

Abstract
Key points
This study represents a first step toward predicting mechanisms of sex‐based arrhythmias that may lead to important developments in risk stratification and may inform future drug design and screening.
We undertook simulations to reveal the conditions (i.e. pacing, drugs, sympathetic stimulation) required for triggering and sustaining reentrant arrhythmias.
Using the recently solved cryo‐EM structure for the Eag‐family channel as a template, we revealed potential interactions of oestrogen with the pore loop hERG mutation (G604S).
Molecular models suggest that oestrogen and dofetilide blockade can concur simultaneously in the hERG channel pore.
Abstract
Female sex is a risk factor for inherited and acquired long‐QT associated torsade de pointes (TdP) arrhythmias, and sympathetic discharge is a major factor in triggering TdP in female long‐QT syndrome patients. We used a combined experimental and computational approach to predict ‘the perfect storm’ of hormone concentration, I Kr block and sympathetic stimulation that induces arrhythmia in females with inherited and acquired long‐QT. More specifically, we developed mathematical models of acquired and inherited long‐QT syndrome in male and female ventricular human myocytes by combining effects of a hormone and a hERG blocker, dofetilide, or hERG mutations. These ‘male’ and ‘female’ model myocytes and tissues then were used to predict how various sex‐based differences underlie arrhythmia risk in the setting of acute sympathetic nervous system discharge. The model predicted increased risk for arrhythmia in females when acute sympathetic nervous system discharge was applied in the settings of both inherited and acquired long‐QT syndrome. Females were predicted to have protection from arrhythmia induction when progesterone is high. Males were protected by the presence of testosterone. Structural modelling points towards two plausible and distinct mechanisms of oestrogen action enhancing torsadogenic effects: oestradiol interaction with hERG mutations in the pore loop containing G604 or with common TdP‐related blockers in the intra‐cavity binding site. Our study presents findings that constitute the first evidence linking structure to function mechanisms underlying female dominance of arousal‐induced arrhythmias.
Keywords: arrhythmias, computational model, dofetilide, hERG mutation, long‐QT, sex‐differences, TdP
Key points
This study represents a first step toward predicting mechanisms of sex‐based arrhythmias that may lead to important developments in risk stratification and may inform future drug design and screening.
We undertook simulations to reveal the conditions (i.e. pacing, drugs, sympathetic stimulation) required for triggering and sustaining reentrant arrhythmias.
Using the recently solved cryo‐EM structure for the Eag‐family channel as a template, we revealed potential interactions of oestrogen with the pore loop hERG mutation (G604S).
Molecular models suggest that oestrogen and dofetilide blockade can concur simultaneously in the hERG channel pore.
Abbreviations
- APD
action potential duration
- BCL
basic cycle length
- DHT
dihydrotestosterone
- E2
oestradiol
- EAD
early afterdepolarization
- hERG
human ether‐à‐go‐go‐related gene
- IKs
slow delayed rectifier K+ current
- ICa‐L
inward Ca2+ current,
- ISO
isoproterenol
- MD
molecular dynamics
- OA
okadaic acid
- Pg
progesterone
- PKA
protein kinase A
- QTc
corrected QT
- SNS
sympathetic nervous system
- TdP
torsade de pointes
Introduction
Cardiac arrhythmia is a primary cause of mortality in both males and females, but the risk factors associated with some types of arrhythmias differ between the sexes. Female sex is the dominant risk factor for acquired long‐QT‐dependent arrhythmias after puberty, with at least 70% incidence in females (Makkar et al. 1993). Despite recognition of this fact, the specific mechanisms underlying sex‐based differences in humans are notably understudied and have not been comprehensively revealed (Tadros et al. 2014; Salama & Bett, 2014). Clinical and experimental data uniformly agree that women have longer QT intervals than men and are more likely to develop long‐QT‐dependent arrhythmias such as torsade de pointes (TdP) (Lehmann et al. 1996; Pham et al. 2001; Rodriguez et al. 2001; Pham et al. 2002; Salama & Bett, 2014) that can degenerate into sudden cardiac death. Women are also susceptible to acquired long‐QT syndrome, resulting from block of the cardiac repolarizing rapidly activating delayed rectifier K+ current (I Kr), through the human ether‐à‐go‐go‐related gene (hERG) channel arising from the KCNH2 gene.
The longer QT interval observed in women is attributed to a reduced repolarization reserve (James et al. 2007). Recent clinical and experimental studies suggest that this is due to the non‐genomic effects that sex steroid hormones have on the electrical properties of the heart (Burke et al. 1997; Rodriguez et al. 2001; Bai et al. 2005; Liu et al. 2005; Nakagawa et al. 2006; Nakamura et al. 2007; Kurokawa et al. 2008, 2009; Sims et al. 2008; Yang et al. 2012; Salama & Bett, 2014; Anneken et al. 2016). Oestrogen and progesterone interact acutely with multiple subcellular targets, altering the resultant emergent electrical activity at the level of cells and tissues (Nakamura et al. 2007; Yang et al. 2010; Kurokawa et al. 2015). During the menstrual follicular phase (prior to ovulation), oestrogen level is the highest, QT interval is longer and susceptibility to drug‐induced arrhythmias is increased. In the luteal phase (following ovulation) progesterone is increased and arrhythmic events associated with acquired and inherited long‐QT syndrome are reduced (Burke et al. 1997; Rodriguez et al. 2001; Nakagawa et al. 2006; Anneken et al. 2016).
The differences in arrhythmia vulnerability during discrete stages of the menstrual cycle are likely to reflect the effects that sex steroid hormones have on cardiac ion channels (Kurokawa et al. 2008; Kurokawa et al. 2009). Oestradiol (E2) has been shown to inhibit I Kr, although the specific interaction sites have not been fully identified (Kurokawa et al. 2008). The result of E2 interaction with hERG is an increase in ventricular action potential duration, consistent with the longer QT interval observed in women during the follicular phase of the menstrual cycle. Progesterone, on the other hand, enhances the slow delayed rectifier K+ current (I Ks), generated by the KvLQT1 channel (encoded by the KCNQ1 gene), through a nitric oxide‐dependent signalling pathway. This causes a decrease in ventricular action potential duration, consistent with the shorter QT interval observed during the luteal phase prolongation (Nakagawa et al. 2006). The effect of progesterone is similar to the effect of the male sex hormone testosterone, which not only enhances I Ks via a NO‐dependent mechanism, but also inhibits the inward Ca2+ current (ICa‐L), both of which contribute to a shortening of the ventricular action potential and the QT interval in males after puberty (Bai et al. 2005).
There are also sex‐based differences in the effects of the autonomic nervous system. Increased sympathetic or decreased parasympathetic stimulation produces a rate‐dependent decrease in ventricular action potential duration (APD), but this effect is greater in men than in women (Magnano et al. 2002; Nakagawa et al. 2005; Hoeker et al. 2014). This correlates with a longer rate‐corrected QT (QTc) interval in women. Changes in the balance of sympathetic and parasympathetic tone contribute to many types of arrhythmias often resulting in sudden cardiac death (Lanfranchi et al. 2010).
Long‐QT syndrome type 2 patients are susceptible to sudden arousal associated with loud noise or emotional stress (Moss et al. 1999; Wilde et al. 1999). A recent population study noted that 82% of the observed arousal‐induced arrhythmias occurred in females (Kim et al. 2010). In order to begin to understand why changes in autonomic tone are more likely to contribute to arousal‐induced arrhythmias in females, we utilized computer models from our previous work that were based on and validated by experimental data to reflect acute actions of physiologically relevant human concentrations of sex steroid hormones progesterone, oestrogen and testosterone (Nakamura et al. 2007; Yang et al. 2010; Yang & Clancy, 2012). Here, we extended and expanded those models, and undertook simulations to reveal the conditions (i.e. pacing, drugs, sympathetic stimulation) required for triggering and sustaining reentrant arrhythmias in females in the setting of acute sympathetic arousal. We also employed a protein structure‐based molecular docking approach to explore the possible interactions between oestrogen, hERG mutations and hERG blocking drugs like dofetilide. Like the hERG blocker ibutilide, which exhibits clear sex‐ and menstrual cycle‐dependent effects (Rodriguez et al. 2001), Pham & Rosen (2002) reported that submicromolar concentrations of dofetilide coupled with physiological oestrogen levels produced a substantial APD90 increase in females compared to males.
The results of this study suggest that specific therapeutic anti‐arrhythmic strategies for women with long‐QT syndrome should be studied. The study also suggests molecular determinants of drugs that might be especially problematic in females. These predictions might inform future drug design and screening.
Methods
Ethics approval
Animal use was approved by the Institutional Animal Care and Use Committee of Tokyo Medical and Dental University, and conformed to the principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology (Grundy, 2015).
For Fig. 1 and Tables 1 and 2, experimental methods have been reported following the MICEE reporting standard (see www.micee.org): type: isolated cardiac ventricular myocytes; sex: male; weight: 251–350 g; species: Hartley guinea pig; supplier: Saitama Experimental Animal Supply Co. Ltd (Saitama, Japan).
Figure 1. Simulated and experimentally recorded I caL data under ISO stimulations.
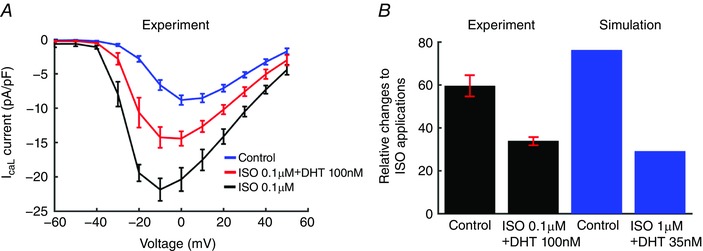
A, effect of DHT (100 nm) on I–V curves from guinea pig ventricular myocytes (n = 5). B, experimental data (error bars indicated by red, n = 5 for each case) compared to simulated relative changes of the peak I CaL to ISO application.
Table 1.
Experimental data for effects of testosterone on I Ca,L for males
| Isoproterenol | I max (pA pF−1) | V 1/2 (mV) | K | n |
|---|---|---|---|---|
| Activation | ||||
| ISO | −24.7 + 1.9 | 4.9 + 0.5 | 5 | |
| ISO + 100 nm DHT | −19.7 ± 2.1 | 5.3 ± 0.7 | ||
| Inactivation | ||||
| ISO | −27.0 ± 3.6 | −26.8 ± 0.6 | 6.3 ± 0.3 | 7 |
| ISO + 100 nm DHT | −19.9 ± 4.5 | −28.9 ± 0.7 | 6.0 ± 0.2 |
Table 2.
Experimental data for effects of DHT with cAMP + OA on I Ca,L current amplitude
| Normalized peak I CaL | |||
|---|---|---|---|
| DHT concentration | Mean (%) | SEM | n |
| 100 nm | 85.6 | 2.2 | 4 |
| 30 nm | 81.3 | 10.6 | 2 |
| 10 nm | 86.7 | 8.3 | 4 |
| 1 nm | 98.8 | 1 | |
Isolation procedure
Adult guinea pigs (white Hartley; 251–350 g) were deeply anaesthetized with intra‐peritoneal injection of sodium pentobarbital (50 mg g−1, i.p.), and then killed by cervical dislocation. Hearts were perfused via the aorta with warm (36 ± 1°C) and oxygenated solutions as follows: (1) Tyrode solution containing (in mmol l−1): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes, pH 7.4, for 5 min; (2) Ca2+‐free Tyrode solution removing CaCl2 from the Tyrode solution; (3) Ca2+‐free Tyrode solution containing collagenase (120 units ml−1) and albumin (1 mg ml−1), for 18 min; and (4) low‐Ca2+ Tyrode solution with 0.2 mmol l−1 CaCl2, for 5 min. At the end of the perfusion, the ventricles were minced in No. 4 solution to release single cells. Only the quiescent myocytes with clear striations were used for this study.
Electrophysiological recordings
For Fig. 1. L‐type Ca2+ channel currents (I Ca,L) were recorded with the whole‐cell configuration of the patch‐clamp technique with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA). Signals were low‐pass filtered at 5 kHz, sampled at 5 kHz, and compensated for cell capacitance (no series resistance compensation). The pCLAMP software (version 9.0, Molecular Devices) was used to generate voltage‐pulse protocols, and for acquisition and analysis of data. The series resistance was 7.2 ± 0.6 MΩ, the capacitance time constant was 2.2 ± 0.3 ms, and the membrane capacitance was 139.3 ± 7.4 pF (n = 22). All experiments were performed at 36 ± 1°C. Pipette solution contained (in mmol l−1): 130 CsCl, 2 MgCl2, 5 adenosine‐5′‐triphosphate disodium salt, 10 Hepes, 20 TEA‐Cl, 10 EGTA (pH 7.25 adjusted with CsOH). External (bath) solution contained (in mmol l−1): 135 NaCl, 5.4 CsCl, 0.53 MgCl2, 2 CaCl2, 5.5 glucose and 5 Hepes (pH 7.4 adjusted with CsOH).
Effects of dihydrotestosterone (DHT) were obtained under sympathetic nervous system (SNS) activation by 10 min dialysis of 3′,5′‐cyclic adenosine monophosphate (cAMP; 0.2 mmol l−1) and okadaic acid (OA; 0.2 μmol l−1) or by 3 min external application of (−)‐isoproterenol (ISO) hydrochloride (0.1 μmol l−1).
To monitor peak inward I Ca,L, a pre‐pulse (100 ms) was applied to –40 mV from a V h of –80 mV to inactivate the Na+ channels and the T‐type Ca2+ channels, followed by a 200 ms V t change to 0 mV at 1 Hz. To obtain I–V relationships for measurement of I Ca,L activation, V t was changed from −60 mV to 50 mV in 10 mV increments. To generate steady state inactivation curves, long pre‐pulses (500 ms) to different voltages (−50 to 20 mV) were applied to allow inactivation to reach a steady state. Then the voltage was set to −40 mV for 10 ms to close the channels, after which a V t change to 10 mV for 200 ms was applied to assess the state of inactivation of the channels.
Model development
We recently developed ‘male’ and ‘female’ computational model representations of human ventricular cardiac myocytes that include genomic‐based differences in expression of key cardiac channels with human physiological concentrations of sex hormones on human action potentials (O'Hara et al. 2011; Yang & Clancy, 2012). The genomic differences were based on measured expression levels of key cardiac ion channel genes as well as connexin43 (underlying the dominant gap‐junction subunit) in epicardial and endocardial ventricular tissue from non‐diseased explanted male and female human hearts (Gaborit et al. 2010). We utilized these data (in construction of ‘male’ and ‘female’ human heart cell computational models by scaling the conductances of the corresponding currents in the O'Hara–Rudy human ventricular cell model (O'Hara et al. 2011) as described in Yang & Clancy (2012) (see also Appendix Table A1). Included in the male model based on new data (shown in Tables 1 and 2 and Fig. 1) is the effect of the acute application of a physiological concentration of testosterone (dihydrotestosterone, DHT) 35 nm, reflecting normal high ranges in post‐pubescent pre‐senescent males (Dorgan et al. 2002).
Simulation of acute sex steroid hormone effects
Three distinct female models were developed that include the hormones 17β‐oestradiol (E2) (Munro et al. 1991; Dighe et al. 2005) and progesterone (Pg) (Munro et al. 1991; Janse de Jonge et al. 2001) during the early follicular, late follicular and luteal phases of the menstrual cycle. The hormone concentrations used in the model simulations and their specific sources are as shown in Table 3.
Table 3.
The female hormone concentrations used in the computational model
| Menstrual cycle | 17β‐Oestradiol | Progesterone |
|---|---|---|
| Early follicular stage | 0.1 nm | 2.5 nm |
| Late follicular stage | 1.0 nm | 2.5 nm |
| Luteal stage | 0.7 nm | 40.6 nm |
The E2 reference ranges are from Munro et al. (1991) and Dighe et al. (2005). The Pg reference ranges (used in our initial study; Nakamura et al. 2007) are from Janse de Jonge et al. (2001) and Munro et al. (1991).
Simulating the effects of dofetilide
An I Kr Markov model (Romero et al. 2014) was incorporated into the O'Hara–Rudy human ventricular action potential (AP) model (O'Hara et al. 2011) to simulate the effects of dofetilide on I Kr.
Simulated G604S mutation
Experiments suggest that the G604S hERG pore loop mutation alters the current amplitude and the gating of the wild‐type (WT) hERG channel. We simulated the mutant I Kr (heterozygous channel: 50% each of WT and G604S) by modifying the conductance of WT current and also the kinetics of WT channel from the O'Hara–Rudy human model (O'Hara et al. 2011) based on the experimental data shown in Table 4.
Table 4.
G604S mutant values
| WT | Mutation | |
|---|---|---|
| G Kr | 0.046 | 0.0147 |
| R kr | 1.0/{1.0 + exp[(v + 55.0)/75.0]} × 1.0/{1.0 + exp[(v − 10.0)/30.0]} | 1.0/{1.0 + exp[(v + 55.0 + 9)/75.0]} × 1.0/{1.0 + exp[(v − 10.0)/30.0]} |
| Reference | O'Hara et al. (2011) | Huo et al. (2008) |
v is membrane potential (mV).
Simulation of sympathetic nervous system: protein kinase A effects
We simulated the effects of 1 μm ISO on I caL, I ks, I kb, I Na, J rel, J up, troponin and I Nak, according to O'Hara & Rudy (2012). In addition, progesterone and testosterone affect the conductance of I Ks, but have no distinguishable effects on its kinetics under SNS stimulations. To model the effects of progesterone and testosterone on I Ks, we modified G Ks by scaling factors as indicated by the experimental data. In order to not overestimate the combined effects of DHT and SNS on I Ks, we assumed that the combined effects reached a saturating level that is less than an additive effect, as was shown for progesterone. If the combination were additive, we would expect even more protection by testosterone in the setting of SNS.
Testosterone and SNS stimulation effects on I Ks, I CaL and the kinetics of the latter are shown in Table 5. DHT application during SNS stimulation affects the kinetics of I Ca,L: the activation curve is less steep and is shifted to depolarized potentials compared to baseline (half‐maximal activation shifted ΔV 1/2 = 5 mV and slope factor Δk = 0.4 for DHT 100 nmol l−1). The inactivation curve is shifted in the hyperpolarized direction and becomes steeper compared to baseline (half‐maximal inactivation shifted ΔV 1/2 = −2.1 mV and slope factor Δk = −0.3).
Table 5.
Simulated effects of testosterone (DHT) on I Ks and I Ca,L
| Channel | DHT | DHT (SNS stimulation) | |
|---|---|---|---|
| 10 nm | 35 nm | 35 nm | |
| I Ks | 1.38 | 1.4 | 3.52 |
| I CaL | 0.94 | 0.82 | 2.05 |
| Kinetics of I CaL | No effects |
|
|
| Reference | Bai et al. (2005) | Table 1 | |
v is membrane potential (mV).
In the simulations, we shifted the I Ca,L activation and inactivation curves by the same amount as above experimental data suggested to account for the different dosages of progesterone (see Table A2). Also the experimentally observed I Ca,L current reduction factor is 0.82 for [DHT] = 35 nm (Table 5). We used experimental data from 100 nm of DHT because 35 nm is a maximally stimulating dose of DHT on I Ca,L (Table 5). We then multiplied the scaling factors for I Ks and I Ca,L when DHT is applied (see Table 5). Simulated results are in good agreement with experimental data shown in Fig. 1. The effects of progesterone on I Ca,L and I Ks are shown in Appendix Table A2. Note that the ISO dose (0.1 μm) used in the experiments is a maximally stimulating dose.
Pacing protocol for arousal arrhythmia conditions
Cells were paced for 400 beats at basic cycle length (BCL) 1200 ms with no protein kinase A (PKA) effects, and followed by 10 beats (BCL = 800 ms) with PKA application.
Transmural tissue simulations
We simulated a transmural fibre composed of 360 ventricular cells (Δx = Δy = 100 μm) connected by resistances to simulate gap junctions (Faber & Rudy, 2000). The fibre contains an endocardial region (cells 1–160) and epicardial region (cells 161–360), with a linear decrease in APD as indicated by experimental data (Glukhov et al. 2010; Lou et al. 2011). G Kr was used as the index value of endocardium in cell no. 1, and the index value of epicardium in cell no. 360. In the female model, G Kr was monotonically increased from 0.036 to 0.042. In the male model, G Kr was linearly increased from 0.046 to 0.05. AP simulations were carried out in epi‐/endocardial cells by changing various ion channel conductances and gap‐junctions (Yang & Clancy, 2012). The fibre was paced at BCL = 1200 ms for 500 beats and simulated arousal arrhythmia conditions (see above).
We simulated a heterogeneous 2D cardiac tissue composed of 360 by 440 cells with Δx = Δy = 150 μm. The tissue contains an endocardial region (fibres 1–160) and epicardial region (fibres 161–360). Channel conductance and gap‐junction parameters are the same as in the one‐dimensional simulations. Current flow is described by the following equation:
where V is the membrane potential, x and y are distances in the longitudinal and transverse directions, respectively, Dx and Dy are diffusion coefficients in the x and y directions, C m is membrane capacitance (C m = 1), and I stim is 180 mA cm−2 for the first 0.5 ms. We also incorporated anisotropic effects by setting Dx and Dy such that the ratio of conduction velocities is 1:2 (Young & Panfilov, 2010).
The tissue was first paced for 500 beats at BCL = 1000 ms on the entire length of one side of tissue prior to application of SNS, and then the 501th beat was paced on the top left corner in an endocardial region with no PKA effects at BCL = 1000 ms followed by PKA additions in the next beat paced in the same region.
Pseudo‐ECG computation
Extracellular unipolar potentials (Φe) generated by the fibre in an extensive medium of conductivity σe, were computed from the transmembrane potential V m using the integral expression as in Gima & Rudy (2002).
In one dimension:
In two dimensions:
where ∇V is the spatial gradient of V m, a is the radius of the fibre, σi is the intracellular conductivity, σe is the extracellular conductivity, and r is the distance from a source point (x, y) to a field point (x′, y′). Φe was computed at an ‘electrode’ site 2.0 cm away from the distal end along the fibre axis.
Action potential duration mapping
We reconstructed ‘human transmural myocardial’ based on data describing transmural action potential heterogeneity mapped from normal human left ventricle (Glukhov et al. 2010) (Fig. 8 A). First, the O'Hara–Rudy human model was used to generate a G Kr lookup table corresponding to APD80. Next, an experimental 2D APD80 map (100 × 100–Fig. 8 A) was used to create a 2D G Kr map using the G Kr lookup table. Then the two‐dimensional G Kr values (100 × 100) were used to simulate APD80. We paced the female heart at 1 Hz and modified the length of APD80 to match the clinically observed QT intervals ∼400 ms (Stramba‐Badiale et al. 1997; Ebert et al. 1998; Nakagawa et al. 2005). We then constructed a 3D wedge of 100 by 100 by 1 with Δx = Δy = 200 μm and Δz = 500 μm using this APD mapping data. Current flow is described by the following equation:
where V is the membrane potential, Dx, Dy and Dz are diffusion coefficients in the x, y and z directions, and I stim is 150 mA cm−2 for 0.5 ms. We also incorporated anisotropic effects by setting Dx, Dy and Dz such that the ratio of conduction velocities is 2:4:1 (Young & Panfilov, 2010).
Figure 8. In silico 3D reconstructed human long‐QT syndrome type 2 female LV wedge based on Glukhov et al. (2010) paced with SNS applied.
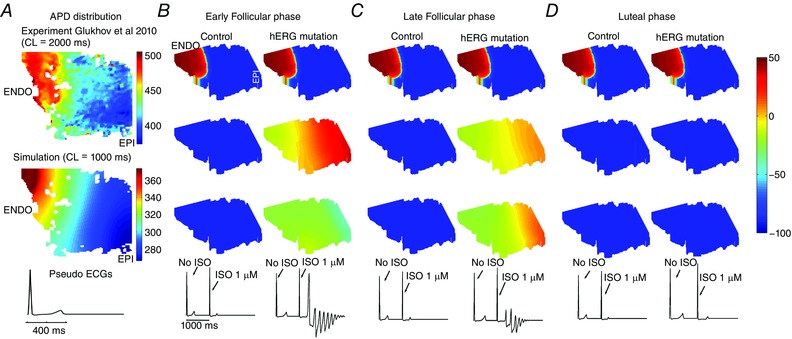
A, reconstruction of a human tissue in silico from normal female explanted heart at a pacing rate of 1 Hz. Experimental data from normal human left ventricle (Glukhov et al. 2010) shown at the top. B–C, simulated tissue and pseudo ECG are shown in the bottom panel. In the early follicular (B) and late follicular (C) phases of the menstrual cycle, SNS application initiates arrhythmia. D, the luteal phase is not predicted to be sensitive to SNS. APD variability is indicated by the colour scale from long APD80 (red) to short (blue).
Structural model development
The SWISS‐MODEL homology modelling program (Kopp & Schwede, 2004) was used for the development of the hERG model from the available Eag1 channel cryo‐EM structure (PDB ID 5K7L), determined at 3.78 Å resolution (Model 1). A multiple sequence alignment was performed using the CLUSTALW algorithm (Thompson et al. 1994). Protein models were generated from the alignment in a stepwise manner. The conserved backbone coordinates of the template were preserved in the final model (Wang et al. 2016). The gap regions in the sequence alignment were modelled either from a loop library or through a conformational space search using constrained element methods. The scoring function used for determining side chain conformations was derived from a backbone‐dependent rotamer library (Benkert et al. 2011) that accounts for favourable interactions, such as hydrogen bonds and disulfide bridges, as well as unfavourable close contacts. A more detailed description of protocols and sequence alignments used for hERG model generation were provided previously (Durdagi et al. 2012; Anwar‐Mohamed et al. 2014; Wang et al. 2016).
Model 1 was used to study all interactions between E2 and extracellular pore loop residues in both WT and G604S mutant. While this model provides a new template for understanding the effects of mutations in this region, it contains a pore domain in the closed state, and the molecular volume available for binding inside the pore is far too small to accommodate any of the common blockers. Therefore, to study the potential interactions between the channel and hERG blockers, we used a previously developed and experimentally validated model of the hERG pore in the open state (Durdagi et al. 2012; Anwar‐Mohamed et al. 2014) based on the Kv1.2 X‐ray crystal structure (PDB ID 2A79), determined at 2.9 Å resolution (Model 2). It is important to stress that this previously published model of the hERG pore displays remarkable structural conservation with the published Eag1 structure for residues critical for drug binding (T623, S624, Y652 and F656), with major differences in the pore loop region far away from the common intra‐cavity binding site (Wang et al. 2016). To further refine both Model 1 and Model 2 of the hERG channel, we performed molecular dynamics (MD) simulations with a protocol described below.
Molecular docking and MD simulations
First, the molecular structure of oestradiol (E2) was downloaded from the ZINC database (Irwin et al. 2012) and the partial charges were recalculated using the GAAMP protocol (Huang & Roux, 2013). Next, molecular docking was performed using the Glide‐XP (extra precision) and Induced Fit Docking (IFD) modules of the Maestro suite from Schrödinger (Gabrielsen et al. 2012) for dofetilide and E2 binding to Model 1 and Model 2. A molecular grid of 36 Å × 36 Å × 36 Å was used to map binding pockets on the hERG surface. We followed the recommended protocol for docking of dofetilide and oestradiol including the following steps: (1) constrained minimization of the receptor with a root mean square deviation cutoff of 0.18 Å; (2) initial Glide docking of each ligand using soft potentials; (3) refinement of the derived docking poses (i.e. minimization of the protein receptor site within 20 Å of the ligand‐bound area) with Schrodinger's Prime module; and (4) Glide re‐docking of the protein–ligand complexes. Clustering analysis was used to evaluate the docking poses in both the poor loop (using Model 1), and intracellular cavity site (using Model 2). The top‐scoring pose from each docking simulation was used for MD simulations. We considered five hERG–substrate complexes in total, including hERG–E2 (pore loop WT), hERG–E2 (pore loop G604S), hERG–E2 (cavity), hERG–dofetilide (cavity), and hERG–E2–dofetilide (cavity). CHARMM‐GUI (Jo et al. 2008) was used to prepare protein–dipalmitoylphosphatidylcholine lipid bilayer complexes solvated in 150 mm KCl aqueous solution using CHARMM‐36 force field and TIP3P water model (Jorgensen et al. 1983; MacKerell et al. 1998; Noskov et al. 2004; Noskov & Roux, 2008; Klauda et al. 2010; Best et al. 2012). The remaining parameters for oestradiol were generated with MATCH (Yesselman et al. 2012). The molecular parameters for a neutral form of dofetilide were reported previously (Wang et al. 2016). The fully assembled systems were equilibrated for 10 ns using NAMD2.10 (Phillips et al. 2005), and production runs were then performed for 50 ns for E2 binding to the WT and G604S mutant pore loop site (Model 1). Both dofetilide and E2 binding to the intracellular cavity (Model 2) were studied with 100 ns MD simulations. All production runs were performed under a constant temperature and pressure (NPT) ensemble, with a pressure of 1 atm and temperature of 315.15 K. Long‐range electrostatic interactions were treated by the particle mesh Ewald (PME) algorithm (Essmann et al. 1995). Non‐bonded interactions were switched off at 10–12 Å. The systems were simulated with a time step of 2 fs under periodic orthorhombic boundary conditions. The ensembles of frames obtained from various MD simulations were used for the calculation of binding enthalpies using a previously developed protocol based on continuum solvent model enthalpies (Robertson et al. 2008).
Results
Combined effects of sex, SNS and I Kr channel blocking drugs
Clinical and experimental studies have shown that females are especially susceptible to QT‐interval prolongation by I Kr blocking drugs (Kurokawa et al. 2008). This finding may partially explain why females are more prone to drug‐induced arrhythmias (Makkar et al. 1993; Lehmann et al. 1996). Hence, we tested the combined effects of hormones and the I Kr channel blocker dofetilide on SNS‐induced arrhythmia triggers.
Dofetilide is a prototype of the pro‐arrhythmic class – associated with hERG block, QT prolongation and TdP (Van Opstal et al. 2001). We recently developed a detailed kinetically based model of the hERG blocker dofetilde by extending the consensus five‐state Markov chain model that includes three closed states (C3, C2 and C1), a conducting open state (O) and an inactivation state (I) (Romero et al. 2014). The expanded I Kr model that includes dofetilide interactions was incorporated into the O'Hara–Rudy model of the human ventricular action potential (AP) (O'Hara et al. 2011).
The dofetilide model, described and validated in detail previously (Romero et al. 2014), includes the experimentally observed 70‐fold preferential binding to the inactivated state relative to the open state (Perrin et al. 2008) and mimics the clinically observed 16% prolongation of the QT interval produced by the therapeutic dose 8.22 nm (Demolis et al. 1996). The effects of the female hormones 17β‐oestradiol (E2) and progesterone (Pg), the male hormone dihydrotestosterone (DHT) and sex‐specific genomic‐based differences in expression of key cardiac channels on human action potentials from this study and O'Hara et al. (2011) and Yang & Clancy (2012) were also incorporated to develop female and male models as described in more details in Methods above.
We utilized these male and female models and added the combined effects of dofetilide with acute effects of β‐adrenergic stimulation by isoproterenol to predict the combined effects of (1) genomic‐based differences in expression of key cardiac channels, (2) human physiological concentrations of sex hormones, (3) an inherited long‐QT syndrome and (4) sympathetic arousal that includes an increase in pacing rate.
Simulation of cell‐level effects of sex‐based arousal arrhythmias
Figure 2 (upper panels) shows the predicted effects of dofetilide to generate arrhythmia triggers in single simulated myocytes in the early follicular phase of the menstrual cycle (Fig. 2 A), the late follicular phase (Fig. 2 B), the luteal phase (Fig. 2 C) and in the presence of testosterone (Fig. 2 D). Shown are the predicted effects of sex differences and SNS in the setting of acquired long‐QT syndrome, modelled as the application of dofetilide. Action potentials are shown following pacing for 400 paced beats at a cycle length of 1200 ms in single simulated endocardial cells. The 399th and 400th paced beats are shown, after which SNS was acutely applied and the pacing cycle length increased to 800 ms to mimic SNS effects on heart rate. Upon acute SNS application arrhythmia triggers were observed in single myocytes (upper panels) in both the early follicular phase of the menstrual cycle (Fig. 2 A) and in the late follicular phase (Fig. 2 B). The follicular phases of the menstrual cycle when oestradiol is highest were especially vulnerable. Progesterone was apparently protective during the luteal phase (Fig. 2 C) as was the presence of testosterone (Fig. 2 D). These simulations predict that the female cardiac myocytes in the presence of a hERG blocker exhibit arrhythmogenic rhythms upon SNS. When no drug or SNS was applied, no arrhythmogenic rhythms were observed (Appendix Fig. A1).
Figure 2. Sex‐based differences in human cardiac electrophysiology predicted in the setting of acquired long‐QT syndrome induced by dofetilide.
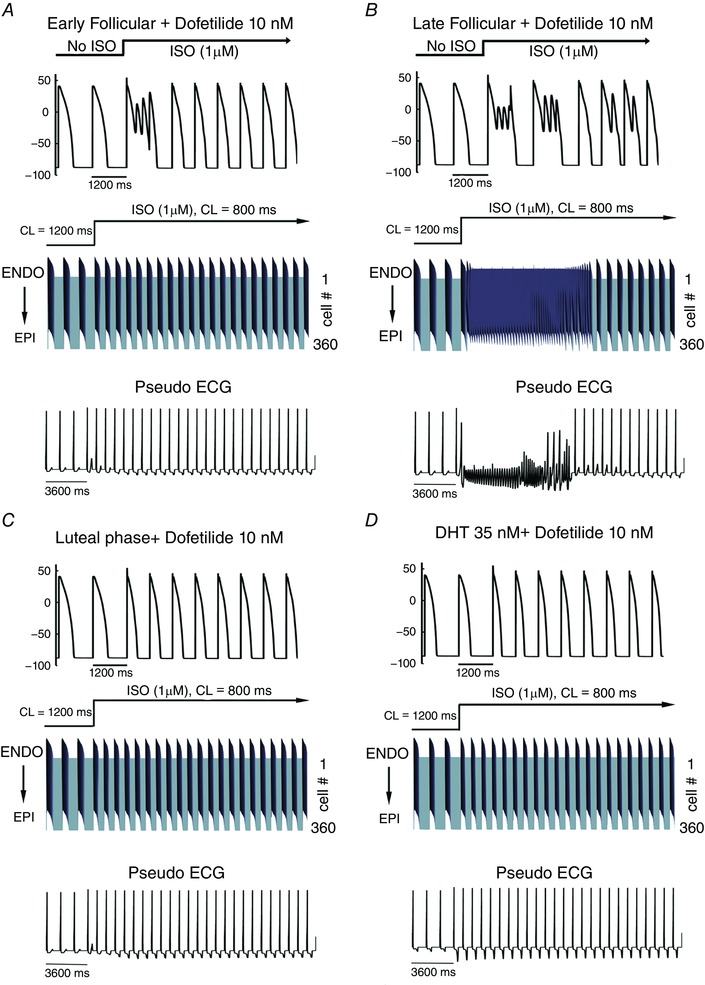
Single cell action potentials were simulated before and during simulated arousal via sympathetic stimulation following pretreatment with dofetilide (top panel A–D). Transmural 1‐dimensional tissue models and pseudo‐ECGs were simulated before and during SNS. The pseudo ECG is shown beneath the tissue simulation in each panel. A, simulated combined effects of oestradiol and progesterone on female tissues during the early follicular phase of the menstrual cycle. B, simulated combined effects of oestradiol and progesterone on female tissues during the late follicular phase of the menstrual cycle. C, simulated combined effects of oestradiol and progesterone on female tissues during the luteal phase of the menstrual cycle. D, simulated effect of testosterone on male tissue during SNS.
Simulation of tissue‐level effects of sex‐based arousal arrhythmias
We also explored the effects of sex‐based differences in the setting of acquired long‐QT syndrome type 2 and acute SNS in a one‐dimensional transmural strand of coupled cells comprising epicardial to endocardial regions (detailed explanation of construction of the tissue is in Methods).
Figure 2 (middle and lower panels) illustrates the predicted sex differences in tissue arrhythmia vulnerability during acquired long‐QT following arousal by SNS. Simulations in one‐dimensional transmural tissue composed of coupled cells (360 cells) are shown in the presence of 10 nm dofetilide during the early follicular phase of the menstrual cycle (Fig. 2 A). Time is shown on the x‐axis and voltage on the z‐axis in the middle panels. The corresponding electrograms are shown in each panel below the space‐time plots. Simulated action potentials (APs) are shown following pacing for 400beats at 1200 ms pacing cycle length. The last 3 beats are shown and then SNS was acutely applied and the cycle length was increased to 800 ms. As shown in Fig. 2 B, the late follicular phases of the menstrual cycle where oestradiol is highest exhibits an arrhythmogenic rhythm triggered by the acute application of SNS. An early afterdepolarization (EAD) initiated in the endocardial region (in the late follicular case shown in Fig. 2 B) generates a sufficient source current to drive full action potential initiation in the downstream excitable (repolarized) epicardial region. The second depolarization during the endocardial EAD results in a self‐sustaining oscillation derived from a spatially discordant alternans that zig‐zags back and forth to re‐excite downstream or upstream cells. The excitatory signal is runaway, as it is a self‐generating oscillation in the form of a one‐dimensional reentry. Both an increase in progesterone during the luteal phase (Fig. 2 C) and testosterone in the male tissue (Fig. 2 D) were predicted to protect against SNS arrhythmia provocation.
We next tested the vulnerability to arrhythmia induced by SNS in models of acquired long‐QT syndrome in simulated human male and female two‐dimensional tissues as shown in Fig. 3 by simulating a heterogeneous cardiac tissue containing an endocardial region (tissue columns 1 to 160) and an epicardial region (tissue columns 161 to 360) based on recordings from human tissue (Glukhov et al. 2010; Lou et al. 2011) also incorporating anisotropic effects as described in detail in Methods above. Following pacing for 500 beats (s1) at BCL = 1000 ms along the entire endocardial length, SNS was then applied with an additional stimulus (s2) after which the simulation ran for 3000 ms without any additional stimulation.
Figure 3. Female sex is predicted to increase susceptibility to reentrant arrhythmia following acute SNS stimulation in the setting of acquired long‐QT syndrome induced by dofetilide application.
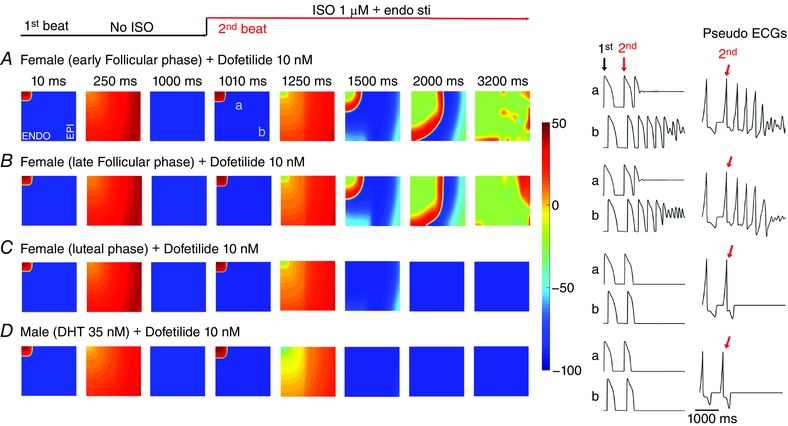
A–B, predicted reentrant wave following SNS on female heterogeneous tissue (5.4 cm × 6.6 cm) containing an endocardial region (tissue columns 1–160) and an epicardial region (tissue columns 161–360) based on recordings from human tissue, and anisotropic conduction in the presence of female hormones during the early follicular (A), late follicular (B) and luteal phases (C) of the menstrual cycle. D, no arrhythmia was predicted in male tissue with 35 nm testosterone. Single APs from site ‘a’ and site ‘b’ in the simulated tissues are shown in the middle panels for each case. Computed electrograms from simulated tissues are shown in the right panels.
Figure 3 shows voltage snapshots in time as indicated prior to and following the application of SNS in two‐dimensional tissue models. The simulated male tissue with testosterone is shown in Fig. 3 D. The 500th paced beat is shown prior to application of SNS. The presence of dofetilide resulted in SNS‐induced self generated arrhythmias in the female simulated tissues during the early follicular (Fig. 3 A) and late follicular (Fig. 3 B) phases of the menstrual cycle. The time course of single cell action potentials from two points in space (labelled as ‘a’ and ‘b’) in the simulated 2D tissue and the computed electrograms are shown in the right columns. Site ‘a’ undergoes two consecutive stimuli (indicated as ‘1st and ‘2nd’) at 10 ms and 1010 ms. Sympathetic activation occurs concurrently with the second stimulus. However, the site of activation (near ‘a’) becomes a site of reactivation via back‐propagating action potential conducted from downstream tissue (near ‘b’). The tissue near site ‘a’ then re‐excites site ‘b’, which continues in a feedback loop (observed as an oscillation on the computed electrogram), during which site ‘a’ maintains depolarization and serves as a persistent current source.
When progesterone was high during the luteal phase (Fig. 3 C), no arrhythmias were predicted. We also did not observe any arrhythmias in the male model when testosterone was present (Fig. 3 D). Application of dofetilide led to development of spontaneous oscillations in the female only – suggesting vulnerability to arrhythmia in the follicular phases of the menstrual cycle. The computed electrograms from the tissue in Fig. 3 A and B exhibit both periodicity and sinusoidal amplitude, consistent with torsade de pointes type arrhythmias.
Simulation of structural molecular‐level effects of sex‐based arousal arrhythmias
The cellular‐ and tissue‐level simulations discussed above suggest that cardiac ion channel hormone interactions may constitute an important component of sex‐based arousal arrhythmias with I Kr current modulation playing a central role. Experimentally the reduction of I Kr resulting from the action of oestradiol on the hERG channel has been demonstrated previously (Kurokawa et al. 2008); however, the molecular‐level interactions that underlie this effect remain uncharacterized. As a first step to identify potential binding sites as well as key stabilizing interactions between ligand and receptor, we carried out molecular docking of dofetilide (Fig. 4 A) and E2 (Fig. 4 B) to the intra‐cellular cavity (IC) of the WT open‐state hERG atomistic model (Model 2) developed previously (Durdagi et al. 2011, 2012).
Figure 4. Top scoring docking poses for dofetilide (A and C) and oestradiol (B and C) in the intracellular cavity (IC) site of hERG.
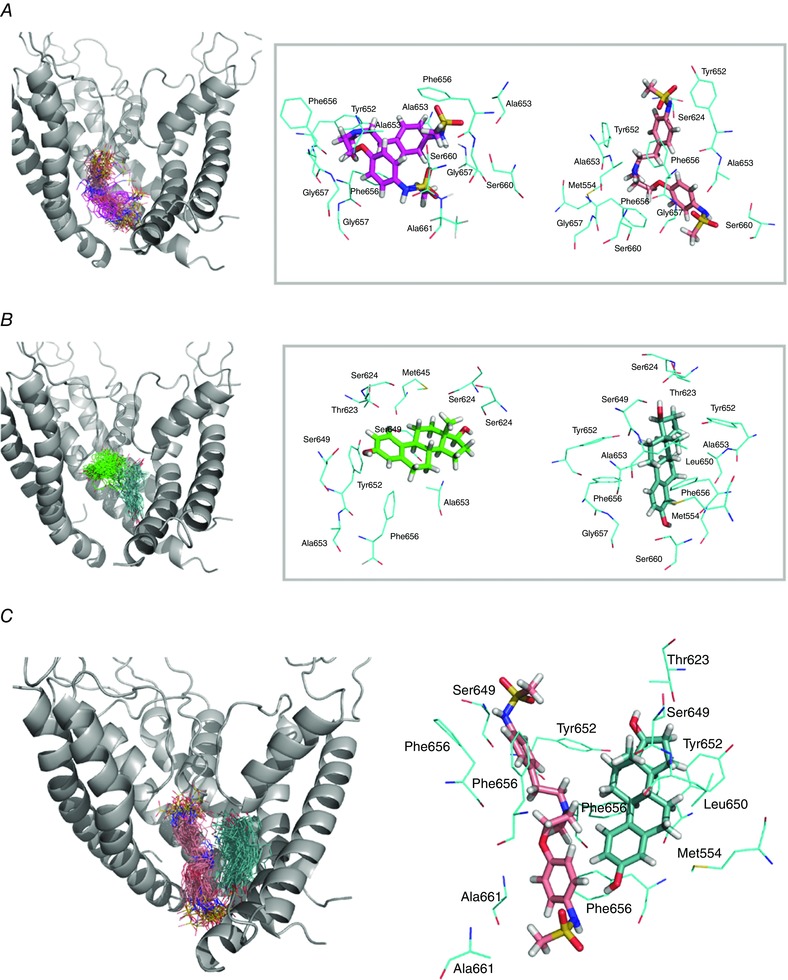
Left panels: pore domain of the hERG open state model and selected frames showing dofetilide (pink) (A), and E2 (green) (B), binding to the IC. Right panels: average position of dofetilide (A), and E2 (B) shown together with coordinating residues within 3.5 Å for two mapped binding sites. Two dominant binding orientations are shown in magenta and pink for dofetilide (A), and in green and light‐blue for E2 (B). The relative orientation of dofetilide (pink) and E2 (light‐blue), as well as the dominant interacting residues in the hERG IC site, are shown in C.
The simulations showed that a clustering of dofetilide poses that provides additional confirmation that dofetilide interacts with binding pocket for common hERG blockers (Fig. 4 A; Vandenberg et al. 2001; Ficker et al. 2001; Durdagi et al. 2012). E2 poses and follow‐up MD simulations led to identification of two tentative binding pockets with favourable binding docking scores for the oestradiol molecule. The high‐affinity site, shown in Fig. 4 B, is located in the intracellular cavity of hERG and overlaps with a binding pocket for common hERG blockers including dofetilide (as shown in Fig. 4 A) (Durdagi et al. 2012). To provide in‐depth analysis of E2 dynamics in this IC site, we performed 100 ns MD simulations of the hERG–E2 complex in explicit lipid bilayers. The oestradiol molecule exhibits substantial flexibility in the IC of hERG, resulting in a broad distribution of binding enthalpies. However, most of them are in the −10 to −15 kcal mol−1 range with a mean value around −12 kcal mol−1, which (without taking into account entropic contribution) corresponds to low micromolar/potent blocking affinity (Zachariae et al. 2009; Anwar‐Mohamed et al. 2014). Similar to many other blockers, oestradiol explores both low‐ and high‐affinity sites in the IC of hERG channel (Duff et al. 1995; Lees‐Miller et al. 2000). Analysis of MD simulations shows that the most dominant binding pocket for E2 relies on a combination of π‐stacking interactions of Y652 and F656 rings with an aromatic centroid of E2, supplemented by hydrogen bonding with the constellation of T623/S624 in the pore helix region and S645 in the S6 helix.
The second binding site is defined by the aromatic F656 side‐chain, supplemented by a number of hydrophobic residues including L650 and A653. Y652 is also transiently involved into coordination of bound oestradiol at this site. It is apparent that the main determinant of binding is whether the F656–E2 interaction can be stabilized by the additional hydrophobic interactions. Kurokawa et al. (2008) have shown that E2 does not inhibit both F656T and F656M mutant hERG currents, which is in agreement with the theoretical results presented here. It is notable that the relatively small size of oestradiol allows for another orientation in the binding pocket, where the bound hormone is oriented along the two distal S6 helices from adjacent subunits.
The E2 IC binding site is adjacent to that of dofetilide (see Fig. 4 C), suggesting a likely interaction. In fact, our binding enthalpy calculations from MD simulations demonstrated that the presence of intracellular cavity bound E2 leads to a modestly more favourable binding of dofetilide (by around 1.2 kcal mol−1). Also, the width of the distribution for dofetilide binding energies is much narrower with E2 bound to IC of hERG channel. All this suggests that a presence of E2 can potentially enhance hERG block by dofetilide, and thus enhance its proclivity to promote arrhythmia. The combination of molecular docking, MD simulations and binding enthalpy computations in conjunction with electrophysiology data provides consistent evidence for IC blocker action of E2.
Simulation of cellular‐level effects of inherited sex‐based arousal arrhythmias
We next extended the O'Hara–Rudy computational model (O'Hara et al. 2011) of I Kr to include the functional effects of the cardiac pore loop mutation G604S. As shown in Fig. 5 A and B, the mutant G604S I Kr model was developed by optimizing parameters to experimental data for current–voltage relationships of peak and tail currents of G604S I Kr recorded from HEK cells. In Fig. 5 A and B, the adjusted, model‐generated I Kr (lines) is shown superimposed on experimental I Kr records (symbols). The normalized I Kr current–voltage relationship for peak current for WT (black) and the hERG‐G604S mutation (red) are shown in panel (A). In panel (B) there are current–voltage relationships from normalized tail currents for WT (blue) and hERG‐G604S mutant channels (red).
Figure 5. Simulated and experimentally recorded I Kr data.
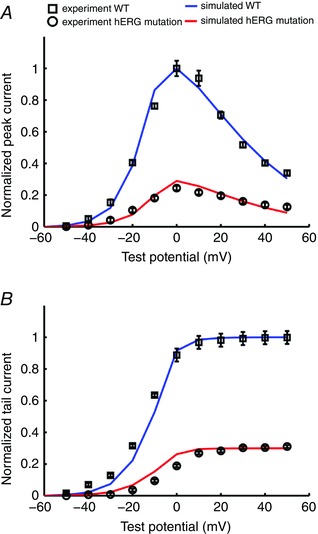
Model‐generated I Kr (lines) is shown superimposed on experimental I Kr records (symbols with error bars, n = 9) from HEK cells (Huo et al. 2008). A, normalized I Kr current–voltage relationship for peak current for WT (blue) and the hERG‐G604S mutation (red). B, current–voltage relationships from normalized tail currents for WT (blue) and hERG‐G604S mutant channels (red).
To determine the sex‐based propensity for arousal‐induced arrhythmias, we added the combined effects of the long‐QT syndrome type 2 pore mutation G604S with acute effects of β‐adrenergic stimulation by isoproterenol. Figure 6 shows the predicted effects on arrhythmia triggers in single simulated myocytes (upper rows) in female and male models. These results indicate that the female cells in the presence of the G604S pore mutation, particularly in the follicular phases of the menstrual cycle when oestradiol is highest, are considerably more vulnerable to the emergence of arrhythmogenic rhythms upon SNS.
Figure 6. Simulated arousal via sympathetic stimulation in the setting of the G604S mutation causing inherited long‐QT syndrome type 2.
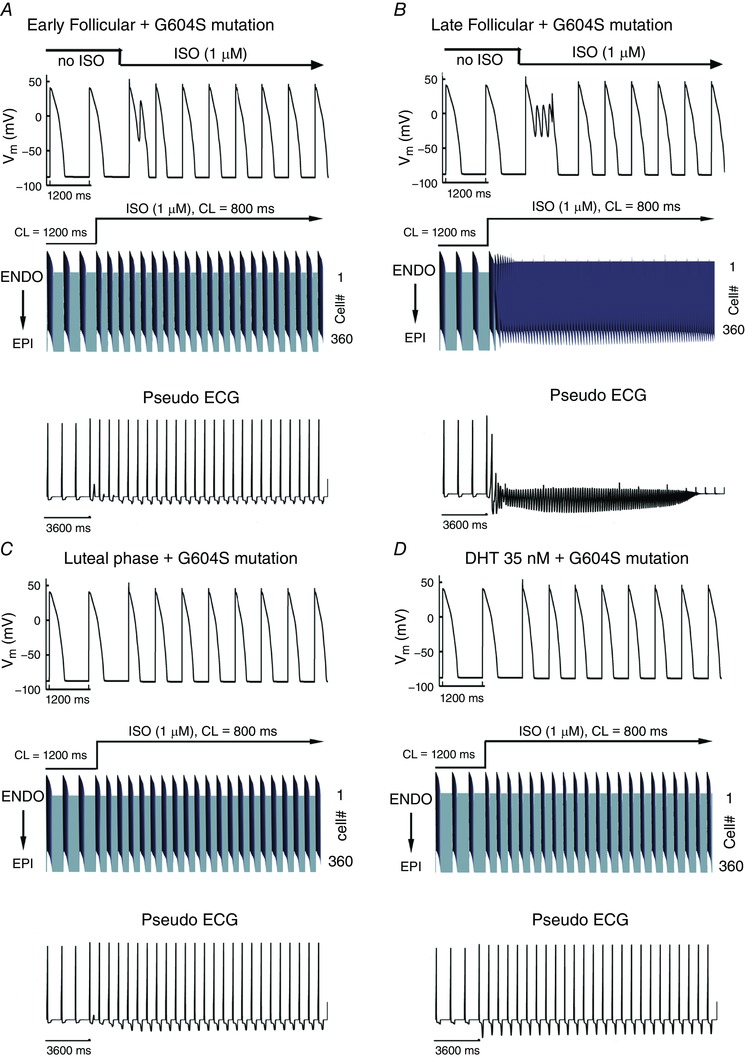
Single cell action potentials are shown at the top of each panel. Transmural 1‐dimensional tissue models are shown in the middle. Time is shown on the x‐axis and voltage on the z‐axis. The pseudo ECG is shown beneath the tissue simulation in each panel. A, simulated combined effects of oestradiol and progesterone on female tissues during the early follicular phase of the menstrual cycle. B, simulated combined effects of oestradiol and progesterone on female tissues during the late follicular phase of the menstrual cycle. C, simulated combined effects of oestradiol and progesterone on female tissues during the luteal phase. D, simulated effect of testosterone on male tissue during SNS.
Simulation of tissue‐level effects of inherited sex‐based arousal arrhythmias
We next explored the effects of sex‐based differences in the setting of inherited long‐QT syndrome type 2 pore mutation G604S and acute SNS in a one‐dimensional transmural strand of coupled cells comprising epicardial to endocardial regions. The middle panels in Fig. 6 A–C (middle rows) show the sex‐based effects in an electrically coupled female tissue during the early follicular (Fig. 6 A), late follicular (Fig. 6 B) and luteal (Fig. 6 C) phases of the menstrual cycle. The male tissue with testosterone present is shown in the middle of Fig. 6 D. We also computed electrograms (lower panels) in female and male virtual tissues (Fig. 6).
Importantly, the simulation shows longer repolarization in the female compared to male tissues following SNS, consistent with the longstanding observations that females have longer corrected QT intervals than males (Jose & Collison, 1970; Huikuri et al. 1996; Burke et al. 1997; Stramba‐Badiale et al. 1997; Bidoggia et al. 2000; Smetana et al. 2002). The prolonged T‐wave in females corresponds to longer APD (as observed in the space–time plot) and is likely to contribute to the arrhythmogenic rhythm that emerges in the late follicular phase (Fig. 6 B). Note that the computed electrogram from the tissue in Fig. 6 B exhibits both periodicity and sinusoidal amplitude, consistent with torsade de pointes type arrhythmias.
We also tested the vulnerability to arrhythmia induced by SNS with the LQT2 G604S hERG pore mutation in simulated human male and female two‐dimensional tissues using the same protocol as described in Fig. 3. Figure 7 shows voltage snapshots in time as indicated prior to and following the application of SNS in female tissue in simulated phases corresponding to early follicular (Fig. 7 A), late follicular (Fig. 7 B) and luteal (Fig. 7 C) phases of the menstrual cycle. The simulated male tissue with testosterone is shown in Fig. 7 D. Simulations are shown following pacing from the endocardium for 500 beats at 1000 ms pacing cycle length. The 500th paced beat is shown prior to application of SNS. In the presence of testosterone (Fig. 7 D) or high progesterone (luteal phase in Fig. 7 C) application of SNS is not predicted to be arrhythmia provoking.
Figure 7. Female sex is predicted to increase susceptibility to reentrant arrhythmia following acute SNS stimulation in the setting of inherited LQT2 syndrome.
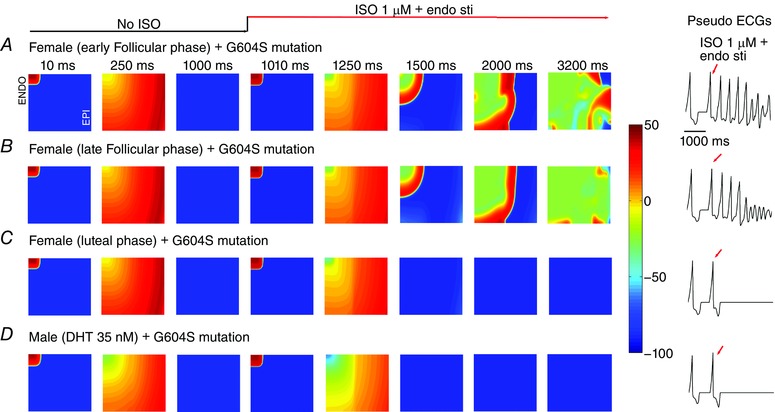
A–C, predicted reentrant wave following SNS on female heterogeneous tissue (5.4 cm × 6.6 cm) containing an endocardial region (tissue columns 1–160) and an epicardial region (tissue columns 161–360) based on recordings from human tissue, and anisotropic conduction in the presence of female hormones during the early follicular (A), late follicular (B) and luteal (C) phases of the menstrual cycle. D, no arrhythmia was predicted in male tissue with 35 nm testosterone. Computed electrograms from simulated tissues are shown in the right panels.
The outcome was starkly different in the female simulated tissue during the early (Fig. 7 A) and late follicular (Fig. 7 B) phases, which following SNS application developed spontaneous focal arrhythmia activity – in these cases, the oscillations persist for more than 3 s. The development of spontaneous sustained oscillations suggests female vulnerability to arrhythmia is especially noticeable during the follicular phases of the menstrual cycle. The computed electrograms from the simulated tissues are shown in the right panels, clearly illustrating the abrupt degeneration of the electrical rhythm following acute SNS in Fig. 7 A and B. Notably, in the absence of either the G604S pore mutation or SNS, no arrhythmia was observed at the cell (Appendix Fig. A1) and tissue levels (Appendix Fig. A2).
Finally, because a three‐dimensional tissue is a larger electrotonic sink, we set out to test if the predictions of female vulnerability to SNS‐induced arrhythmia in the setting of long‐QT syndrome type 2 would persist in higher dimensions. We developed an in silico left ventricular 3D wedge reconstruction of the human female based on the experimental data from Glukhov et al. (2010) with the pore loop G604S mutation (Fig. 8). Even in the 3D wedge reconstruction, we observed arousal‐induced spontaneous arrhythmias in the early follicular (Fig. 8 A) and late follicular (Fig. 8 B) phases of menstrual cycle by acute SNS application. Just as in lower dimensions, the luteal phase is not predicted to be sensitive to SNS (Fig. 8 C).
Ionic mechanisms of arrhythmia vulnerability in females
We next utilized a component dissection approach to determine the mechanism of self‐generated arrhythmia triggers in the late follicular phase of the menstrual cycle. Because PKA activity results in increases in two inward currents, I Na and I CaL, it is difficult to assess which effect is required to promote arrhythmogenic early afterdepolarizations. Thus, we selectively blocked the individual effects of PKA facilitation of I Na and I Ca, while maintaining all other effects. The 399th–400th beats (red arrows) are shown prior to application of SNS. Upon SNS application, the pacing cycle length was increased to 800 ms for 100 beats. The last 11 of 100 beats (BCL = 800 ms, under SNS stimulations) are shown in the right panels. As can be seen in Fig. 9 A–C, the blocking PKA effects on I CaL alone was sufficient to prevent arrhythmia triggers, which did not emerge even after 100 beats (right panels). On the other hand, blocking the effects of PKA on I Na (Fig. 9 D–F) did not prevent afterdepolarizations, which persisted after 100 additional beats (right panels). Finally, based on recent data from the Salama group, we examined the effect of increasing I NCX (NCX is a sodium–calcium exchanger) density by 2‐fold (Chen et al. 2011) in the epicaridum as shown in Fig. 9 G–I. The cell (Fig. 9 G, red line) and coupled tissue (Fig. 9 H) exhibited an exacerbation of proarrhythmia when the I NCX density was increased compared with no changes in I NCX (black line). The effect on the computed electrogram is shown as a tachycardic rhythm in Fig. 9 I.
Figure 9. Ionic mechanisms of simulated arousal via sympathetic stimulation in the setting of inherited or acquired LQT during late follicular phase.
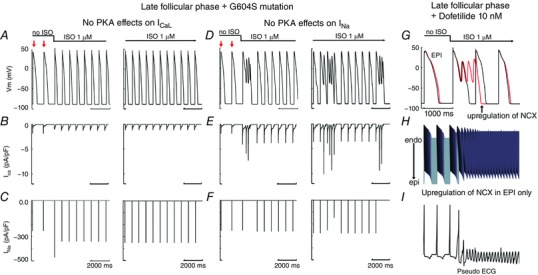
Since ISO increased I caL and I Na, here we investigated which current induced EAD. A–C suggested that when we removed PKA effects on I caL (B), there was no EAD induction. The 399th–400th (red arrows) are shown prior to application of SNS. Upon SNS application, the pacing cycle length was increased to 800 ms for 100 beats. The last 11 beats (BCL = 800 ms, under SNS stimulations) are shown in the right panels. D–F show that eliminating PKA effects on I Na (F) does not suppress EAD‐triggered activity. G, I NCX densities were increased by 2‐fold (Chen et al. 2011) in epi cell (red line) compared with no changes in I NCX (black line). H, transmural 1‐dimensional tissue models with upregulation of NCX in epicardium. I, the pseudo ECG is shown beneath the tissue simulation.
The explanation for this mechanism can be traced back in part to the effects of hormones on the I CaL window current. Progesterone and testosterone cause a dose‐dependent reduction in I CaL window current (Appendix Fig. A3). During the follicular phases of the menstrual cycle, low progesterone (2.5 nm) does not sufficiently reduce the I CaL window current, which when combined with reduced hERG current due to long‐QT syndrome type 2 mutations or drugs, resulted in increased susceptibility to EADs in female myocytes during the follicular phase. Our simulations suggest that EADs were initiated by a perfect storm of PKA effects, acute hormone effects on I CaL and I Kr suppression by the long‐QT syndrome type 2 mutation or dofetilide (Appendix Fig. A4). At the tissue level, there is an additional effect in the female of reduced gap junction conductance, incorporated in our model based on the observation of reduced Cx43 in female versus male hearts (Gaborit et al. 2010; Yang & Clancy, 2012). Reduced gap junction coupling (Xie et al. 2010) in the female relative to male (Appendix Table A1) is predicted to promote the development of EADs, by creating a source–sink mismatch that allows for triggered activity in groups of cells to initiate reentry in tissue.
Structure‐based mechanisms of arrhythmia vulnerability in females
Similar to female predominance of arrhythmia in the acquired setting, which suggests interactions between hERG, dofetilide and oestradiol, the female predominance of inherited arrhythmia linked the the G604S mutations, suggests that oestradiol may differentially interact with the mutant hERG channel compared to WT. To probe a possible molecular mechanism, we utilized the molecular docking of oestrogen throughout hERG and identified a second E2 binding site located near the pore loop region discovered in molecular docking studies performed with hERG Model 1 (based on the Eag1 template) described above that involves a number of residues implicated in gating dynamics. In the WT, this site is exposed to the lipid bilayer, allowing for easy access to oestradiol (Fig. 10 A, left). The estimated binding affinity of −5.4 kcal mol−1 places E2 in the cohort of weak binders with the estimated pIC50 (a negative log of 50% inhibition coefficient) <4.5, comparable to carvedilol block of WT hERG (Durdagi et al. 2012; Anwar‐Mohamed et al. 2014). The MD simulations of E2 to this binding site show that oestradiol displays a very broad range of docking poses, and can rapidly unbind and diffuse away from this low‐affinity site to lipid bilayer. Notably, this site hosts the G604S pore loop mutation, implicated in female proclivity to arousal arrhythmia.
Figure 10. Oestradiol (E2) binding to a pore loop site of hERG from molecular docking (A), and molecular dynamics (B and C) simulations for wild‐type (WT; left) and G604S mutant hERG channel (right).
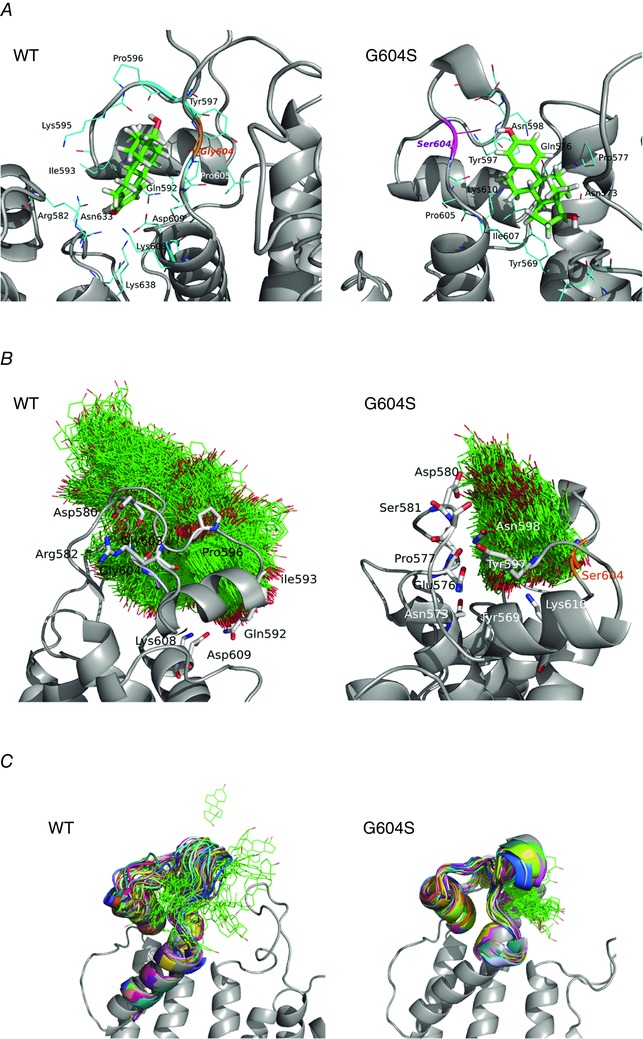
A, top scoring docking poses for dofetilide in the pore loop site shown together with coordinating residues within 3.5 Å for mapped binding sites. B, E2 positions (shown in green wireframe representation) are mapped from the frames collected in 40 ns MD for WT and G604S in order to illustrate the conformational space explored by the ligand in the two systems. It is apparent that E2 is kinetically stabilized (trapped) in the G604S mutant, while it is unable to bind stably in WT and leaves the binding pocket within 20 ns of MD simulation. C, a G604S mutation is predicted to enhance stability of the bound E2 (green wireframe) in the binding pocket by controlling binding site flexibility: the mutation stabilizes the binding site allowing for E2 to remain in the pocket. In contrast, E2 readily leaves binding pocket in the WT hERG protein and diffuses away to the lipid bilayer. Shown are 32 selected frames from the first 15 ns of molecular dynamics simulation for E2/WT and E2/G604S are shown in different colours.
This pore loop region appears to be a unique structural feature of both Eag and Erg channels. MD simulations suggest that E2 dissociates rapidly from this site in WT and hence is unlikely to impair function of WT hERG channel. In order to investigate how this specific mutation G604S affects oestradiol binding at the pore loop receptor site, we mutated our WT hERG model accordingly, and again used molecular docking to identify key binding motifs, shown in the left and right panels of Fig. 10 A for WT and G604S, respectively.
The MD simulations of the G604S–E2 complex are in stark contrast with results we obtained for WT channel. Molecular docking alone results in indistinguishable binding enthalpies of E2 between WT and G604S: −6.1 kcal mol−1 vs. −5.4 kcal mol−1, respectively. However, MD simulations reveal major differences in conformational dynamics of the pore loop region with E2 bound in WT and G604S. The clustering of E2 poses on the hERG extracellular loop in WT and mutant proteins is shown in Fig. 10 B. The RMS fluctuations show that the mobility/flexibility is much lower when E2 is bound to the G604S mutant compared to WT (Fig. 10 C and Appendix Fig. A5). When E2 is bound to the G604S mutant, interaction remains stable throughout the 50 ns MD, whereas E2 departs from the WT channel within 20 ns (Fig. 10 C).
These results suggest that the pore loop mutation enhances binding of E2 to the hERG channel. They also suggest that the enhanced affinity is not due to key interactions between the G604 mutated residue and E2, but rather the interplay between conformational and flexibility changes resulting from the mutation. Vandenberg and colleagues (Pages et al. 2009) suggested that dynamics and flexibility of this region is critical for the stability of the selectivity filter and therefore is expected to play a substantial role in inactivation dynamics.
Discussion
‘It has only been possible to collect a few figures for normal women, but so many of these fall outside the range for normal men,’ wrote Bazett in his 1920 analysis of cardiac electrical activity (Bazett, 1997). Nearly a century has passed since the initial recognition of sex‐based differences in cardiac electrical activity. Yet, even now, there is a little known about the structural and functional mechanisms underlying the sex‐based differences that predispose human women to inherited and acquired long‐QT syndrome and associated torsade de pointes (TdP) arrhythmias (Makkar et al. 1993; Locati et al. 1998; Salama & Bett, 2014).
Recent experimental studies in a rabbit model of acquired long‐QT syndrome show an increased risk for arrhythmia and TdP in adult female hearts that is not observed in pre‐pubescent female or adult male hearts (Liu et al. 2005; Sims et al. 2008). In these studies, oestrogen‐dependent genomic upregulation of I Ca,L and I NCX were implicated as possible contributors to TdP in females (Chen et al. 2011; Yang et al. 2012). However, these studies did not address the impact of the autonomic nervous system that apparently impacts arrhythmia risk in females. Our predictions (Fig. 9) do support the idea that upregulation of NCX may promote arrhythmias in females. A population study by Kim et al. (2010) showed that 82% of observed arousal‐induced arrhythmias occurred in females.
Because sympathetic discharge is a well‐recognized factor in triggering TdP in long‐QT syndrome patients, the combined effects of protein kinase A (PKA) phosphorylation and sex hormones on ion channels have recently been studied experimentally (Furukawa & Kurokawa, 2007; Nakamura et al. 2007; Vaseghi & Shivkumar, 2008). Progesterone was shown to prevent PKA‐mediated increases in depolarizing current through the I Ca,L, while enhancing the effect of PKA‐induced repolarizing I Ks (Nakamura et al. 2007). The net effect of these two changes is to increase repolarizing current and reduce APD. In this study, we also included new data showing the effects of testosterone on the L‐type Ca2+ current in the presence of isoproterenol (Fig. 1). Just as has been shown for progesterone in a previous study (Nakamura et al. 2007), testosterone application blunted the isoproterenol‐induced enhancement of the L‐type Ca2+ current. This provides a plausible mechanism for testosterone protection against arousal‐induced arrhythmias: a reduction in L‐type Ca2+ current allows for greater rate‐dependent shortening of the male action potential and reduces arrhythmia likelihood.
Women are disproportionately affected by inherited forms of long‐QT syndrome associated with hERG pore loop mutations as well as by acquired long‐QT syndrome, which results from unintended off‐target block of the major cardiac repolarizing current I Kr, encoded by the hERG gene and characterized by prolongation of the QT interval. The basis of our study was the hypothesis that oestrogen will acutely increase arrhythmia vulnerability and that progesterone will protect against arrhythmia initiation, especially in the setting of sympathetic stimulation, a major factor in triggering TdP. This hypothesis is based on published data showing that oestrogen interacts directly with the promiscuous hERG channel, reduces repolarizing I Kr current and increases the rate of channel deactivation (Kurokawa et al. 2008). Kurokawa and co‐authors proposed that aromatic centroid of E2 may be responsible for increasing the sensitivity of hERG block by E4031 via interaction with the aromatic side chain of Phe‐656 and aromatic rings of the hERG blocker.
Kurokawa and co‐authors have also shown that in the presence of E2, hERG is markedly more sensitive to block by drugs and that the concentration of E2 fluctuates throughout the menstrual cycle, from the peak follicular phase serum E2 level of 1 nm to 0.7 nm in the luteal phase (Kurokawa et al. 2008). Since E2 has dramatic effects on sensitivity to hERG block within this range, it stands to reason that susceptibility to drug‐induced arrhythmia by hERG block may also vary through the menstrual cycle, but the exact mechanism of potential concomitant effects on common blocker action remain unknown. The predictions from molecular modelling studies based on bacterial KcsA and MthK (Perry et al. 2004; Stansfeld et al. 2007) channels or the mammalian channel Kv1.2 (Durdagi et al. 2010; Stary et al. 2010) concur with the experimental findings that have revealed two key residues responsible for drug stabilization in the hERG1 cavity, e.g. Y652 and F656 (Lees‐Miller et al. 2000; Mitcheson et al. 2000; Ficker et al. 2001; Vandenberg et al. 2001; Fernandez et al. 2004; Durdagi et al. 2012). Model studies reproduced this feature in studies of hERG blockers such as dofetilide, KN‐93 and other common high‐affinity blockers (Durdagi et al. 2011, 2012).
Previous studies of high‐ and low‐affinity blockers such as dofetilide, astemizole and haloperidole suggest that an ‘ideal’ high‐affinity blocker should simultaneously interact with F656, Y652 and/or A653 at the inner cavity of hERG, and form hydrogen bonds with T623 and S624 from the pore helix (Mitcheson et al. 2000; Lees‐Miller et al. 2000; Ficker et al. 2001; Fernandez et al. 2004; Durdagi et al. 2011, 2012; Vandenberg et al. 2001). The binding data collected from our MD simulations shown in Fig. 4 endorse the presence of these sites accommodating favourable and synergistic binding of both E2 and dofetilide. The distribution of binding energies and binding pockets for E2 and dofetilide suggest a plausible scenario of hormone modification of common blocker action. In the intracellular cavity (IC) of hERG, the presence of E2 only modestly enhances the binding affinity of dofetilide; however, there is less variance in the distribution of binding energies for dofetilide with E2 bound. That is, bound hormone stabilizes the high‐affinity site for dofetilide and virtually eliminates binding to a low‐affinity site.
The hERG channel exhibits a high degree of homology with the Eag1 channel, the structure of which has recently been determined (Whicher & MacKinnon, 2016). The availability of the Eag1 structure provided us with a new and unique structural template for understanding arrhythmia‐associated mutations.
A key feature revealed by the structure is the extended ‘extracellular turret’. Indeed, this region is home to the G406S mutation studied here, and we found that it appears to provide a pocket for oestrogen binding that is stabilized by the mutation. The new structural data allowed the revelation of interactions between physiologically important and tightly controlled lipophilic substances such as E2 and functionally important lipid‐facing regions of the hERG channel. It is also predicted in our study that E2 binding may impact the channel through two distinct mechanisms. The first mechanism involves a high‐affinity binding site that is located near the pore loop region, which is involved channel kinetics and plays an important role in normal physiology. Post‐adolescent females show more than nine times the number of arousal‐triggered arrhythmias than men of the same age group, and arousal‐triggered events are more than twice as high among female patients exhibiting mutations in the pore loop region of hERG, but not mutations in other regions (Kim et al. 2010). We hypothesized that pore loop mutations may modify the oestrogen interaction with hERG. Indeed, this notion is corroborated by our findings, which suggest that E2 binds with higher affinity in the hERG model with the pore loop G604S mutation.
The second mechanism involves another high‐affinity binding site for E2, located in the intracellular cavity of hERG in the vicinity of Y652, a residue crucial for the channel block. This suggests that E2 may enhance the action of common blockers, such as dofetilide, as predicted by our binding enthalpy calculations. It is also probable that the mechanisms co‐exist, depending on the physiologically driven concentration fluctuations of E2 in cell membranes. Establishing a connection between these two mechanisms of action is a goal for our future studies.
The present study focuses on the role that changes in sympathetic tone play in triggering arousal‐induced arrhythmia. It should be noted that there are also sex‐related differences in parasympathetic tone that we have not considered in this study. It has been demonstrated that parasympathetic influence on cardiac function is greater in females than males (Dart et al. 2002). Elevated parasympathetic tone, which is associated with greater heart rate variability, was found to be greater in premenopausal women and postmenopausal women receiving hormone replacement therapy, suggesting that oestrogen may be responsible (Liu et al. 2003). Because of this, it is conceivable that abrupt termination of parasympathetic input also contributes to arousal‐induced arrhythmias, since vagal withdrawal has been shown to result in an increase in cAMP production that potentiates sympathetic responses (Belevych et al. 2001; Harvey & Belevych, 2003).
Limitations of the current study
There are several limitations in our study. While the O'Hara–Rudy model was extensively validated in reproducing experimentally observed rate dependence in single cells, the model overestimates the rate dependence of APD in human tissue compared to experiments reported by Franz et al. (1988). We also did not consider the presence of other prevalent disease states such as diabetes that have a sex difference component (Shimoni et al. 2009).
An important limitation in the interpretation of structural modes of E2 binding to hERG is an absence of a high‐resolution experimental structure of the channel. In this study, we resorted to using homology models in our simulations, which depend on the availability of high‐resolution experimental template structures, in the desired conformational state, and with high sequence identity. We therefore chose suitable templates (Eag1 and Kv1.2) for our models that allowed us to study two distinct regions of interest in hERG: the pore loop region and the intracellular cavity. While we recognize the inherent limitations of using homology models, there is, importantly, remarkable consensus for crucial drug binding sites in both models that we used.
We also acknowledge that our molecular simulation approach returns only binding enthalpy distributions, which, in the absence of the entropic and diffusion data that are crucial for quantitative drug–channel thermodynamic and kinetic predictions, are not able to unambiguously predict quantitative indices of current inhibition. We are planning further studies now with mapping free energy and diffusion profiles to compute ‘on’ and ‘off’ channel‐hormone interaction rates for the kinetic models to evaluate impact of hormone binding on the channel current blockade. Our current study reveals the first mechanistic models of how E2 could potentially enhance hERG blockade. However, dofetilide is known to exhibit several modes of binding to hERG and our first simulation here suggests that oestradiol helps to stabilize one of them in the open pore of the hERG channel. Furthermore, based on work of Sanguinetti (Chen et al. 2002) and studies that followed (Rodriguez‐Menchaca et al. 2006; Stork et al. 2007; Lees‐Miller et al. 2015), we may expect a state‐dependent blockade by dofetilide and thus will need to evaluate binding of a hormone to different states. Whether these are separate mechanisms or ones that can coexist are the questions we intend to study in the near future. This work will refine different E2 binding modes, their affinities and kinetics through a combination of experimental and computational studies.
The computational modelling and simulation approach has allowed us to explore the complex non‐linear interplay between processes that underlie differences in risk to acquired long‐QT‐dependent arrhythmias. They also allow us to predict the ‘perfect storm’ of hormone concentration, I Kr derangement and sympathetic stimulation that may explain one mechanism of increased proclivity to arousal arrhythmia in females. The results of this study suggest specific therapeutic anti‐arrhythmic strategies for women with long‐QT syndrome, which may include specific hormone replacement therapy, as well as suggested and counter‐indicated drugs. To realize the most basic biological mechanisms underlying sex‐based differences in long‐QT syndrome risk and susceptibility to TdP arrhythmias is the critical first step to set the stage for specific sex‐based diagnosis and therapeutic targeting of cardiac disease.
Additional information
Competing interests
None.
Author contributions
P.‐C.Y., L.L.P., and S.N. designed and performed simulations, analysed data, and prepared the manuscript; Y.W. and M.‐T.J. designed and performed simulations; F.L‐R. and J.K. designed and performed experiments; K.R.D. and I.V. analysed data, and revised the manuscript; R.D.H drafted the manuscript; C.E.C. designed simulations and experiments, analysed data, coordinated and oversaw the project, and prepared the manuscript. All authors approved the final submitted version.
Funding
American Heart Association (GIAs (13GRNT14370019), Western States Affiliate), the National Institutes of Health NHLBI R01HL128170‐02 (to CEC), NHLBI U01HL126273‐02 (CEC and RDH), National Institutes of Health R01GM101928‐01 (to RDH and CEC) and R01HL128537‐01A1 (to CEC, RDH and SYN), Canadian Institutes for Health Research (MOP‐186232) and Heart and Stroke Foundation, Alberta (GIA) (to SYN). American Heart Association Predoctoral Fellowship 16PRE27260295 (to KRD). Japan AMED16mk0104027h1102, AMED16mk0104007h9903, MEXT/JSPS KAKENHI 15H04684 (to JK).
Simulation of genomic sex steroid hormone effects
Female hearts had reduced expression of the K(C)‐channel subunits HERG, minK, Kir2.3, Kv1.4, KChIP2, SUR2, and Kir6.2, and lower mRNA expression of connexin43 and phospho‐lamban compared to males. Experiments demonstrated concurrence of changes between gene and protein expression for HERG, minK, Kv1.4, KChIP2, IRX5, Nav1.5 and connexin43 (Nav1.5‐expression showed no male–female differences) (Gaborit et al. 2010). Details are given in Table A1.
Table A1.
Sex‐based differences in ion channel subunit expression from non‐diseased ventricles used in the computational model
| Epi | Endo | ||||
|---|---|---|---|---|---|
| Channels in the model | Gene | Male | Female | Male | Female |
| I Ks | KvLQT1 | 104.2 | 77.5 | 90.4 | 109.6 |
|
MinK (KCNE1) |
13.6 ± 1.4 m*↑ |
7.3 ± 5 f*↓ |
11.9 ± 4.2 m ** ↑ |
5.8 ± 2.6 f ** ↓ |
|
| Ratio | 1.04 ± 0.04 | 0.87 ± 0.14 | 1 ± 0.12 | 0.83 ± 0.07 | |
| Functions | Co‐expression of KvLQT1 and MinK shifts the voltage to more positive voltages. Also it increases amplitude of expressed current. Only MinK shows significant differences between male and female. The stoichiometry of KCNE1:KCNQ1 in I Ks channels is a fixed 2:4 (Nakajo et al. 2010). The ratio was calculated by modified the 1/3 of current activity depending on MinK. | ||||
| I Kr |
hERG (Kv11.1) |
179.5 ± 6.4 m*↑ |
144.2 ± 41.1 f*↓ |
164.8 ± 54.9 ↑ |
130.5 ± 65 ↓ |
| Ratio | 1.09 ± 0.039 | 0.875 ± 0.25 | 1 ± 0.33 | 0.79 ± 0.39 | |
| I K1 | Kir2.1 | 94.5 | 93.8 | 104.1 | 79.7 |
| Kir2.2 | 111.1 | 115.3 | 93.6 | 104.5 | |
| Kir2.3 |
91.2 ± 31.8 m*↑ |
21.4 ± 10.6 f*↓ |
92.7 ± 26.8 m**↑ |
55.2 ± 21.5 f**↓ |
|
| Ratio | 0.98 ± 0.12 | 0.74 ± 0.04 | 1 ± 0.09 | 0.86 ± 0.077 | |
| Functions | Kir2.x channels mediate cardiac I K1 (Dhamoon et al. 2004); however, only Kir2.3 is significantly different. The ratio was calculated by changing the 1/3 of Kir2.3 channel activity. | ||||
| I to,s | Kv1.4 |
12.1 ± 3.3 m*↑ |
5.4 ± 3.8 f* ↓ |
20.2 ± 4.0 m ** ↑ |
12.9 ± 5.2 f ** ↓ |
| Ratio | 0.6 ± 0.16 | 0.26 ± 0.19 | 1 ± 0.2 | 0.64 ± 0.26 | |
| I NaK |
ATPase a1 |
207.7 ± 67 m* ↓ |
513.4 ± 134.6 f* ↑ |
269.0 ± 70.3 m** ↓ |
622.5 ± 287.7 f ** ↑ |
| a3 |
1481 ± 267 m* ↑ |
917.8 ± 416.7 f*↓ |
1547.6 ± 299.5 m**↑ |
1014.2 ± 294.5 f** ↓ |
|
| Ratio | 0.94 ± 0.18 | 0.7 ± 0.3 | 1.0 ± 0.2 | 0.79 ± 0.26 | |
| Functions | a3 is about 2‐fold more active than a1 in LV (Gaborit et al. 2007). The ratio was calculated depending on 1/3 of a1 and 2/3 of a3. | ||||
| I pCa | PMCA1 | 31.7 | 46.4 | 44.1 | 48.1 |
| PMCA4 |
377.0 ± 57.2 m* ↓ |
682.1 ± 265.9 f* ↑ |
426.8 ± 116.6 m↓ |
685.2 ± 379.7 f↑ |
|
| Ratio | 0.88 ± 0.13 | 1.6 ± 0.6 | 1.± 0.27 | 1.6± 0.89 | |
| Functions | PMCA1 serves a critical housekeeping function that is required for the maintenance of basic cellular function (Brini, 2009). PMCA4 nearly ubiquitous distribution has a similar role to that of PMCA1. PMCA4 was reported much more activity than PMCA1 in LV (Gaborit et al. 2007), and there are no differences between sexes in PMCA1. The ratio was calculated depended only on the PMCA4. | ||||
| I up | SERCA2 |
4850.5 ± 146 m* ↓ |
6728.4 ± 1876.1 f* ↑ |
3410.4 ± 982.1 m |
3921.9 ± 1760.7 f |
| Ratio | 1.42 ± 0.04 | 1.97 ± 0.55 | 1 ± 0.28 | 1.15 ± 0.5 | |
| Calmodulin | CALM1 | 1329.5 | 991.9 | 879.8 | 1122.7 |
| CALM3 |
1326.9 ± 220 m* ↓ |
1955.5 ± 372.2 f* ↑ |
1206.9 ± 187.7 m ** ↓ |
1600.5 ± 242.9 f** ↑ |
|
| Ratio | 1.07 ± 0.12 | 1.41 ± 0.2 | 1 ± 0.1 | 1.21 ± 0.14 | |
| Functions | CALM3 is more activity than CALM1 (∼2‐fold in LV) (Gaborit et al. 2007). The ratio was obtained by changing the 2/3 of activity depends on CALM3. | ||||
| Gap‐junction | Cx43 |
1124.1 ± 357 m* ↑ |
728.6 ± 274.1 f* ↓ |
1196.3 ± 311.7 m** ↑ |
810.9 ± 333.9 f** ↓ |
| Ratio | 0.94 ± 0.3 | 0.61 ± 0.24 | 1.0 ± 0.26 | 0.68 ± 0.28 | |
Ratios are relative to the male endocardial cell as we published previously (Yang & Clancy, 2012). *Differences between male and female in Epi is statistically significant. **Differences between male and female in Endo are statistically significant.
Table A2.
Simulated effects of progesterone on I Ks and I Ca,L
| Channel | Progesterone (baseline) | Progesterone (SNS stimulations) | ||
|---|---|---|---|---|
| 2.5 nm | 40.6 nm | 2.5 nm | 40.6 nm | |
| I Ks | 1.19 | 1.4 | 3.488 | 3.52 |
| I CaL | 1.0 | 1.0 | 2.5 | 2.5 |
| Kinetics of I CaL | No effects |
d
ss = 1.0/{1.0 + exp[(−(v + 3.94 + 16 − 1.5))/4.73]} f ss = 1.0/{1.0 + exp[(v + 19.58 + 8.0 + 1.7)/3.596]} |
d
ss = 1.0/{1.0 + exp[(−(v + 3.94 + 16−4.4))/5.23]} f ss = 1.0/{1.0 + exp[(v + 19.58 + 8.0 + 3.7)/3.096]} |
|
| Reference | Nakamura et al. (2007) | |||
Figure A1. Single cell action potentials during simulated arousal via sympathetic stimulation in the setting of control or the G604S mutation.
Action potentials are shown following pacing for 400 paced beats at a cycle length of 1000 ms in single simulated endocardial cells. The 399th and 400th paced beats are shown, after the pacing frequency was increased to 800 ms with or without SNS applied. A, control case with no SNS applications. B, dofetilide with no SNS applications. C, G604S mutation with no SNS applications. D, control case with SNS applied after 400 beats.
Figure A2. Human cardiac electrophysiology predicted in female late follicular phase (no mutation) transmural tissue models and simulated pseudo‐ECGs during SNS.
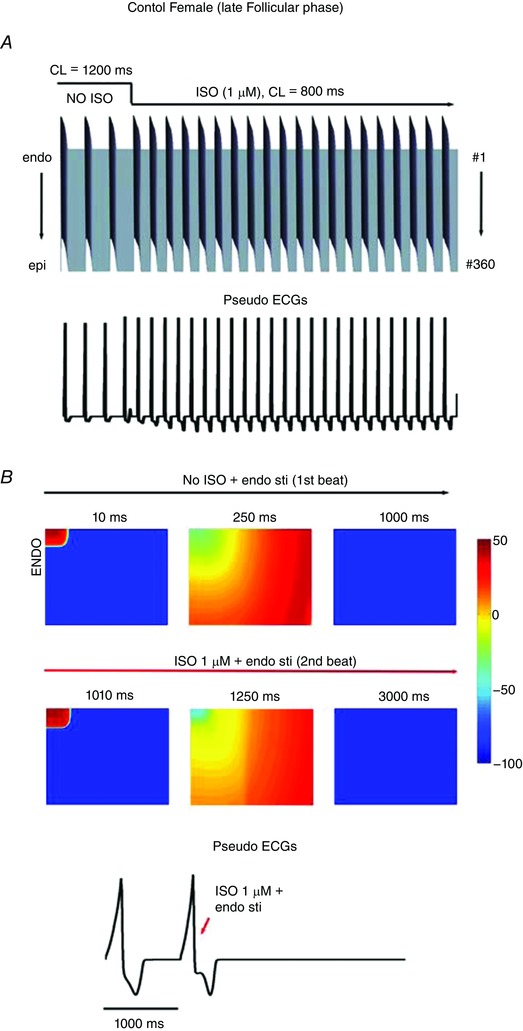
A, simulated 1‐dimensional action potentials (APs) are shown following pacing for 400 beats at 1200 ms pacing frequency. The 398th–400th paced beats are shown prior to application of SNS. Upon SNS application, the pacing frequency was increased to 800 ms. The pseudo ECG is shown beneath the tissue simulation. Time is shown on the x‐axis and voltage on the z‐axis. B, simulated combined effects of oestradiol and progesterone on female (no mutation) 2‐dimensional tissues during the late follicular phase of the menstrual cycle.
Figure A3. The L‐type Ca2+ window current.
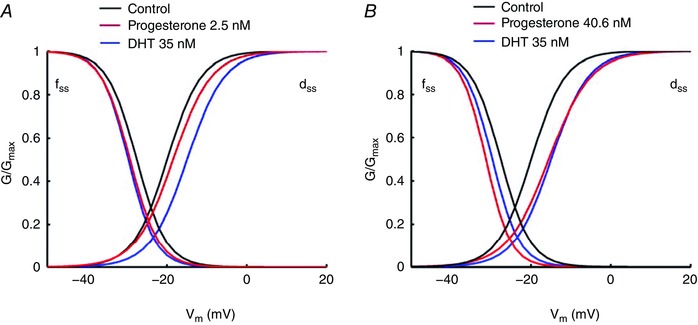
Steady‐state activation (d ss) and steady‐state inactivation (f ss) show the I CaL window current during SNS via ISO. A, DHT 35 nm (blue lines) indicated a smaller window current compared to progesterone 2.5 nm (red lines). B, high concentration of progesterone (40.6 nm, red lines) reduced the I CaL window current.
Figure A4. Ionic mechanisms of simulated arousal via sympathetic stimulation in the setting of the G604S mutation causing inherited LQT2 syndrome.
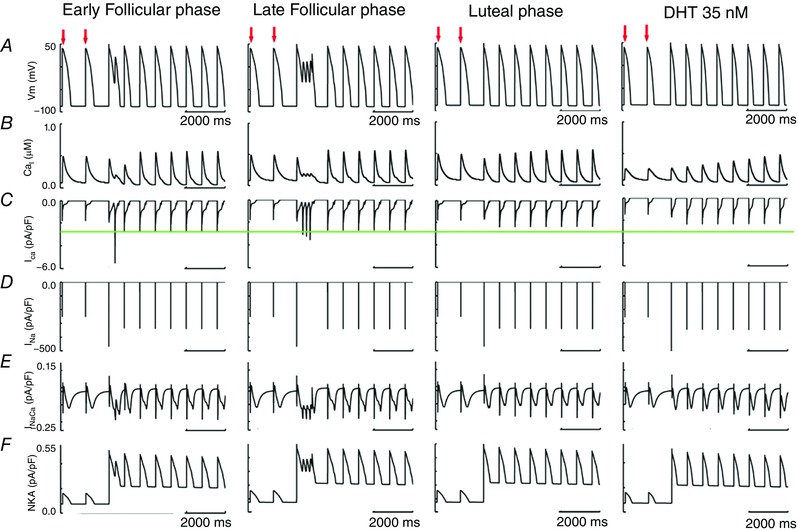
A, cellular APs. B, intracellular Ca2+ concentration. C, L‐type Ca2+ current. D, Na+ current. E, Na+/Ca2+ exchange current. F, Na+/K+‐ATPase (NKA). Same pacing protocol was used as in Fig. 2. The 399th–400th paced beats (red arrows) are shown prior to application of SNS. Upon SNS application, the pacing frequency was increased to 800 ms for 100 beats. C shows the peak of I CaL was lower in luteal phase or with DHT 35 nm.
Figure A5. RMS fluctuations per residue (550–670) for all the subunits of WT (thick grey line) and G604S (thick red line), E2 bound to WT chain C (black circles) and E2 bound to G604S chain C (red squares).
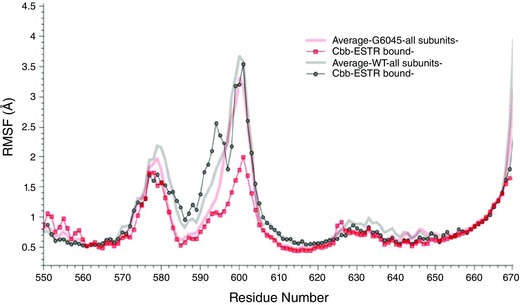
Figure A6. Action potential voltage clamp of simulated arousal via sympathetic stimulation in the setting of the G604S mutation causing inherited LQT2 Syndrome.
A, cellular action potentials were clamped to the voltages shown in Fig. A4. B, L‐type Ca2+ current. C, intracellular Ca2+ concentration was clamped to the diastolic rest concentration. Same pacing protocol was used as in Fig. A4. When Ca2+‐dependent inactivation is reduced (Ca2+ concentration clamped to diastolic level), with an EAD voltage clamp protocol, the Ca2+ current is even larger, suggesting a strong effect of ISO that is usually tempered by Ca2+‐dependent inactivation. In the absence of ISO, the effect is much smaller (right panels).
This is an Editor's Choice article from the 15 July 2017 issue.
Contributor Information
Robert D. Harvey, Email: rdharvey@medicine.nevada.edu
Junko Kurokawa, Email: junkokuro@u-shizuoka-ken.ac.jp.
Sergei Y. Noskov, Email: snoskov@ucalgary.ca
Colleen E. Clancy, Email: ceclancy@ucdavis.edu.
References
- Anneken L, Baumann S, Vigneault P, Biliczki P, Friedrich C, Xiao L, Girmatsion Z, Takac I, Brandes RP, Kissler S, Wiegratz I, Zumhagen S, Stallmeyer B, Hohnloser SH, Klingenheben T, Schulze‐Bahr E, Nattel S & Ehrlich JR (2016). Estradiol regulates human QT‐interval: acceleration of cardiac repolarization by enhanced KCNH2 membrane trafficking. Eur Heart J 37, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar‐Mohamed A, Barakat KH, Bhat R, Noskov SY, Tyrrell DL, Tuszynski JA & Houghton M (2014). A human ether‐á‐go‐go‐related (hERG) ion channel atomistic model generated by long supercomputer molecular dynamics simulations and its use in predicting drug cardiotoxicity. Toxicol Lett 230, 382–392. [DOI] [PubMed] [Google Scholar]
- Bai CX, Kurokawa J, Tamagawa M, Nakaya H & Furukawa T (2005). Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation 112, 1701–1710. [DOI] [PubMed] [Google Scholar]
- Bazett HC (1997). An analysis of the time‐relations of electrocardiograms. Ann Noninvasive Electrocardiol 2, 177–194. [Google Scholar]
- Belevych AE, Sims C & Harvey RD (2001). ACh‐induced rebound stimulation of L‐type Ca2+ current in guinea‐pig ventricular myocytes, mediated by Gβγ‐dependent activation of adenylyl cyclase. J Physiol 536, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P, Biasini M & Schwede T (2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M & Mackerell AD Jr (2012). Optimization of the additive CHARMM all‐atom protein force field targeting improved sampling of the backbone ϕ, ψ and side‐chain χ1 and χ2 dihedral angles. J Chem Theory Comput 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidoggia H, Maciel JP, Capalozza N, Mosca S, Blaksley EJ, Valverde E, Bertran G, Arini P, Biagetti MO & Quinteiro RA (2000). Sex differences on the electrocardiographic pattern of cardiac repolarization: possible role of testosterone. Am Heart J 140, 678–683. [DOI] [PubMed] [Google Scholar]
- Brini M (2009). Plasma membrane Ca2+‐ATPase: from a housekeeping function to a versatile signalling role. Pflugers Archiv 457, 657–664. [DOI] [PubMed] [Google Scholar]
- Burke JH, Ehlert FA, Kruse JT, Parker MA, Goldberger JJ & Kadish AH (1997). Gender‐specific differences in the QT interval and the effect of autonomic tone and menstrual cycle in healthy adults. Am J Cardiol 79, 178–181. [DOI] [PubMed] [Google Scholar]
- Chen G, Yang X, Alber S, Shusterman V & Salama G (2011). Regional genomic regulation of cardiac sodium‐calcium exchanger by oestrogen. J Physiol 589, 1061–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Seebohm G & Sanguinetti MC (2002). Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci USA 99, 12461–12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart AM, Du XJ & Kingwell BA (2002). Gender, sex hormones and autonomic nervous control of the cardiovascular system. Cardiovasc Res 53, 678–687. [DOI] [PubMed] [Google Scholar]
- Demolis JL, Funck‐Brentano C, Ropers J, Ghadanfar M, Nichols DJ & Jaillon P (1996). Influence of dofetilide on QT‐interval duration and dispersion at various heart rates during exercise in humans. Circulation 94, 1592–1599. [DOI] [PubMed] [Google Scholar]
- Dhamoon AS, Pandit SV, Sarmast F, Parisian KR, Guha P, Li Y, Bagwe S, Taffet SM & Anumonwo JM (2004). Unique Kir2.x properties determine regional and species differences in the cardiac inward rectifier K+ current. Circ Res 94, 1332–1339. [DOI] [PubMed] [Google Scholar]
- Dighe AS, Moy JM, Hayes FJ & Sluss PM (2005). High‐resolution reference ranges for estradiol, luteinizing hormone, and follicle‐stimulating hormone in men and women using the AxSYM assay system. Clin Biochem 38, 175–179. [DOI] [PubMed] [Google Scholar]
- Dorgan JF, Fears TR, McMahon RP, Aronson Friedman L, Patterson BH & Greenhut SF (2002). Measurement of steroid sex hormones in serum: a comparison of radioimmunoassay and mass spectrometry. Steroids 67, 151–158. [DOI] [PubMed] [Google Scholar]
- Duff HJ, Feng ZP & Sheldon RS (1995). High‐ and low‐affinity sites for [3H]dofetilide binding to guinea pig myocytes. Circ Res 77, 718–725. [DOI] [PubMed] [Google Scholar]
- Durdagi S, Deshpande S, Duff HJ & Noskov SY (2012). Modeling of open, closed, and open‐inactivated states of the hERG1 channel: structural mechanisms of the state‐dependent drug binding. J Chem Inf Model 52, 2760–2774. [DOI] [PubMed] [Google Scholar]
- Durdagi S, Duff HJ & Noskov SY (2011). Combined receptor and ligand‐based approach to the universal pharmacophore model development for studies of drug blockade to the hERG1 pore domain. J Chem Inf Model 51, 463–474. [DOI] [PubMed] [Google Scholar]
- Durdagi S, Subbotina J, Lees‐Miller J, Guo J, Duff HJ & Noskov SY (2010). Insights into the molecular mechanism of hERG1 channel activation and blockade by drugs. Curr Med Chem 17, 3514–3532. [DOI] [PubMed] [Google Scholar]
- Ebert SN, Liu XK & Woosley RL (1998). Female gender as a risk factor for drug‐induced cardiac arrhythmias: evaluation of clinical and experimental evidence. J Womens Health 7, 547–557. [DOI] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H & Pedersen LG (1995). A smooth particle mesh ewald method. J Chem Phys 103, 8577–8593. [Google Scholar]
- Faber GM & Rudy Y (2000). Action potential and contractility changes in [Na+]i overloaded cardiac myocytes: a simulation study. Biophys J 78, 2392–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Ghanta A, Kauffman GW & Sanguinetti MC (2004). Physicochemical features of the HERG channel drug binding site. J Biol Chem 279, 10120–10127. [DOI] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W & Brown AM (2001). Molecular determinants of inactivation and dofetilide block in ether a‐go‐go (EAG) channels and EAG‐related K+ channels. Mol Pharmacol 60, 1343–1348. [DOI] [PubMed] [Google Scholar]
- Franz MR, Swerdlow CD, Liem LB & Schaefer J (1988). Cycle length dependence of human action potential duration in vivo. Effects of single extrastimuli, sudden sustained rate acceleration and deceleration, and different steady‐state frequencies. J Clin Invest 82, 972–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T & Kurokawa J (2007). Regulation of cardiac ion channels via non‐genomic action of sex steroid hormones: implication for the gender difference in cardiac arrhythmias. Pharmacol Ther 115, 106–115. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S & Demolombe S (2007). Regional and tissue specific transcript signatures of ion channel genes in the non‐diseased human heart. J Physiol 582, 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaborit N, Varro A, Le Bouter S, Szuts V, Escande D, Nattel S & Demolombe S (2010). Gender‐related differences in ion‐channel and transporter subunit expression in non‐diseased human hearts. J Mol Cell Cardiol 49, 639–646. [DOI] [PubMed] [Google Scholar]
- Gabrielsen M, Ravna AW, Kristiansen K & Sylte I (2012). Substrate binding and translocation of the serotonin transporter studied by docking and molecular dynamics simulations. J Mol Model 18, 1073–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gima K & Rudy Y (2002). Ionic current basis of electrocardiographic waveforms: a model study. Circ Res 90, 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N & Efimov IR (2010). Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res 106, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in the journal of physiology and experimental physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RD & Belevych AE (2003). Muscarinic regulation of cardiac ion channels. Br J Pharmacol 139, 1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeker GS, Hood AR, Katra RP, Poelzing S & Pogwizd SM (2014). Sex differences in beta‐adrenergic responsiveness of action potentials and intracellular calcium handling in isolated rabbit hearts. PLoS One 9, e111411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L & Roux B (2013). Automated force field parameterization for nonpolarizable and polarizable models based on ab initio target data. J Chem Theory Comput 9, 3543–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huikuri HV, Pikkujamsa SM, Airaksinen KE, Ikaheimo MJ, Rantala AO, Kauma H, Lilja M & Kesaniemi YA (1996). Sex‐related differences in autonomic modulation of heart rate in middle‐aged subjects. Circulation 94, 122–125. [DOI] [PubMed] [Google Scholar]
- Huo J, Zhang Y, Huang N, Liu P, Huang C, Guo X, Jiang W, Zhou N, Grace A, Huang CL & Ma A (2008). The G604S‐hERG mutation alters the biophysical properties and exerts a dominant‐negative effect on expression of hERG channels in HEK293 cells. Pflugers Archiv 456, 917–928. [DOI] [PubMed] [Google Scholar]
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES & Coleman RG (2012). ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AF, Choisy SCM & Hancox JC (2007). Recent advances in understanding sex differences in cardiac repolarization. Prog Biophys Mol Biol 94, 265–319. [DOI] [PubMed] [Google Scholar]
- Janse de Jonge XA, Boot CR, Thom JM, Ruell PA & Thompson MW (2001). The influence of menstrual cycle phase on skeletal muscle contractile characteristics in humans. J Physiol 530, 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG & Im W (2008). CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW & Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J Chem Phys 79, 926–935. [Google Scholar]
- Jose AD & Collison D (1970). The normal range and determinants of the intrinsic heart rate in man. Cardiovasc Res 4, 160–167. [DOI] [PubMed] [Google Scholar]
- Kim JA, Lopes CM, Moss AJ, McNitt S, Barsheshet A, Robinson JL, Zareba W, Ackerman MJ, Kaufman ES, Towbin JA, Vincent M & Goldenberg I (2010). Trigger‐specific risk factors and response to therapy in long QT syndrome type 2. Heart Rhythm 7, 1797–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon‐Ramirez C, Vorobyov I, MacKerell AD Jr & Pastor RW (2010). Update of the CHARMM all‐atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp J & Schwede T (2004). The SWISS‐MODEL Repository of annotated three‐dimensional protein structure homology models. Nucleic Acids Res 32, D230–D234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa J, Sasano T, Kodama M, Li M, Ebana Y, Harada N, Honda S, Nakaya H & Furukawa T (2015). Aromatase knockout mice reveal an impact of estrogen on drug‐induced alternation of murine electrocardiography parameters. J Toxicol Sci 40, 339–348. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Suzuki T & Furukawa T (2009). New aspects for the treatment of cardiac diseases based on the diversity of functional controls on cardiac muscles: acute effects of female hormones on cardiac ion channels and cardiac repolarization. J Pharmacol Sci 109, 334–340. [DOI] [PubMed] [Google Scholar]
- Kurokawa J, Tamagawa M, Harada N, Honda S, Bai CX, Nakaya H & Furukawa T (2008). Acute effects of oestrogen on the guinea pig and human IKr channels and drug‐induced prolongation of cardiac repolarization. J Physiol 586, 2961–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranchi PA, Ackerman MJ, Kara T, Shamsuzzaman AS, Wolk R, Jurak P, Amin R & Somers VK (2010). Gene‐specific paradoxical QT responses during rapid eye movement sleep in women with congenital long QT syndrome. Heart Rhythm 7, 1067–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees‐Miller JP, Duan Y, Teng GQ & Duff HJ (2000). Molecular determinant of high‐affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol 57, 367–374. [PubMed] [Google Scholar]
- Lees‐Miller JP, Guo J, Wang Y, Perissinotti LL, Noskov SY & Duff HJ (2015). Ivabradine prolongs phase 3 of cardiac repolarization and blocks the hERG1 (KCNH2) current over a concentration‐range overlapping with that required to block HCN4. J Mol Cell Cardiol 85, 71–78. [DOI] [PubMed] [Google Scholar]
- Lehmann MH, Hardy S, Archibald D, Quart B & MacNeil DJ (1996). Sex difference in risk of torsade de pointes with d,l‐sotalol. Circulation 94, 2535–2541. [DOI] [PubMed] [Google Scholar]
- Liu CC, Kuo TB & Yang CC (2003). Effects of estrogen on gender‐related autonomic differences in humans. Am J Physiol Heart Circ Physiol 285, H2188–H2193. [DOI] [PubMed] [Google Scholar]
- Liu T, Choi B‐R, Drici M‐D & Salama G (2005). Sex modulates the arrhythmogenic substrate in prepubertal rabbit hearts with Long QT 2. J Cardiovasc Electrophysiol 16, 516–524. [DOI] [PubMed] [Google Scholar]
- Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, Towbin JA, Priori SG, Napolitano C, Robinson JL, Andrews M, Timothy K & Hall WJ (1998). Age‐ and sex‐related differences in clinical manifestations in patients with congenital long‐QT syndrome: findings from the International LQTS Registry. Circulation 97, 2237–2244. [DOI] [PubMed] [Google Scholar]
- Lou Q, Fedorov VV, Glukhov AV, Moazami N, Fast VG & Efimov IR (2011). Transmural heterogeneity and remodeling of ventricular excitation‐contraction coupling in human heart failure. Circulation 123, 1881–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph‐McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz‐Kuczera J, Yin D & Karplus M (1998). All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Magnano AR, Holleran S, Ramakrishnan R, Reiffel JA & Bloomfield DM (2002). Autonomic nervous system influences on QT interval in normal subjects. J Am Coll Cardiol 39, 1820–1826. [DOI] [PubMed] [Google Scholar]
- Makkar RR, Fromm BS, Steinman RT, Meissner MD & Lehmann MH (1993). Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. J Am Med Assoc 270, 2590–2597. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J & Sanguinetti MC (2000). Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J Gen Physiol 115, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss AJ, Robinson JL, Gessman L, Gillespie R, Zareba W, Schwartz PJ, Vincent GM, Benhorin J, Heilbron EL, Towbin JA, Priori SG, Napolitano C, Zhang L, Medina A, Andrews ML & Timothy K (1999). Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol 84, 876–879. [DOI] [PubMed] [Google Scholar]
- Munro CJ, Stabenfeldt GH, Cragun JR, Addiego LA, Overstreet JW & Lasley BL (1991). Relationship of serum estradiol and progesterone concentrations to the excretion profiles of their major urinary metabolites as measured by enzyme immunoassay and radioimmunoassay. Clin Chem 37, 838–844. [PubMed] [Google Scholar]
- Nakagawa M, Ooie T, Ou B, Ichinose M, Takahashi N, Hara M, Yonemochi H & Saikawa T (2005). Gender differences in autonomic modulation of ventricular repolarization in humans. J Cardiovasc Electrophysiol 16, 278–284. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Ooie T, Takahashi N, Taniguchi Y, Anan F, Yonemochi H & Saikawa T (2006). Influence of menstrual cycle on QT interval dynamics. Pacing Clin Electrophysiol 29, 607–613. [DOI] [PubMed] [Google Scholar]
- Nakajo K, Ulbrich MH, Kubo Y & Isacoff EY (2010). Stoichiometry of the KCNQ1 – KCNE1 ion channel complex. Proc Natl Acad Sci USA 107, 18862–18867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Kurokawa J, Bai C‐X, Asada K, Xu J, Oren RV, Zhu ZI, Clancy CE, Isobe M & Furukawa T (2007). Progesterone regulates cardiac repolarization through a nongenomic pathway: an in vitro patch‐clamp and computational modeling study. Circulation 116, 2913–2922. [DOI] [PubMed] [Google Scholar]
- Noskov SY, Berneche S & Roux B (2004). Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature 431, 830–834. [DOI] [PubMed] [Google Scholar]
- Noskov SY & Roux B (2008). Control of ion selectivity in LeuT: Two Na+ binding sites with two different mechanisms. J Mol Biol 377, 804–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara T & Rudy Y (2012). Arrhythmia formation in subclinical (“silent”) long QT syndrome requires multiple insults: quantitative mechanistic study using the KCNQ1 mutation Q357R as example. Heart Rhythm 9, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara T, Virag L, Varro A & Rudy Y (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7, e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages G, Torres AM, Ju P, Bansal PS, Alewood PF, Kuchel PW & Vandenberg JI (2009). Structure of the pore‐helix of the hERG K+ channel. Eur Biophys J 39, 111–120. [DOI] [PubMed] [Google Scholar]
- Perrin MJ, Kuchel PW, Campbell TJ & Vandenberg JI (2008). Drug binding to the inactivated state is necessary but not sufficient for high‐affinity binding to human ether‐à‐go‐go‐related gene channels. Mol Pharmacol 74, 1443–1452. [DOI] [PubMed] [Google Scholar]
- Perry M, de Greoot MJ, Helliwell R, Leishman D, Tristani‐Firouzi M, Sanguinetti MC & Mitcheson J (2004). Structural determinants of HERG channel block by clofilium and ibutilide. Mol Pharmacol 66, 240–249. [DOI] [PubMed] [Google Scholar]
- Pham TV, Robinson RB, Danilo JP & Rosen MR (2002). Effects of gonadal steroids on gender‐related differences in transmural dispersion of L‐type calcium current. Cardiovasc Res 53, 752. [DOI] [PubMed] [Google Scholar]
- Pham TV & Rosen MR (2002). Sex, hormones, and repolarization. Cardiovasc Res 53, 740–751. [DOI] [PubMed] [Google Scholar]
- Pham TV, Sosunov EA, Gainullin RZ, Danilo P Jr & Rosen MR (2001). Impact of sex and gonadal steroids on prolongation of ventricular repolarization and arrhythmias induced by IK‐blocking drugs. Circulation 103, 2207–2212. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L & Schulten K (2005). Scalable molecular dynamics with NAMD. J Comput Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JL, Palmer LG & Roux B (2008). Long‐pore electrostatics in inward‐rectifier potassium channels. J Gen Physiol 132, 613–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez I, Kilborn MJ, Liu XK, Pezzullo JC & Woosley RL (2001). Drug‐induced QT prolongation in women during the menstrual cycle. J Am Med Assoc 285, 1322–1326. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Menchaca A, Ferrer‐Villada T, Lara J, Fernandez D, Navarro‐Polanco RA & Sanchez‐Chapula JA (2006). Block of HERG channels by berberine: mechanisms of voltage‐ and state‐dependence probed with site‐directed mutant channels. J Cardiovasc Pharmacol 47, 21–29. [DOI] [PubMed] [Google Scholar]
- Romero L, Trenor B, Yang PC, Saiz J & Clancy CE (2014). In silico screening of the impact of hERG channel kinetic abnormalities on channel block and susceptibility to acquired long QT syndrome. J Mol Cell Cardiol 72, 126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama G & Bett GC (2014). Sex differences in the mechanisms underlying long QT syndrome. Am J Physiol Heart Circ Physiol 307, H640–H648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoni Y, Emmett T, Schmidt R, Nygren A & Kargacin G (2009). Sex‐dependent impairment of cardiac action potential conduction in type 1 diabetic rats. Am J Physiol Heart Circ Physiol 296, H1442–H1450. [DOI] [PubMed] [Google Scholar]
- Sims C, Reisenweber S, Viswanathan PC, Choi B‐R, Walker WH & Salama G (2008). Sex, age, and regional differences in L‐type calcium current are important determinants of arrhythmia phenotype in rabbit hearts with drug‐induced long QT type 2. Circ Res 102, e86–e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smetana P, Batchvarov VN, Hnatkova K, Camm AJ & Malik M (2002). Sex differences in repolarization homogeneity and its circadian pattern. Am J Physiol Heart Circ Physiol 282, H1889–H1897. [DOI] [PubMed] [Google Scholar]
- Stansfeld PJ, Gedeck P, Gosling M, Cox B, Mitcheson JS & Sutcliffe MJ (2007). Drug block of the hERG potassium channel: Insight from modeling. Proteins 68, 568–580. [DOI] [PubMed] [Google Scholar]
- Stary A, Wacker SJ, Boukharta L, Zachariae U, Karimi‐Nejad Y, Aqvist J, Vriend G & de Groot BL (2010). Toward a consensus model of the HERG potassium channel. ChemMedChem 5, 455–467. [DOI] [PubMed] [Google Scholar]
- Stork D, Timin EN, Berjukow S, Huber C, Hohaus A, Auer M & Hering S (2007). State dependent dissociation of HERG channel inhibitors. Br J Pharmacol 151, 1368–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stramba‐Badiale M, Locati EH, Martinelli A, Courville J & Schwartz PJ (1997). Gender and the relationship between ventricular repolarization and cardiac cycle length during 24‐h Holter recordings. Eur Heart J 18, 1000–1006. [DOI] [PubMed] [Google Scholar]
- Tadros R, Ton AT, Fiset C & Nattel S (2014). Sex differences in cardiac electrophysiology and clinical arrhythmias: epidemiology, therapeutics, and mechanisms. Can J Cardiol 30, 783–792. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG & Gibson TJ (1994). CLUSTAL‐W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position‐specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg JI, Walker BD & Campbell TJ (2001). HERG K+ channels: friend and foe. Trends Pharmacol Sci 22, 240–246. [DOI] [PubMed] [Google Scholar]
- Van Opstal JM, Leunissen JD, Wellens HJ & Vos MA (2001). Azimilide and dofetilide produce similar electrophysiological and proarrhythmic effects in a canine model of Torsade de Pointes arrhythmias. Eur J Pharmacol 412, 67–76. [DOI] [PubMed] [Google Scholar]
- Vaseghi M & Shivkumar K (2008). The role of the autonomic nervous system in sudden cardiac death. Prog Cardiovasc Dis 50, 404–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YB, Guo JQ, Perissinotti LL, Lees‐Miller J, Teng GQ, Durdagi S, Duff HJ & Noskov SY (2016). Role of the pH in state‐dependent blockade of hERG currents. Sci Rep 6, 32536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher JR & MacKinnon R (2016). Structure of the voltage‐gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 353, 664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde AA, Jongbloed RJ, Doevendans PA, Duren DR, Hauer RN, van Langen IM, van Tintelen JP, Smeets HJ, Meyer H & Geelen JL (1999). Auditory stimuli as a trigger for arrhythmic events differentiate HERG‐related (LQTS2) patients from KVLQT1‐related patients (LQTS1). J Am Coll Cardiol 33, 327–332. [DOI] [PubMed] [Google Scholar]
- Xie Y, Sato D, Garfinkel A, Qu Z & Weiss JN (2010). So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J 99, 1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PC & Clancy CE (2012). In silico prediction of sex‐based differences in human susceptibility to cardiac ventricular tachyarrhythmias. Front Physiol 3, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang PC, Kurokawa J, Furukawa T & Clancy CE (2010). Acute effects of sex steroid hormones on susceptibility to cardiac arrhythmias: a simulation study. PLoS Comput Biol 6, e1000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Chen G, Papp R, Defranco DB, Zeng F & Salama G (2012). Oestrogen upregulates L‐type Ca²⁺ channels via oestrogen‐receptor‐α by a regional genomic mechanism in female rabbit hearts. J Physiol 590, 493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesselman JD, Price DJ, Knight JL & Brooks CL (2012). MATCH: an atom‐typing toolset for molecular mechanics force fields. J Comput Chem 33, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RJ & Panfilov AV (2010). Anisotropy of wave propagation in the heart can be modeled by a Riemannian electrophysiological metric. Proc Natl Acad Sci USA 107, 15063–15068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae U, Giordanetto F & Leach AG (2009). Side chain flexibilities in the human ether‐a‐go‐go related gene potassium channel (hERG) together with matched‐pair binding studies suggest a new binding mode for channel blockers. J Med Chem 52, 4266–4276. [DOI] [PubMed] [Google Scholar]