hernia. American journal of medical genetics. Part A. 2016;170:2457–2461. doi: 10.1002/ajmg.a.37830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kantarci S, Donahoe PK. Congenital diaphragmatic hernia (CDH) etiology as revealed by pathway genetics. American journal of medical genetics. Part C, Seminars in medical genetics. 2007;145C:217–226. doi: 10.1002/ajmg.c.30132. [DOI] [PubMed] [Google Scholar]
- 35.Al Turki S, et al. Rare variants in NR2F2 cause congenital heart defects in humans. American journal of human genetics. 2014;94:574–585. doi: 10.1016/j.ajhg.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes & development. 1999;13:1037–1049. doi: 10.1101/gad.13.8.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You LR, et al. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16351–16356. doi: 10.1073/pnas.0507832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swindell EC, et al. Rx-Cre, a tool for inactivation of gene expression in the developing retina. Genesis. 2006;44:361–363. doi: 10.1002/dvg.20225. [DOI] [PubMed] [Google Scholar]
- 39.Tang K, Rubenstein JL, Tsai SY, Tsai MJ. COUP-TFII controls amygdala patterning by regulating neuropilin expression. Development. 2012;139:1630–1639. doi: 10.1242/dev.075564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muller EE, Locatelli V, Cocchi D. Neuroendocrine control of growth hormone secretion. Physiological reviews. 1999;79:511–607. doi: 10.1152/physrev.1999.79.2.511. [DOI] [PubMed] [Google Scholar]
- 41.Kappeler L, et al. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS biology. 2008;6:e254. doi: 10.1371/journal.pbio.0060254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kappeler L, et al. Early postnatal nutrition determines somatotropic function in mice. Endocrinology. 2009;150:314–323. doi: 10.1210/en.2008-0981. [DOI] [PubMed] [Google Scholar]
- 43.Sabra-Makke L, et al. Hypothalamic ventromedial COUP-TFII protects against hypoglycemia-associated autonomic failure. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4333–4338. doi: 10.1073/pnas.1219262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Enderlin S, Norman AW, Celio MR. Ontogeny of the calcium binding protein calbindin D-28k in the rat nervous system. Anatomy and embryology. 1987;177:15–28. doi: 10.1007/BF00325286. [DOI] [PubMed] [Google Scholar]
- 45.Alvarez-Bolado G, Rosenfeld MG, Swanson LW. Model of forebrain regionalization based on spatiotemporal patterns of POU-III homeobox gene expression, birthdates, and morphological features. The Journal of comparative neurology. 1995;355:237–295. doi: 10.1002/cne.903550207. [DOI] [PubMed] [Google Scholar]
- 46.Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nature reviews. Neuroscience. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 47.Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nature reviews. Neuroscience. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- 48.Ip NY, Yancopoulos GD. The neurotrophins and CNTF: two families of collaborative neurotrophic factors. Annual review of neuroscience. 1996;19:491–515. doi: 10.1146/annurev.ne.19.030196.002423. [DOI] [PubMed] [Google Scholar]
- 49.Xu B, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nature neuroscience. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pipaon C, Tsai SY, Tsai MJ. COUP-TF upregulates NGFI-A gene expression through an Sp1 binding site. Molecular and cellular biology. 1999;19:2734–2745. doi: 10.1128/MCB.19.4.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang K, Tsai SY, Tsai MJ. COUP-TFs and eye development. Biochimica et biophysica acta. 2015;1849:201–209. doi: 10.1016/j.bbagrm.2014.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galabov P, Schiebler TH. The ultrastructure of the developing neural lobe. Cell and tissue research. 1978;189:313–329. doi: 10.1007/BF00209280. [DOI] [PubMed] [Google Scholar]
- 53.Abuzzahab MJ, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. The New England journal of medicine. 2003;349:2211–2222. doi: 10.1056/NEJMoa010107. [DOI] [PubMed] [Google Scholar]
- 54.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 55.Holzenberger M, et al. A targeted partial invalidation of the insulin-like growth factor I receptor gene in mice causes a postnatal growth deficit. Endocrinology. 2000;141:2557–2566. doi: 10.1210/endo.141.7.7550. [DOI] [PubMed] [Google Scholar]
- 56.Markakis EA. Development of the neuroendocrine hypothalamus. Frontiers in neuroendocrinology. 2002;23:257–291. doi: 10.1016/S0091-3022(02)00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takamoto N, et al. Haploinsufficiency of chicken ovalbumin upstream promoter transcription factor II in female reproduction. Molecular endocrinology. 2005;19:2299–2308. doi: 10.1210/me.2005-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewin GR, Barde YA. Physiology of the neurotrophins. Annual review of neuroscience. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 59.Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang K, et al. COUP-TFs regulate eye development by controlling factors essential for optic vesicle morphogenesis. Development. 2010;137:725–734. doi: 10.1242/dev.040568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang, X. F., S., Tang, K. COUP-TF genes, human diseases, and the development of the central nervous system in murine models. Current Topics in Developmental Biology, doi:10.1016/bs.ctdb.2016.12.002 (2017). [DOI] [PubMed]
- 62.Wu SP, et al. Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nature communications. 2015;6:8245. doi: 10.1038/ncomms9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin J, et al. COUP-TFII inhibits TGF-beta-induced growth barrier to promote prostate tumorigenesis. Nature. 2013;493:236–240. doi: 10.1038/nature11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.