
Figure 2.
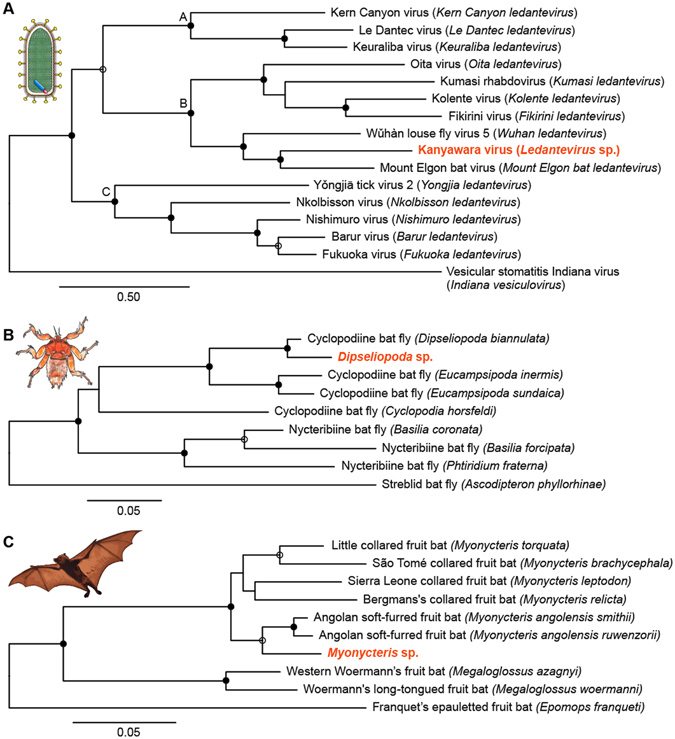
Maximum likelihood phylogenetic trees of rhabdoviruses (A), bat flies (B), and pteropodid bats (C). The rhabdovirus phylogeny is based on concatenated codon-based alignments (8,256 positions) of nucleotide sequences of the canonical rhabdovirus nucleoprotein (N), phosphoprotein (P), matrix (M), glycoprotein (G), and RNA-dependent RNA polymerase (L) genes of 15 viruses of the subgroups A–C of the genus Ledantevirus with vesicular stomatitis Indiana virus (genus Vesiculovirus) as the outgroup. The bat fly phylogeny is based on concatenated codon-based alignments (876 positions) of mitochondrial cytochrome oxidase subunit II (COII) and cytochrome B (CYTB) nucleotide sequences of eight nycteribiids (subfamilies Cyclopodiinae and Nycteribiinae) with the streblid Ascodipteron phyllorhinae as the outgroup. The bat phylogeny is based on concatenated codon-based alignments (1,820 positions) of mitochondrial cytochrome C subunit I (COI) and CYTB nucleotide sequences of nine pteropodids of the subfamily Epomophorinae, with eight of them belonging to the tribe Myonycterini and Franquet’s epauletted fruit bats (tribe Epomophorini) as the outgroup. Circles on nodes indicate statistical confidence based on 1,000 bootstrap replicates of the data (closed circles = 100%; open circles ≥75%); scale bars indicate nucleotide substitutions per site.