
Figure 10. Oestradiol (E2) binding to a pore loop site of hERG from molecular docking (A), and molecular dynamics (B and C) simulations for wild‐type (WT; left) and G604S mutant hERG channel (right).
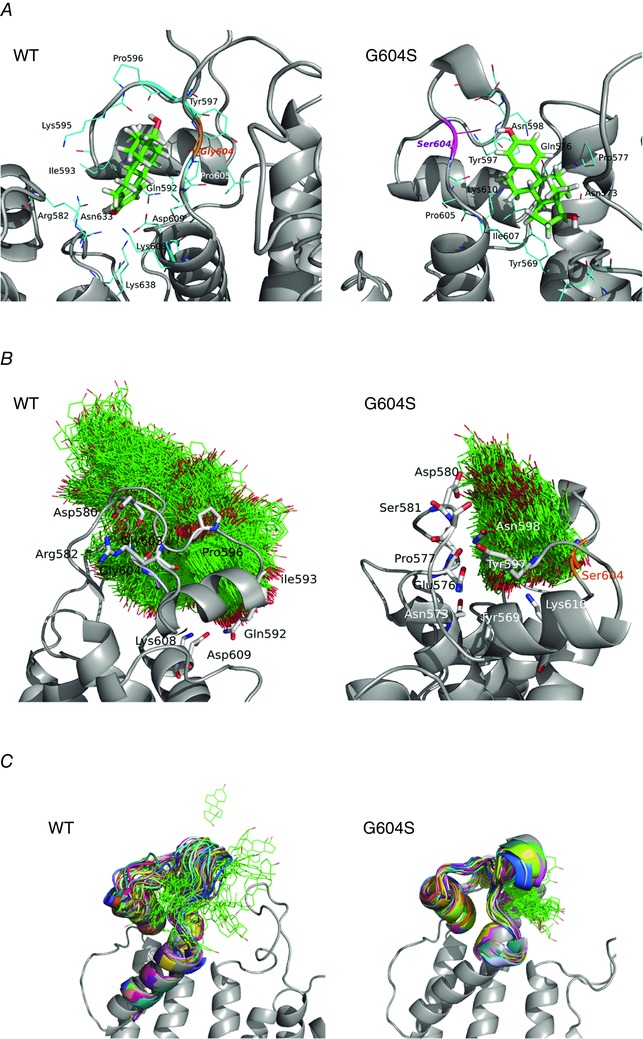
A, top scoring docking poses for dofetilide in the pore loop site shown together with coordinating residues within 3.5 Å for mapped binding sites. B, E2 positions (shown in green wireframe representation) are mapped from the frames collected in 40 ns MD for WT and G604S in order to illustrate the conformational space explored by the ligand in the two systems. It is apparent that E2 is kinetically stabilized (trapped) in the G604S mutant, while it is unable to bind stably in WT and leaves the binding pocket within 20 ns of MD simulation. C, a G604S mutation is predicted to enhance stability of the bound E2 (green wireframe) in the binding pocket by controlling binding site flexibility: the mutation stabilizes the binding site allowing for E2 to remain in the pocket. In contrast, E2 readily leaves binding pocket in the WT hERG protein and diffuses away to the lipid bilayer. Shown are 32 selected frames from the first 15 ns of molecular dynamics simulation for E2/WT and E2/G604S are shown in different colours.