

Abstract
Objectives
This review surveys trialed therapies and molecular defects in adenoid cystic carcinoma (ACC), with an emphasis on neural crest‐like stemness characteristics of newly discovered cancer stem cells (CSCs) and therapies that may target these CSCs.
Data Sources
Articles available on Pubmed or OVID MEDLINE databases and unpublished data.
Review Methods
Systematic review of articles pertaining to ACC and neural crest‐like stem cells.
Results
Adenoid cystic carcinoma of the salivary gland is a slowly growing but relentless cancer that is prone to nerve invasion and metastases. A lack of understanding of molecular etiology and absence of targetable drivers has limited therapy for patients with ACC to surgery and radiation. Currently, no curative treatments are available for patients with metastatic disease, which highlights the need for effective new therapies. Research in this area has been inhibited by the lack of validated cell lines and a paucity of clinically useful markers. The ACC research environment has recently improved, thanks to the introduction of novel tools, technologies, approaches, and models. Improved understanding of ACC suggests that neural crest‐like stemness is a major target in this rare tumor. New cell culture techniques and patient‐derived xenografts provide tools for preclinical testing.
Conclusion
Preclinical research has not identified effective targets in ACC, as confirmed by the large number of failed clinical trials. New molecular data suggest that drivers of neural crest‐like stemness may be required for maintenance of ACC; as such, CSCs are a target for therapy of ACC.
INTRODUCTION
Salivary Adenoid Cystic Carcinoma (ACC) is a slow growing but relentlessly recurring and progressive tumor. Until recently, little has been understood about molecular drivers or therapeutic targets. Cell cultures and models have only recently emerged. Insights from new sequencing data and cultures suggest that ACC possesses characteristics of neural crest stem cells and that targeting drivers of neural crest stemness may result in therapeutic benefit.
Clinical Presentation, Histopathology, and Current Therapy
Adenoid cystic carcinoma (ACC) is the second most frequent malignancy of salivary glands, accounting for more than 20% of all salivary cancers with an incidence of ∼1.5 per 100,000.1, 2 Familial history, toxic exposures, or environmental exposures are not associated with increased risk of ACC.2, 3 In the parotid gland, ACC is the second most common cancer type, occurring in 7% to 18% of cases; however, ACC is most common cancer type in the sublingual, submandibular, and minor salivary glands.4, 5, 6, 7Whereas most often found in the minor salivary glands of the tongue, buccal mucosa, and palate,4, 6 ACC also occurs in other airway sites, including the paranasal sinuses, lips, larynx, trachea, and nasolacrimal tract.8 Outside of the aerodigestive tract, ACC arises in diverse sites that contain glandular structures, such as the breast,9 prostate,10 skin,11 and female genital tract.12 The classical behavior of ACC is somewhat paradoxical: growth is slow and indolent, but the cancer is relentless and progressive.8 Adenoid cystic carcinoma normally presents as an asymptomatic mass, but can be associated with numbness, paresthesia, or pain. Although the 5‐year survival rate of ACC patients is relatively high at ∼70%, 15‐year survival rate drops to ∼40%, and 20‐year survival is as low as 20%.13, 14 Due to this indolent course, ACC affects about 10,000 lives in the United States alone. The major reasons for poor survival are frequent local recurrences and distant metastases to the lungs, brain, and bone.15, 16
Adenoid cystic carcinoma is composed of two cell types: myoepithelial and ductal cells.6 Within the tumor, myoepithelial‐type cells are typically arranged at the periphery of nests or tubules, whereas cells with ductal/epithelial differentiation are arranged toward the pseudo‐lumens. Myoepithelial cells share immunophenotypic features with smooth muscle and basal/stem cells.17, 18, 19 Of the three histological subtypes of ACC (cribriform, tubular, and solid), cribriform is the most common, and ACC grade is based solely on the growth pattern: grade 1 = tubular, grade 2 = cribriform, and grade 3 = solid.20, 21 Solid histology has historically been associated with a worse prognosis22; however, more recent studies reveal that tumor stage rather than histologic grade is a more accurate prognostic indicator.23, 24 In all subtypes, perineural invasion is very common (60% of cases), even in early stage tumors.25, 26, 27
Treatment of ACC is currently limited to surgery with or without postoperative radiation. Positive margin status is a significant predictor of local recurrence, but interestingly, margin status does not alter the rate of distant metastasis.28 High recurrence rates and dismal survival of ACC may be explained in part by its intrinsic resistance to radiation and chemotherapy, emphasizing the need for new targeted therapies29; however, a myriad of clinical trials have not been successful (see below).
MATERIALS AND METHODS
Articles available on PubMed or OVID MEDLINE database were reviewed and combined with recent unpublished observations.
RESULTS AND DISCUSSION
Clinical Trials
Systemic Single‐Agent Cytotoxics
Because recurrence and metastasis occur frequently despite surgery and radiation, multiple clinical trials have been performed to determine utility of systemic therapy in ACC. Despite over 30 phase II clinical trials enrolling patients with ACC since 1985, no drug therapies have shown efficacy or been approved for ACC treatment.30
Single‐agent treatments with traditional cytotoxic drugs have largely failed to achieve objective responses. In eight trials of single‐agent cytotoxic regimens of gemcitabine,31 paclitaxel,32 cisplatin,33, 34 mitotraxone,35, 36 epirubicin,37 and vinorelbine,38 objective responses occurred in only 18 of 141 (12.8%) patients with recurrent and/or metastatic disease.30 For those cases that did respond to treatment, durations of response were short, ranging from 5 to 20 months. Disease stabilization was a more common finding in these trials, reported in 64 of 111 (57.7%) patients30; however, durations of effect were relatively short, with a maximum reported stabilization period of 30 months. Given its variably indolent nature, treatment‐induced disease stabilization versus natural course of disease is difficult to distinguish in ACC.
Combinatorial Cytotoxic Regimens
Combinations of traditional cytotoxic agents in ACC produced a higher response rate compared to single agents, but once again failed to attain long‐term responses. A review of 17 trials of combinatorial chemotherapy found that 35 of 143 (24.5%) patients demonstrated an objective response.30 Cisplatin and doxorubicin were most commonly tested, and they were employed in combination with cyclophosphamide in four trials.39, 40, 41, 42 These trials reported the highest objective response rates occurring in nine of 36 (25%) patients; however, duration of response was only from 5 to 16 months.40, 42 Given the short duration of response and accompanying increased toxicities, no combinations were recommended as a standard.38
Despite the low rate of objective responses, chemotherapeutic agents are used for palliation in ACC management, with symptomatic relief reported in 30% to 70% of patients treated with single‐agents and 25% to 64% of those treated with combinatorial regimens.30
Targeted Therapies
Given the discovery of high c‐Kit expression in 90% of ACC tumors, imatinib, a tyrosine kinase inhibitor (TKI) with activity blocking c‐Kit activity, has been the agent of multiple trials. Six trials of have assessed imatinib activity in a cumulative 71 ACC patients, five of which found no objective responses43, 44, 45, 46, 47 and one of which saw two objective responses.46 One trial also assessed the effect of adding cisplatin to imatinib, reporting three patients with partial responses in the combination group versus none in the imatinib‐only group.48 As a result of these trials, targeting c‐Kit therapy has currently been abandoned in ACC.
As in other epithelial cancers, members of the epidermal growth factor receptor (EGFR) family are expressed/overexpressed in ACC,49 prompting trials of EGFR inhibitors. Gefitinib, the first inhibitor of EGFR's tyrosine kinase domain; Lapatinib, a TKI targeting EGFR and human epidermal growth factor receptor 2 (HER2/neu or ERBB2); and cetuximab, a monoclonal antibody inhibitor of EGFR, each failed to elicit objective responses in ACC patients.50, 51, 52 Combinatorial therapy using cetuximab as a backbone with cisplatin, and 5‐fluorouracil in 12 patients with metastatic EGFR‐positive ACC or cisplatin, and concomitant radiotherapy in nine patients with locally advanced EGFR‐positive ACC53, revealed favorable results compared to historical outcomes. Objective responses were found in four (44%) patients with locally advanced disease and five (42%) patients with metastatic disease.53 Although the toxicity of these regimens can be high, regimens combining EGFR inhibitors; platinum‐based chemotherapy; and radiotherapy, when indicated, may be a viable treatment approach in ACC.
A notable proportion of ACCs highly express fibroblast growth factor receptor (FGFR) pathway components, and mutations of FGFRs have been found in a small percentage of tumors,54, 55 providing impetus for clinical trials of fibroblast growth factor receptor inhibition. A phase II study of dovitinib, a multi‐kinase inhibitor of FGFR, vascular endothelial growth factor receptors (VEGF), and platelet‐derived growth factor receptors (PDGFR) revealed only one partial response (3%) in patients with unresectable or metastatic ACC.56 Similarly, sunitinib, which inhibits of multiple targets expressed in ACC (e.g., c‐Kit, VEGFR, PDGFR, and the rearranged during transfection [RET] proto‐oncogene) also failed to deliver objective responses in patients with progressive, recurrent, or metastatic disease.57
Trials of bortezomib, a proteasome inhibitor, alone or in combination, delivered only one partial response in a patient treated concurrently with doxorubicin.58 Inhibition of the mammalian target of rapamycin with everolimus was studied a phase II trial enrolling ACC patients, but no responses were observed.59
Current Trials
Development of drugs inhibiting potential molecular targets in ACC is prompting more phase II clinical trials that are currently ongoing. Agents under investigation include selinexor (inhibitor of nuclear export), eribulin (inhibitor of microtubule dynamics), and pembrolizumab (PD‐1 inhibitor).
Cytotoxic and targeted therapies have dismally failed to impact disease progression in ACC. To improve clinical trial success, a more complete understanding of molecular changes in ACC—accompanied by biological studies to identify important molecular drivers as well as preclinical models—are needed.
Mutational Landscape of Adenoid Cystic Carcinoma
The poor track record of clinical trials using cytotoxic and targeted agents in ACC reflects inherent or rapid development of acquired resistance and highlights the inadequacy or lack of preclinical studies. Fortunately, advances in sequencing and in cell culture techniques are beginning to provide improved insight into potential therapeutic targets. Three reports of genome and whole exome sequencing of a combined 112 specimens54, 55, 60 have catalogued genetic defects in ACC.
Chromosomal Aberrations and Myeloblastosis and Myeloblastosis‐like 1 Fusions
Recurrent chromosomal loss of 1p36, 6q, 9p, and 12q occur in ACC,54, 61, 62 and ACC‐intrinsic chromosomal aberrations in 6q16‐25 have been known for almost 30 years63, 64; however, only recently has the product of the translocation between chromosomes 6 and 9 t(6;9) been shown to produce a fusion between the myeloblastosis (MYB) and the nuclear factor 1 B‐type (NFIB) transcription factors.65, 66 Myeloblastosis‐NFIB fusions are found in 50% of ACC cases; however, other MYB translocations identified by whole genome sequencing raise the total of MYB translocations in ACC to approximately 75%, making them by far the most frequent recurrent ACC‐intrinsic genetic alteration.54, 55, 67, 68, 69, 70, 71
As a transcription factor, MYB's oncogenic activity is well established, primarily due to its clear role as a driver of many hematopoietic cancers. Myeloblastosis was initially found as the cellular homologue of the viral transforming viral‐MYB (v‐MYB) gene, which induces leukemia following infection of birds with the avian retroviruses: avian myeloblastosis virus and E26 leukemia virus (reviewed in72). The MYB protein contains three motifs: an N‐terminal helix‐loop‐helix DNA binding domain that binds to PyAACG/T, a central transcription activation domain, and a C‐terminal negative regulatory domain (reviewed in72). In some human tumors, MYB mutations result in truncated proteins reminiscent of truncations that activate v‐MYB. Tumor‐associated MYB mutations increase MYB protein production or stability through disruption of MYB gene sequences that inhibit its transcription or through loss of the C‐terminal negative regulatory domain. However, MYB lacking mutations is also amplified in solid tumors, including about 30% of BRCA‐1 mutated hereditary triple‐negative breast cancers and approximately 10% of pancreatic cancers. Curiously, MYB mutations are not described in ACC.
As an alternative means of MYB activation, gene fusions or rearrangements involving MYB are found in several tumor types. A subset of T‐cell acute lymphoblastic leukemia (T‐ALL) is defined by an intrachromosomal fusion placing MYB adjacent to the T‐cell receptor β gene. Intrachromosomal rearrangements placing MYB adjacent to sequences within chromosome 6 and creating fusions of C‐terminally truncated MYB are found in angiocentric gliomas, a subset of pediatric low‐grade gliomas.73 In ACC, the most frequent translocation between chromosomes 6 and 9 t(6;9) links exon 14 of MYB with exon 8c or 9 of NFIB67, 72; but other MYB‐NFIB breakpoints are described. We recently identified an intrachromosomal 6q rearrangement that truncates MYB in exon 14, producing a C‐terminal MYB truncation65 and adjoining the truncated MYB gene to a long noncoding RNA gene (unpublished data, Yale Laboratory for Head and Neck Oncology). Regardless of the exact breakpoint, all fusions in ACC maintain both the DNA binding and transcription activation domains of MYB.67, 72
The C‐terminal MYB truncations, as frequently produced by MYB translocations in cancer, stabilize the MYB protein or mRNA by diminishing its C‐terminal ubiquitination or by excluding MYB sequences that bind to negatively regulating miRNAs.74, 75, 76 In addition to its activation through C‐terminal truncation, translocations or intrachromosomal rearrangements can place enhancer elements close to the MYB gene. In ACC, translocations juxtapose the MYB gene and superenhancers flanking NFIB, and these enhancers interact with the MYB promoters to drive MYB expression.71 Translocations that adjoin the MYB gene to NFIB‐enhancer sequences establish a positive‐feedback loop because the MYB protein binds to the errantly adjacent superenhancer, thereby increasing its own transcription.
Somewhat surprisingly, a subset of known fusion‐negative ACCs highly express MYB, suggesting that this overexpression occurs through other mechanisms.68, 77 Adenoid cystic carcinoma gene expression profiling reveals a common intrinsic gene signature, with MYB being the top scoring gene, even among those who do not contain MYB fusions.78 These findings suggest that the known MYB‐NFIB fusions are not the only mechanism driving MYB activity. As a potential alternative mechanism for MYB stabilization, homozygous deletion or mutation of the F‐Box and WD repeat domain containing 7 (FBXW7) gene is described in ACC79, 80 (unpublished data, Yale Laboratory for Head and Neck Oncology). Given that FBXW7 functions to ubiquitinate the MYB protein, leading to its destruction, loss of FBXW7 is expected to increase MYB expression through stabilization of the MYB protein.
Mechanisms driving MYB expression, such as loss of FBXW7 or MYB fusions to partners other than NFIB, easily explain detection of the MYB transcriptional signature in ACCs that lack MYB‐NFIB fusions. However, the MYB expression signature is also found in tumors lacking MYB expression, suggesting that another mechanism drives a MYB‐like expression pattern.78 Myeloblastosis has two homologues, MYB‐like 1 (MYBL1) and MYB‐like 2 (MYBL2). Defects in MYBL1 are associated with pediatric low‐grade gliomas.81 Whole genome sequencing of ACC lacking MYB‐NFIB fusions found that approximately 30% contained a fusion between MYBL1 and NFIB, or MYBL1 and other loci.82 As with MYB fusions, MYBL1 fusions retain the DNA binding and transcription activation domains and express the fusion protein. Expression of MYB and MYBL1 is inversely correlated,82 suggesting that MYB or MYBL1 likely serve identical functions as independent drivers of ACC. It is possible that all ACCs must have MYB or MYBL1 expressed or activated. Finding that gene expression profiles did not differ between tumors with MYB or MYBL1 fusions further supports this hypothesis.82
As outlined above, it is becoming clear that MYB or MYB family members are highly expressed in the majority ACC regardless of fusion status,65, 67 but the potential role of fusion partners is not known. Angiocentric gliomas contain a characteristic fusion between MYB and Quaking (QKI),73 and the in‐frame fusion with QKI inactivates this tumor suppressor gene. These findings suggest that, although MYB activation is required, the fusion partner may also be important for tumor development with certain tumors being dependent on certain MYB fusion partners. If activation of MYB signaling alone is sufficient to drive ACC development, it is curious that NFIB is such a favored partner. Whereas the favored partner status of NFIB in ACC may be related to unknown factors favoring interaction of MYB and NFIB genes in this cell type, the preference of NFIB may relate to the findings that NFIB fusion proteins are more stable compared to truncated, unfused MYB, and that MYB‐NFIB fusions in untransformed Schwann cells supports a more transformed phenotype compared to the expression of truncated MYB in the absence of fused NFIB sequences.78 Plausible explanations of the preference for NFIB as the fusion partner of MYB have been offered; however, a role for NFIB has yet to be described.
Although MYB signaling is clearly a driver and likely required in ACC, the absence of cell lines and MYB targeting drugs has prevented exploration of MYB's role in ACC carcinogenesis and/or tumor maintenance.
Mutations
Following advancements in sequencing technologies, the complement of gene defects in many cancers has been described. For ACC, three sequencing studies of an aggregate 112 tumors revealed one of the lowest mutation rates in human cancer of less than or equal to 13 mutations per exome or 0.4 mutations per megabase (Table 1).54, 55, 83 Previously, the lowest rate of ∼1 mutation per Mb was detected in chronic lymphocytic leukemia, whereas the highest rate was found in melanoma (∼15 mutations per megabase).84 A large variety of genes are mutated in ACC, with few recurrent mutations identified in any single study—and despite the haphazard appearance of mutations in ACC, recurrent alterations can be grouped into those involved in NOTCH and FGF signaling as well as epigenetic modification. Fibroblast growth factor receptor and NOTCH defects will be discussed later as drivers of neural crest‐like stemness. Epigenetic states required for stemness place chromatin in a state that allows for transcription widely across genes while simultaneously allowing for suppression of genes involved in differentiation. The role of epigenetic modifiers in normal and cancer stem cell (CSC) maintenance has been reviewed.85, 86, 87 In ACC, the histone 3 lysine 27 demethylase (KDM6A) is recurrently the target of splice and frameshift mutations, with the mutation rate ranging from 7% in mixed‐grade ACC55 to 18% in high‐grade ACC.83 In addition to defects involving KDM6A, mutations in other histone modifiers (SMARCA2, CREBBP, EP300, and ARID1A) are found in ACC. The low mutational burden, combined with alterations in histone modifiers, suggests an important role for epigenetic regulation of gene expression in ACC. Perhaps because of their role in cancer stem‐cell maintenance, or their wide‐ranging effect on gene expression, this group of chromatin modifiers is implicated in many cancers. Inactivating KDM6A mutations occur in pancreatic, bladder, esophageal squamous cell carcinomas, medulloblastoma, glioblastoma, lung, endometrial, colon, kidney, prostate cancers, and hematopoietic tumors.88, 89, 90, 91, 92 Individual genetic alterations and mutations that affect signaling more directly linked to neural crest‐like stemness (e.g., FGFR, MYB, NOTCH) are discussed below.
Table 1.
Whole‐Exome ACC Sequencing From Three Studies Shows Low Overall Somatic Mutation Rate and Reveals Mutations in NOTCH Signaling, FGF Signaling, and Chromatin Remodeling Genes.
| Ho et al., Nat Genet 201355 | Stephens et al., J Clin Invest 201354 | Ross et al., Am J Surg Pathol 201483 | |
|---|---|---|---|
| Tumor specimens, N | 60 | 24 | 28 |
| Mean somatic mutation, rate per exome (per megabase) | 11.8 (0.4) | 13 (0.4) | 1.57 (0.05) |
| MYB‐NFIB fusions | 33 (55%) | 19 (79%) | Not determined |
| Mutations (≥ 5%) | KDM6A (8.3%), RYR3 (6.7%), CREBBP (6.7%), MUC16 (6.7%), NOTCH1 (6.7%), PIK3CA (5%), SMARCA2 (5%), TP53 (5%) | SPEN (25%), SMARCA (12.5%), FGFR2 (8.3%), NOTCH1 (8.3%), KDM6A (8.3%), CREBBP (8.3%), EP300 (8.3%) | KDM6A (18%), NOTCH1 (11%), ARID1A (14%), RUNX1 (7%), MYC (7%) |
ACC = adenoid cystic carcinoma; FGF = fibroblast growth factor; MYB = myeloblastosis; NFIB = nuclear factor 1 B‐type.
Drivers of Neural Crest‐like Stemness in Adenoid Cystic Carcinoma
The marked resistance of ACC to radiation and cytotoxic chemotherapy may be attributed, at least in part, to stem cell populations within the tumor because a hallmark of CSCs is resistance to a variety of therapies.93 A role for stem cells in ACC was predicted in a single study that relied on aldehyde dehydrogenase (ALDH) activity as a marker. Aldefluor measurement of ALDH activity in cells that were derived from ACC xenografts revealed that cells with higher ALDH activity had increased tumorigenesis in nonobese diabetic/severe combined immunodeficiency mice, as well as increased invasion, migration, and spheroid formation in culture.94 Interestingly, measurement of ALDH1 protein expression by immunohistochemistry in 216 ACCs revealed no expression in ACC tumor cells.95 The differing results in these studies may relate to measuring ALDH activity versus measuring ALDH1 expression because ALDH isoforms in addition to ALDH1 contribute to Aldefluor positivity. Despite correlation of stem‐like behaviors with Aldefluor activity, drivers of stemness in ACC were not explored.94
Using Affymetrix expression array analysis, we recently characterized an ACC‐intrinsic gene signature enriched with neural crest stem cell genes and markers.96, 97 This SOX10‐centered expression signature is found in greater than 90% of ACCs. Several of these genes drive critical oncogenic pathways in multiple cancers thought to arise from neural crest, including ACC, glioblastoma, neuroblastoma, melanoma, and basal‐like breast carcinoma (Fig. 1). Key components of the signature and additional genes critical in stem cell maintenance will be discussed.
Figure 1.

Critical genes in neural stem cells that drive formation of adenoid cystic carcinoma, glioblastoma, neuroblastoma, melanoma, and basal‐like breast carcinoma.
Neural Crest Stem Cells
Excluding embryonic stem cells, the neural crest contains perhaps the most plastic cells in human development, serving as precursors to diverse tissue types of both ectodermal and mesodermal designation, including: bone and cartilage of the face and skull, dentin and pulp of the teeth, catecholaminergic cells of adrenal glands, calcitonin‐producing cells of the thyroid, Schwann cells, glia, melanocytes, myofibroblasts, myoepithelial cells, carotid body and other glomus cells, keratocytes in the cornea, smooth muscle cells, neurons, and connective components of the aorticopulmonary septum/outflow tracts of the heart.98, 99 The stem cell nature, ability to differentiate into epithelial and mesenchymal cells, and migratory predisposition are intrinsic to neural crest cells. These stem cell characteristics are co‐opted during carcinogenesis of tissues derived from neural crest and likely contribute to tumor behavior.
The role of neural crest cells in melanomagenesis has long been suspected based on the neural crest origin of melanocytes and gene expression patterns found in these tumors. Elegant experiments now reveal that reversion of a single melanocyte to a neural crest stem cell state defines the origin of melanoma in a zebrafish model system.100 SOX10 is a major transcription factor in neural crest cells that is essential for their maintenance, migration, and development.101 Discovery of a SOX10 coexpression profile containing several genes associated with neural crest stemness led us to propose that neural crest signaling is critical in ACC.96, 97 Myoepithelial cells derived from neural crest surround acini and proximal ducts of several exocrine glands, possibly explaining that ACC is found in these otherwise distinct glands.
SOX10
The SOX family of transcription factors in general, and SOX10 in particular, play key roles in neurogenesis and maintenance of embryonic and adult neural crest stem cells, where SOX10 is essential.99, 101, 102 In agreement with their neural crest origin, SOX10 is also expressed in adult myoepithelial cells97, 103 and is critical for maintenance of adult neural stem cells that reside in the subventricular zone of the brain.104 The role of neural crest stem cell derivatives and SOX10 in cancer is perhaps most clearly understood in melanoma, where SOX10 is almost universally expressed,103 and in a mouse model. The loss of only one SOX10 allele prevented melanoma despite expression of mutant N‐Ras.105 In human cells, depletion of SOX10 suppressed proliferation and abolished melanoma xenograft tumor formation, indicating that SOX10 is required for CSC maintenance in melanoma.105
Interestingly, we found that genes coexpressed with SOX10 in ACC, basal‐like breast carcinoma, melanoma, glioblastoma, and neuroblastoma shared common elements, suggesting that these cancers may rely on neural crest stem cell‐supporting transcription programs.97 Immunohistochemistry experiments performed on a normal and malignant salivary and breast gland tissue confirmed SOX10 expression in each of these tissues.80, 97, 106 Comparative in silico analyses performed by our lab revealed additional neural stem cell markers and regulators coexpressed with SOX10, such as NOTCH1, fatty acid‐binding protein 7 (FABP7), and CD133.96, 97, 107
Myeloblastosis
Very early in the study of MYB, its critical role in hematopoiesis and maintenance of undifferentiated cellular states was identified.108 In normal hematopoiesis, MYB expression inversely correlates with differentiation109, 110 and is required for proliferation and prevention of lineage differentiation.108 Myeloblastosis is also required for maintenance of adult stem cells of the colon and brain. In the colon crypts and in colon cancer, it stimulates stem cells required for tissue maintenance and tumor repopulation, respectively.111, 112 Using a mouse model with inducible tissue specific loss, MYB was also found to be critical for maintenance of neural stem cell progenitors in the adult brain.113 Interestingly, of MYB's two closely related family members, only MYBL1 expression patterns link it to proliferation of immature neuronal precursor cells.114 In chick development, MYB is expressed in the early neural plate border region, which is the progenitor region of the neural crest. Its expression is maintained in early migratory neural crest cells.115
The majority of evidence supporting MYB as a necessary driver in CSCs derives from hematologic malignancies. Both acute myeloid leukemia and chronic myelogenous leukemia blasts are sensitive to MYB loss116; in T‐ALL, MYB knockdown promotes differentiation. Interestingly, the effects of MYB loss on differentiation and proliferation is synergized by NOTCH inhibition in T‐ALL, which also harbor NOTCH mutations.117 NOTCH and MYB may also both support ACC given that ACC CSCs are dependent on NOTCH activity107; however, synergy with MYB has yet to be tested.
Like MYC, MYB initiates a transcriptional program by binding to enhancer binding (E‐box) sites within target genes. E‐box binding by MYB is necessary for neural crest‐like phenotypic emergence recently found to be required for melanoma formation.100 Myeloblastosis fusions in ACC place the enhancer elements close to the MYB gene, leading to a positive feedback loop whereby MYB supports its own expression. Importantly, unrestrained MYB expression likely contributes to a massive shift in ACC gene expression given that 2,400 genes with adjacent MYB‐enhancer elements are highly expressed in ACC but not expressed in normal salivary glands. These MYB binding sites map to many genes that are associated with neural crest stemness, including: translocated MYB itself, neurotrophin receptors (TRKB, TRKC), NOTCH signaling (NOTCH1, NOTCH2, JAG1, JAG2), FGF signaling (FGFR1, FGFR2, FGF2, FGF12), and the BCL2 antiapoptotic pathway (BCL2, BCL2L1, MCL1).71 The critical roles of MYB in maintenance of neural crest progenitors and CSC from hematopoietic and colon cancers, as well as the number of genes expressed in ACC that have enhancer‐binding sites for MYB—combined with the extremely low genetic alteration rate in ACC—suggest that MYB activity is also critical for maintaining an undifferentiated state in ACC. Cell cultures have recently been developed and stem cells subpopulations identified in ACC to test this hypothesis.103
NOTCH
NOTCH signaling is an evolutionary conserved pathway that regulates stem cells during embryogenesis and in a variety of adult tissues.118 In the developing and adult neural system, NOTCH is fundamental for stem cell maintenance during neurogenesis and in the subventricular zone of the adult brain.119, 120 In spite of the overlap between NOTCH and SOX10 activities, mechanisms of their cooperation are not well understood. NOTCH was one of the first genes discovered whose mutation disrupts neural crest formation,121 and NOTCH signaling is critically involved in development of salivary and breast glands that originate from the neuroectoderm.122 In Drosophila, NOTCH deficiency produced multiple abnormalities during salivary gland development123; in humans, expression of all NOTCH paralogs (NOTCH1‐4) was detected in salivary ductal and acinar cells.124 Although NOTCH serves as a tumor suppressor in some tumors, in other tumors such as neural and breast cancers the NOTCH signaling is activated supporting its oncogenic role and attracting great therapeutic interest toward NOTCH signaling as a principal regulator of CSC.125, 126, 127, 128, 129
NOTCH cell surface receptors are activated by proteolytic cleavage upon binding to Delta or Jagged ligands to generate a transcriptionally active NOTCH‐intercellular domain. NOTCH receptor activation requires the enzyme, γ‐secretase, and NOTCH signaling can be inhibited with small molecule γ‐secretase inhibitors.
The role of NOTCH in carcinogenesis has been explored in a mouse model in which an activated NOTCH4 transgene induces neoplastic transformation of mammary and all three major salivary glands.130 Overexpression of NOTCH1 in ACC has been recently described and associated with cancer progression.131 Finally, NOTCH1‐activating mutations recently reported in high‐grade ACC specimens71, 83 support NOTCH1 as a driver of aggressiveness in ACC. Similar activating mutations in NOTCH1‐3 occur in basal‐like breast cancer,132, 133 for which NOTCH1 activation is also implicated in breast cancer progression, chemotherapy resistance, and stimulation of breast CSCs.134 Altogether, these reports strongly support involvement of activated NOTCH in ACC and triple‐negative breast cancers.
Fatty Acid‐Binding Protein 7
Fatty acid‐binding protein 7, formerly known as brain lipid‐binding protein, was characterized as a brain‐specific signaling regulator more than 20 years ago.135 More recently, FABP7 has been recognized as a NOTCH1 target with a key role in the maintenance of radial glia cells with NSC properties.136, 137, 138, 139, 140 Interestingly, FABP7 is a shared NOTCH1/SOX10 target136, 141 that marks neural stem cells in the subventricular CNS zone, as well as CSC in glioblastoma.142, 143, 144 Fatty acid‐binding protein 7 is coexpressed with SOX10 in ACC and basal‐like breast cancers,97 and in these tumor types FABP7 expression serves as a diagnostic marker associated with poor prognosis.79, 145 Although the mechanism of FABP7 activity in cancer or CSC maintenance is not well described, activation of FABP7 in cancers increases fatty acid uptake resulting in enhanced cell survival and protection against reactive oxygen species.146
Receptor Tyrosine Kinases Signaling Driving Neural Crest Stemness in Adenoid Cystic Carcinoma
Transcription factors such as SOX9, SOX10, MYB, and NOTCH are active in or required for neural crest development and survival; these transcription factors drive expression of many genes, including receptor tyrosine kinases (RTKs). Neural crest proliferation, migration, and survival are linked to expression of RTKs, but different neural crest subpopulations are associated with expression of distinct RTKs. For example, TrkC is essential for neurogenic precursors; FGFR is required for precursors of cranial mesenchyme; c‐Kit is implicated in melanoma precursors; and RET is expressed in cells destined for the enteric ganglion147 (reviewed in148). Although c‐Kit and EGFR family members are expressed in and may be important for ACC CSC maintenance, here we will focus on Trk and FGF receptors because trials with c‐Kit and EGF family members have not shown efficacy as single agents in ACC.
Neurotropic Signaling
Neurotrophic tyrosine kinases—TrkA (NTRK1), TrkB (NTRK2), and TrkC (NTRK3)—are cell surface receptors activated by their ligands, neurotrophins NGF, BDNF, NT‐3, and NT‐4. These receptor tyrosine kinases play important roles in neurogenesis in both the central and peripheral nervous systems.149, 150 In normal tissues, TrkB and TrkC receptors contribute to human embryonic pluripotent stem cell survival by providing anti‐apoptotic signaling through activation of the phosphatidylinositol‐3‐kinase pathway151; forced expression of TrkC in neural stem cells increases their survival and migration when transplanted into spinal cords.152 The role of neurotrophin signaling in neural crest stem cell maintenance is demonstrated in TrkC null mice, which have increased numbers of fate‐restricted cells and an equivalent decrease in pluripotent neural crest stem cells.153
Oncogenic properties of Trk receptors are apparent from gene fusions found in various cancers, including lung adenocarcinoma, papillary thyroid cancer, spitzoid neoplasms, glioblastoma, astrocytoma, pediatric glioma, secretory breast carcinoma, mammary analogue secretory carcinoma of salivary glands, and many more.154 As an alternate mechanism for Trk activation, TrkB and TrkC are overexpressed in cancers in which autocrine signaling loops with their ligands (BDNF and NT‐3, respectively) are linked to poor prognosis.155, 156, 157, 158, 159, 160 The great majority of ACC cases (∼94%) express high TrkC levels in conjunction with its ligand, NT‐3. In ACC, TrkC is selectively expressed on neoplastic myoepithelial cells and is often coexpressed with Erk1/2 and BCL2.96 Autocrine TrkC/NT‐3 signaling may promote cell survival, motility, and invasion in ACC, although this has not been directly tested. However, Trk inhibition suppresses ACC xenograft tumor growth and cellular proliferation, supporting a role for TrkC in ACC.96
Fibroblast Growth Factor Signaling
Along with NOTCH, basic fibroblast growth factor (FGF2) was among first molecules identified as critical for neural crest development.161 Since that time, other components of FGF signaling, including fibroblast growth factor receptor 1 (FGFR1), have also been implicated in neural crest development.162 Amplification, gene fusions, or activating mutations of FGF signaling components drives several tumor types.163 Like TrkC, FGFR1 is highly expressed in the majority of ACC specimens (Fig. 2). Expression of FGFR1 and its ligands (FGF1 and FGF2), amplifications of FGFR1 and FGFR3, and mutations in FGF signaling components (FGFR1, FGFR2, FGFR4, FGF16, and PIK3CA) are described in ACC, suggesting that FGF signaling is important for ACC carcinogenesis or maintenance.54, 55, 60, 83, 164, 165 Similarly, autocrine loops that activate FGFRs are active in many tumors, including those with neural crest derivation such as melanoma and basal‐like breast cancers.166, 167, 168, 169, 170, 171 Moreover, based on the study that linked MYB to FGF2 stimulation in melanoma,172 it was suggested that similar mechanism may be involved in ACC.60 Our group recently identified an FGFR2‐activating mutation (S252W) in a case of ACC (unpublished data, Yale Laboratory for Head and Neck Oncology). The S252W mutation found occurs in ∼4% of endometrial cancers.173, 174 This mutation is associated with Apert syndrome, a craniosynostosis syndrome distinguished by syndactyly of the hands and feet and central nervous system malformations.175 Interestingly, inactivating mutations of FGFR2 in melanoma have been described.176 Mice that lack an FGFR2 isoform are sensitive to carcinogen‐induced skin cancer,177 suggesting that FGFR‐signaling may protect against tumor formation. The majority of this evidence suggests that tumor‐suppressive effects of FGF signaling function through FGFR2, however; no distinctions of intracellular signaling between FGFR1 and FGFR2 have been found, suggesting that opposing roles of FGFR2 as a tumor suppressor and oncogene may be cell context‐dependent.178
Figure 2.

Overexpression of FGFR1 and NTRK3 in primary clinical (ACC) and grafted (ACCX) tumor specimens, as well as in salivary AD, salivary MEC, HNSCC, and NORM based on Affymetrix gene expression analysis performed in Ivanov et al.96 ACC = adenoid cystic carcinoma; ACCX = ACC xenograft; AD = adenocarcinoma; FGFR = fibroblast growth factor receptor; HNSCC = head and neck squamous cell carcinoma; MEC = mucoepidermoid carcinoma; NORM = normal salivary tissue; NTRK3 = neurotrophic receptor tyrosine kinase 3.
Given activation of FGF signaling in ACC, pharmacological suppression of FGFRs has been explored.179 Unfortunately, clinical trials have been disappointing. Currently, a clinical trial testing regorafinib, a multikinase inhibitor targeting FGFR, VEGFR, PDGFR, and c‐Kit, is recruiting ACC patients (NCT02098538).
Characterization of Cancer Stem Cells in Adenoid Cystic Carcinoma
Cancer stem cells depend on distinct signaling pathways and harbor expression profiles that are different than the bulk cancer cells. Characterization of CSC derived from several tumor types has found that they depend on pathways identified in many non‐CSCs, including Hedgehog, Wnt, Nanog, SOX, and NOTCH. By definition, CSCs must have enhanced ability to form xenograft tumors compared to bulk tumor cells; however, their isolation can be challenging because they represent a minority of cancer cells in a tumor. In many tumor types, CSC isolation relies on cell surface markers or aldehyde dehydrogenase expression or activity. CD133 is expressed on the cell surface of normal neural stem cells and CSCs from brain tumors.180 Although expression of CD133 has been described in ACC,181, 182 its connection with stem cell phenotypes and the molecular identity of CSCs in ACC has only recently been explored.
Starting with ACC patient‐derived xenografts or with primary ACC tissue, CD133‐positve ACC cells were enriched from cell cultures or disaggregated tumor tissue and found to possess behaviors that define CSCs.107 Specifically, cultured CD133‐positive cells regenerate both CD133‐positive and CD133‐negative cells, form spheroids in vitro, and compared to CD133‐negative cells display enhanced tumorigenesis in immunodeficient mice. On the other hand, cultured CD133‐negative ACC cells do not produce CD133‐positive cells and do not form spheroids in culture. As expected, gene expression profiles of CD133‐positive and CD133‐negative cells are distinct. Adenoid cystic carcinomas express neural crest stem cell markers SOX10, NOTCH1, and FABP7, whereas CD133‐negative cells express neural differentiation markers NR2F1 and NR2F2183 as well as p27Kip1, a cyclin‐dependent kinase inhibitor and key regulator of neural differentiation.184, 185, 186 Perhaps most intriguingly, CD133‐negative cells express JAG1, a membrane‐associated NOTCH ligand, and immunofluorescent staining of cocultured CD133‐positve and CD133‐negative cells places cells expressing NOTCH receptors and ligands adjacent to one another.107 These data suggest an ACC model whereby CSCs and more differentiated tumor cells are interdependent, with expression of JAG1 on the more differentiated cells activating NOTCH signaling within CSCs (Fig. 3).
Figure 3.

ACC‐CSC (bottom) express CD133, SOX10, and NOTCH1/N1ICD; re‐produce themselves; and generate CD133‐negative cells. More differentiated CD133‐negative cells express differentiation markers and activate NOTCH1 on ACC‐CSC through expression of the NOTCH ligand, JAG1.
ACC‐CSC = adenoid cystic carcinoma cancer stem cells.
NOTCH signaling is required for survival of many neural CSCs, and depletion of NOTCH1 in ACC cultures inhibits spheroid formation and CSC survival. Cancer stem cells in ACC are also dependent on SOX10 and FABP7 expression, showing that NOTCH1, SOX10, and FABP7 are each required for maintenance of these cells. Blocking NOTCH signaling using γ‐secretase inhibitors selectively kills ACC cancer stem and sensitizes them to irradiation. Inhibition of NOTCH was accompanied by downregulation of SKP2, an E3 ubiquitin ligase that leads to degradation of p27Kip1. SKP2 is associated with poor prognosis in various cancers, including ACC,187, 188, 189 suggesting that p27Kip1 or other SKP2 targets may inhibit survival of ACC CSCs. Importantly, NOTCH inhibition with γ‐secretase suppresses xenograft tumor growth in nude mice, further supporting a major role for NOTCH1 in maintenance of CSC and ACC progression.
Although individual roles for SOX10 and NOTCH1 in stem cells maintenance are well established, and loss of NOTCH signaling decreases SOX10 expression in neural progenitors,190, 191 cooperation between SOX10 and NOTCH1 in stem cell maintenance has not been previously described. Implication of both of these transcription factors as ACC stem cell drivers that signal through common targets, such as FABP7 and SKP2, provides a novel molecular mechanism and potentially new targets for elimination of CSCs.
New Adenoid Cystic Carcinoma Models for In Vitro and In Vivo Research
The generation of cell lines or cultures, as well as robust animal models, are needed to accelerate discovery of new therapies in ACC.73 Modeling of ACC is improving, with several xenografts being maintained,192 and robust cell cultures are emerging that can be used for preclinical testing, genetic manipulation for hypothesis testing, and drug screening.
Adenoid Cystic Carcinoma Xenografts
Recent evidence suggest that patient‐derived xenografts (PDXs) may be the most reliable preclinical model because they maintain the complexity of tumor structure and more closely mimic drug response.193 Given the paucity of clinical material and lack of validated cells lines in ACC, generation of (PDX) has been perhaps the most critical achievement to advance research in this tumor.192 Adenoid cystic carcinoma derivation of 12 PDXs was confirmed by the findings that 11 have MYB locus rearrangements and 10 contain MYB‐NFIB fusions. Importantly for studies of CSCs, PDX models, unlike cancer cell lines, maintain stem cells in a three‐dimensional tumor environment and can serve as a reliable source for derivation of cells for CSC research.194
Adenoid Cystic Carcinoma Cell Cultures
In routine cell culture media, both with and without serum, ACC cells grow very slowly, senesce after a few passages, and eventually die (unpublished data, Yale Laboratory for Head and Neck Oncology). Attesting to the difficulty in establishing ACC cultures, there are no validated ACC cell lines available from ATCC, and genetic profiling of six ACC cell lines shared between laboratories (ACC2, ACC3, ACCM, ACCNS, ACCS, and CAC2) revealed cross‐contamination and misidentification195 and effectively invalidated research that used them. These six cell lines can now be found in the list of cross‐contaminated or misidentified lines available at http://iclac.org/wp-content/uploads/Cross-Contaminations-v7_2.pdf.
In 2013, a cell line was derived from a minor salivary gland ACC, SACC‐83. Serial intravenous injections and re‐derivation from lung deposits created SACC‐LM, a daughter cell line with increased metastatic potential.196, 197 Although these cell lines have not been compared to the original patient or the primary tumor, short‐tandem repeat (STR) analyses confirm that these two cell lines are of the same derivation (16 of 16 match) and distinct from HeLa cells. However, deeper comparison with publically available databases of a broad range of cell lines has not been reported.198
Recently, an additional cell line, MDACC‐ACC‐01, was developed from a base of tongue ACC.199 Unfortunately, the population doubling time (PDT) increased with passage (p5 PDT = 64 hours, p18 PDT = 102 hours). To decrease doubling time and establish a cell line, passage 7 cells were immortalized with human telomerase reverse transcriptase (hTERT), resulting in PDT of approximately 60 hours and stabilizing the culture to grow to 100 passages without signs of senescence. Unfortunately, G‐banding and spectral karyotyping found that a t(6q25;14q13) translocation identified in the primary tumor and in early passages of the culture were lost as early as passage 5 and disappeared entirely by passage 10 of the immortalized cells.199
Our group reported development of several cell cultures from ACC patient‐derived xenografts or from primary ACC tumors using a conditionally reprogrammed cell‐culture protocol reliant on Rho‐associate kinase (ROCK) inhibition.107 Cultures were validated by short‐tandem repeat genotyping, and MYB fusions were confirmed in all cultures tested (Table 2). The best performing culture was derived from the adenoid cystic carcinoma xenograft 11 and has been passaged more than 30 times. Cultured cells from ACCX11 retained the MYB fusion found in the primary tumor, and cultured cells readily formed xenograft tumors in nude mice with histology that recapitulated the primary and xenografted tumors (Fig. 4).107
Table 2.
Validation of ACC Cultures by Microsatellite and MYB‐NFIB Fusion.
| Sample | Specimen Type | MYB‐NFIB Fusion | TH01 | D21S11 | D5S818 | D13S317 | D7S820 | D16S539 | CSF1PO | Amelogenin |
|---|---|---|---|---|---|---|---|---|---|---|
| ACCXM51 | Xenograft | Confirmeda | 9.1, 10 | 28, 29.3, 32, 32.1 | 12.3 | 10, 14 | 7, 11 | 9, 11 | 9.3, 11 | X, Y |
| Accx5m1 | Culture | Confirmed | 9.1, 10 | 29.3, 32.1 | 12.3 | 11, 14 | 7, 11 | 9, 13.1 | 9.3, 10.3 | X, Y |
| ACCX11 | Xenograft | Confirmeda | 6.1, 10 | 28.3, 31.1 | 11.1 | 13, 14 | 8, 9.1 | 11, 12.1 | 10, 12 | X |
| Accx11:9 | Culture | Confirmed | 6.1, 10 | 28.3, 31.1 | 11.1 | 13, 14 | 8, 9.1 | 11, 12.1 | 10, 12 | X |
| ACCX14:9 | Xenograft | Confirmeda | 6.1, 10 | 28.3 | 10.1, 11 | 15 | 9, 10 | 12.1 | 11, 12 | X |
| Accx14:9 | Culture | Confirmed | 6.1, 10 | 28.3 | 10.1, 11.1 | 14 | 9.1, 10 | 11, 12.1 | 11, 12 | X |
| ACCX19 | Xenograft | Confirmeda | 7.1, 8 | 27.3 | 12.3 | 13, 15 | 8, 11 | 10, 11 | 6.3, 12 | ‐ |
| Accx19 | Culture | Not tested | 7.1, 8 | 27.3 | 12.3 | 13, 15 | 8, 11 | 10, 11 | 6.3, 12 | ‐ |
| ACCX29 | Xenograft | Confirmeda | 6.1, 7.1 | 29.3, 31 | 10.1, 11 | 13, 14 | 10 | 9, 11 | 12 | X |
| Accx29 | Culture | Not tested | 6.1, 7.1 | 29.3, 31.1 | 10.1, 11.1 | 13, 15 | 9.3 | 9, 11 | 12 | X |
| ACC33 | Original Tumor | Not tested | 9.1, 10 | 28.3, 29.3 | 9 | 11, 15 | 9.3, 11 | 11, 12.1 | 11, 14 | X |
| Acc33 | Culture | Not tested | 9.1, 10 | 28.3, 29.3 | 9 | 10, 14 | 10, 11 | 11, 12.1 | 11, 14 | X |
MYB‐NFIB confirmation of xenografts from Moskaluk et al. Lab Invest 2011.192
ACC = adenoid cystic carcinoma; ACCX = adenoid cystic carcinoma xenograft; MYB = myeloblastosis; NFIB = nuclear factor 1 B‐type.
Figure 4.
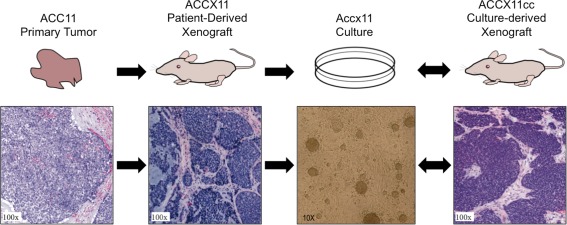
Tumor cells from patient‐derived xenograft ACCX11 are cultured (Accx11); and cells from these cultures, when injected into mice, form tumors (ACCX11cc) histologically resembling the original and patient‐derived xenograft tumors.
ACC = adenoid cystic carcinoma; ACCX = ACC xenograft.
To advance ACC research, it is critical that cell cultures, cell lines, and patient‐derived xenografts be rigorously validated and meticulously maintained. Short‐tandem repeat genotyping is recommended by ATCC (https://www.atcc.org/~/media/PDFs/STR_Profiling.ashx) for all cultures. Short‐tandem repeat testing assures that cell lines or short‐term cultures are not derived from or contaminated with known cell lines. In addition, because STR is based on human sequences, absence of signal following amplification suggests that cells are derived from or contaminated with cells from other species. If the tumor, normal tissue, or blood can be obtained from the patient from which the culture is derived, STR assessment of both the parent tissue and derivative cell culture will assure the provenance of the cultured cells. Cell cultures using ROCK inhibitors and murine fibroblast feeder layers should be subjected to extensive validation before distribution. Although fibroblasts to be used as feeder cells are irradiated, rare fibroblasts acquire proliferative capacity with the potential to contaminate or replace the cells of interest (unpublished data, Yale Laboratory for Head and Neck Oncology). Another issue unique to this culture system is that the ROCK inhibitor potentiates growth of both normal and tumor cells. After STR testing confirms the culture's uniqueness and derivation, the cancerous nature of the cells must be confirmed by tumor formation after xenografting and/or identification of identical gene alterations in the parent tumor and derivative cells.
The lack of centralized resources for distribution of ACC cell lines has not been an issue in the absence of such cultures; however, cultures are starting to be reported, as outlined above. The Salivary Gland Tumor Biorepository is a central distribution source for salivary gland tumors that is supported by the National Institute of Dental and Craniofacial Research (National Institutes of Health, Bethesda, MD). The biorepository currently offers only the MDACC‐ACC‐01 hTERT‐immortalized line discussed above (https://research.mdacc.tmc.edu/Salivary_DB/index.html), but expansion of this resource to include viably frozen tissue and short‐term cultures is being proposed.
Targeting Stemness in Adenoid Cystic Carcinoma
The CSC theory asserts that these cells are at the heart of tumor growth and spread, and that the tumor would lose regenerative capacity if destroyed. In experimental models, injection of very few CSCs recreates the entire tumor architecture and cell variety; however, CSCs have slower proliferative rates and are more resistant to cytotoxic therapies. Experience with other solid tumors demonstrates that targeting CSC inhibits not only tumor growth but also metastatic and invasive behaviors.200 Neural crest stem cell properties of ACC CSCs may be central for their metastatic behavior, which requires cells to migrate, invade, adapt, and grow in a new environment. Targeting stemness in cancer is not a new idea, but there was little data supporting the existence of stem cells in ACC, and until recently, no data identifying critical signaling pathways involved in ACC CSC maintenance. Several lines of data are converging to indicate that stem cells in ACC may be among the best target in this tumor type. As described above, key components of the neural expression signature intrinsic to ACC, including MYB, SOX10, NOTCH, and FABP7, have been closely associated with maintenance of progenitor or undifferentiated cell states. Although stem cells may be targeted by inhibition of transcriptional regulators required for stemness, such as SOX10, MYB, and NOTCH; other targets, such as epigenetic modifiers and immune inhibitory pathways, may also target stem cells.
Myeloblastosis as a Stemness Target in Adenoid Cystic Carcinoma
The role of MYB in carcinogenesis is well established,201, 202 and the recent discovery of MYBL1 fusions in a subset of tumors lacking MYB suggests that MYB‐like signaling may be required for development or maintenance of ACC. Because ACC cell lines have only recently been developed, the effect of MYB loss in ACC or ACC CSCs has not been described. In ACCs containing the MYB‐NFIB translocation, it can be detected in all tumor cells; however, MYB is expressed only in a subset of ACC cells, namely basal cells where it is coexpressed with the stemness marker, p63.70 This pattern of MYB expression suggests that it is confined to less differentiated cells, and as such could be a major driver of the CSC subpopulation within ACC.
Direct targeting to inhibit transcription factors, such as MYB, has been difficult. Chromatin immunoprecipitation analyses revealed that MYB is a master transcription factor altering expression of a plethora of genes,73 suggesting that therapies that target genes whose expression is driven by MYB may not be effective. Regulating epigenetic modification or epigenetic‐driven gene transcription (see below) or inhibiting interactions between MYB and required transcriptional cofactors are potential strategies for indirect inhibition of MYB. The protein acetylase p300 directly binds MYB, altering its transcriptional activity, and loss of p300‐MYB interaction results in hematopoietic defects in mice.203 In addition, expression of mutant MYB incapable of interacting with p300 prevented myeloid leukemia induction by several oncogenes. Because MYB activity is critical for maintenance of leukemia, targeting the MYB/p300 interaction domain has been proposed, but no drugs yet exist (reviewed in204).
Currently, direct targeting of MYB has been difficult, but new approaches may soon make direct degradation of MYB possible. Small‐molecules that bind to a target protein and attract endogenously expressed E3 ubiquitin ligases to initiate proteasomal degradation of the target protein have been termed proteolysis‐targeting chimeras (PROTACs). A PROTAC targeting the androgen receptor effectively decreased androgen receptor expression in prostate cancer cells,205 and “untargetable” proteins have also been degraded using PROTACs (reviewed in 206). Creation of a MYB‐targeting PROTAC relies on discovery of a small molecule that binds to MYB, but this should be possible using high‐throughput screening techniques.
NOTCH as a Stemness Target in Adenoid Cystic Carcinoma
Cancer stem cells with high SOX10, NOTCH1, and FABP7 expression are present in ACC, and although all three gene products are essential for CSC maintenance, only NOTCH1 activation can be currently inhibited. Clinical trials with γ‐secretase alone or in combination with other drugs have shown promising results in solid tumors, including breast cancers,207, 208 but have not been yet tested on ACC patients. Despite signals of activity, trials of γ‐secretase inhibitors have been plagued by gastrointestinal side effects attributed to the broad effects of γ‐secretase inhibition that is not directly related to NOTCH inhibition. To minimize side effects, more specific inhibitors of NOTCH signaling, which include monoclonal antibodies against specific NOTCH receptors such as tareztumab (anti‐NOTCH2/3) and brontictuzumab (anti‐NOTCH1), as well as antibodies against activating ligands of NOTCH such as demcizumab and OMP‐305B83 (both anti‐DLL4) (OncoMed Pharmaceuticals, Inc., Redwood City, CA), have been developed. Two independent trials are assessing the efficacy of BMS‐906024, a new pan‐NOTCH inhibitor, with one study utilizing it as a single‐agent and the other in combination with traditional chemotherapy.209
Receptor Tyrosine Kinases as Stemness Targets in Adenoid Cystic Carcinoma
Intrinsically high expression levels of TrkC and FGFR1 in ACC specimens,96 as well as somatic FGFR2 mutations reported in a fraction of ACC patients (Table 1) (Fig. 2) (unpublished data, Yale Laboratory for Head and Neck Oncology), suggest that these RTKs can be targeted to cripple CSCs. A recent study suggests that neurotrophins may be key to salivary gland development,210 and potential involvement of other neurotrophins and Trk receptors in this process is supported by detection of TrkC/NTRK3 and NGFR expression in the salivary gland primordium (Fig. 5).
Figure 5.

Expression of TrkC/NTRK3 and NGFR in the developing salivary glands (black arrows) of mouse embryos (http://genepaint.org).
TrkC/NTRK3 = neurotrophic tyrosine kinase 3.
Fibroblast growth factor signaling is also engaged in neural stem cell regulation, neurogenesis, and cross talk with neurotrophic signaling.211, 212, 213, 214 Remarkably, FGF2 can transform embryonic stem cells to neural crest stem cells, further confirming the role of FGF signaling in neural crest development.102 Among the four FGFR receptors, only FGFR1 and FGFR2 are expressed in the salivary glands, where they regulate salivary branching morphogenesis and cell survival.215, 216 Two FGFR2 isoforms, IIIb and IIIc, have been implicated in salivary gland development, together with their ligands FGF10 and FGF8, respectively.217, 218, 219
Ample experimental evidence links neurotrophic signaling to maintenance of CSC in a large variety of cancers (reviewed in220). Importantly, existing data suggest that these CSCs hijack embryonic signaling, interconvert between different neurotrophic receptors, and benefit from cross talk between neurotrophic and other signaling pathways. Better understanding of these complex signaling interactions is essential for designing effective combination strategies that could take advantage of recently produced NOTCH, FGFR, Trk, and other RTK inhibitors to thwart acquired and intrinsic drug and radiation resistance.126, 221, 222
Epigenetic Targeting of Stemness in Adenoid Cystic Carcinoma
In addition to inhibition of transcriptional regulators required for ACC stemness (e.g., SOX10, MYB, and NOTCH), epigenetic modifiers can also be targeted. In ACC, chromatin‐modifying enzymes are frequently altered by mutation. Like master transcriptional regulators, aberrant activation of epigenetic modifiers may also prevent differentiation by altering transcription of large numbers of genes. New drugs to alter modification or downstream effects of chromatin modification are emerging.
Targeting of epigenetic modifiers in cancer has shown some success; identification of mutations in CREBBP and EP300 suggest that histone acetylation is dysregulated in ACC.55 HDAC inhibitors (HDACi) have been used in clinical phase 1 to 3 trials on patients with advanced solid tumors, leukemias, and lymphomas.90, 223, 224 A recent phase 2 trial of the HDAC inhibitor, vorinostat, in ACC has completed enrollment, but results have not be published; however, preliminary data was promising with three responses (2 partial and 1 minor), as well as improvement in symptoms in three additional patients out of the 30 patients enrolled.225
Bromodomain and extraterminal (BET) family proteins recruit transcriptional regulator complexes to promoters associated with chromatin acetylation.226 Bromodomain and extraterminal family members bind to acetylated proteins and through protein‐protein interactions nucleate a transcriptional complex that contains CDK9. The kinase activity of CDK9 is important for Pol II transcriptional elongation, and CDK9 activity is enabled in complex with BET family members. Bromodomain and extraterminal family members also contain intrinsic kinase activity that phosphorylates Pol II, but the full impact of this activity is unknown. In 2010, inhibitors of BET proteins that competitively block binding of the bromodomain pocket to acetylated proteins were described and now have now progressed to clinical trials in human cancer patients.227, 228
Many tumor types are sensitive to BET inhibitors, and interestingly, susceptible tumors included hematopoietic tumors where BET inhibition downregulated MYC and BCL2 expression. Likewise, MYC amplified neuroblastomas are also sensitive to BET inhibitors (reviewed in 226). As with MYC, genes transcribed in response to MYB in T‐ALL and angiocentric gliomas are highly correlated with H3K27 acetylation of enhancer sequences73, 229; in angiocentric gliomas, MYB drives a positive‐feedback loop, enforcing its expression based on enhancer H3K27 acetylation of the 3' fusion partner of MYB.73
Changes in gene transcription in response to BET inhibitors is cell type‐specific, with marked suppression of MYC in leukemia but little suppression in fibroblasts.230 Given that BET proteins bind to MYC and other transcription factors,231 it is likely that this binding may also alter gene transcription and/or transcription factor stability. In many tumors, expression of MYB target genes depend on enhancer acetylation, suggesting that inhibition of BET proteins is a promising means of inhibiting MYB‐driven gene expression; however, cell type specific responses to BET inhibitors indicate that preclinical testing to determine effects of BET inhibitors are first needed in ACC.
Through its interaction with CDK9, a CDK family member, CDK7 regulates transcription through phosphorylation of the C‐terminal domain of Pol II232 in areas marked by acetylated chromatin. A covalent inhibitor of CDK7 was described in 2014.32 As with BET inhibitors, T‐ALL cells were more sensitive to CDK7 inhibitors than other lines; in these cells, expression of genes driven by super‐enhancers was disproportionally inhibited.232 At higher concentrations, CDK7 inhibition globally suppresses transcription, suggesting that clinical utility will rely on determining heightened sensitivity, possibly based on transcriptional dysregulation driven by super‐enhancers.
Given the key role of epigenetic modification in stemness, epigenetic modifiers hold great promise for therapy of ACC stemness.
Immune Modulatory Therapy and Stemness
Immune modulatory therapy has had great success in many tumor types, such as melanoma, bladder cancer, and lung cancer, and there are ongoing studies in the most common solid tumor types. Breakthroughs initially came in melanoma patients through inhibition of cytotoxic T‐lymphocyte‐associated protein‐4 (CTLA‐4) and the programmed death‐ligand 1/programmed cell death protein 1 (PD‐L1/PD‐1) immune checkpoint proteins. Studies are now focused on identification of markers that may predict response to different inhibitors based on expression of inhibitory molecules, tumor infiltrating lymphocytes, or other markers.
There is limited data assessing the immune response to ACC tumors. A study of 20 primary and metastatic tumors revealed that ACC tumor cells do not express PD‐L1 and that the majority of tumors have few infiltrating immune cells.233 Interestingly, the majority of ACCs express PD‐L2, and chemo‐radiation therapy is associated with increased CD8+ T‐cells, decreased FOXP3+ regulatory cells, and increased circulating antibodies targeting potential tumor antigens.233 Expression analyses reveal that another immune inhibitory molecule, B7‐H4, is expressed in the majority of ACC, and that expression is greater in ACC cancer stem cells (unpublished data, Yale Laboratory for Head and Neck Oncology). Although not widely expressed in normal tissues, B7‐H4 is expressed in brain tumors, the majority of ovarian papillary carcinoma, ovarian serous adenocarcinomas, and ductal lobular breast cancer.234 When expressed, B7‐H4 inhibits T‐cell proliferation, cytokine production, and production of activated cytotoxic T‐lymphocytes. Consistent with this immune inhibitor function, blockade of B7‐H4 with antibodies elicits T‐cell responses but is not associated with autoimmunity in mice.234 Antibodies targeting this immune inhibitor protein are currently being developed for clinical use and may be trialed in ACC once available.
CONCLUSION
Targeting of stem cells may be particularly effective for ACC given the early evidence suggesting that neural crest‐derived stem cells may be required in ACC.96, 97 Currently, many stem cell drivers, such as SOX10, FABP7, and MYB, cannot be directly targeted; however, NOTCH targeting with γ‐secretase inhibitors has progressed to clinic, and more specific inhibitors of NOTCH signaling are being developed. Targeting of receptor tyrosine kinases associated with neural crest stemness is now possible, but experience with c‐Kit, EGFR, and FGFR inhibitors suggests that combinatorial therapies may be needed for efficacy. Epigenetic targeting of molecules needed for super‐enhancer activity is an intriguing direction given that ACCs express MYB or MYBL1 under the control of super‐enhancers; and of course the direct targeting of MYB, as the most frequently altered gene, is a future goal. Success of immune modulatory therapy in many tumors suggests that such therapy targeting stem cells in ACC may be effective.
Driven by genomic and proteomic advancements, we are entering a new phase in understanding ACC. Suspected molecular drivers in ACC can now be tested in preclinical models that are available as xenografts, as well as in newly developed cell cultures and cell lines. New platforms for preclinical studies may lead to new therapies and decrease the high failure rates that have come to define clinical trials in ACC.30
Financial Disclosure: Supported by the Division of Otolaryngology and the Department of Surgery at Yale School of Medicine. The authors have no other funding, financial relationships, or conflicts of interest to disclose.
BIBLIOGRAPHY
- 1. Speight PM, Barrett AW. Salivary gland tumours. Oral Dis 2002;8:229–240. [DOI] [PubMed] [Google Scholar]
- 2. Ellington CL, Goodman M, Kono SA, et al. Adenoid cystic carcinoma of the head and neck. Cancer 2012;118:4444–4451. doi: 10.1002/cncr.27408. [DOI] [PubMed] [Google Scholar]
- 3. foundation Accr . Understanding ACC Needham, MA2015 [cited 2015 December 22].
- 4. Boukheris H CR, Land CE, Dores GM. Incidence of carcinoma of the major salivary glands according to the WHO classification, 1992 to 2006. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2009;91:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu T GQ, Wang XY, Wen YM, Li LJ. Malignant sublingual gland tumors: a retrospective clinicopathologic study of 28 cases. Oncology 2007;72:39–44. [DOI] [PubMed] [Google Scholar]
- 6. Hellquist H, Skalova A. Adenoid cystic carcinoma Histopathology of the Salivary Glands. Berlin, Germany: Springer‐Verlag; 2014:221–260. [Google Scholar]
- 7. Ascani G, Messi M, Lupi E, et al. Salivary gland tumours: a retrospective study of 454 patients. Minerva Stomatol 2006;55:209–214. [PubMed] [Google Scholar]
- 8.Bradley PJ. Adenoid cystic carcinoma of the head and neck: a review. Curr Opin Otolaryngol Head Neck Surg 2004;12:127–132. [DOI] [PubMed] [Google Scholar]
- 9. Bennett AK, Mills SE, Wick MR. Salivary‐type neoplasms of the breast and lung. Semin Diagn Pathol 2003;20:279–304. [DOI] [PubMed] [Google Scholar]
- 10. Iczkowski KA, Grier DD, Hossain D, et al. Adenoid cystic/basal cell carcinoma of the prostate: clinicopathologic findings in 19 cases. Am J Surg Pathol 2003;27:1523–1529. [DOI] [PubMed] [Google Scholar]
- 11. Fueston JC, Gloster HM, Mutasim DF. Primary cutaneous adenoid cystic carcinoma: a case report and review of the literature. Cutis 2006;77:157–160. [PubMed] [Google Scholar]
- 12. Elhassani LK, Ismaili N, Bensouda Y, et al. Advanced adenoid cystic carcinoma of the cervix: a case report and review of the literature. Cases 2009;2:6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conley J, Dingman DL. Adenoid cystic carcinoma in the head and neck (cylindroma). Arch Otolaryngol 1974;100:81–90. [DOI] [PubMed] [Google Scholar]
- 14. Hickman RE, Cawson RA, Duffy SW. The prognosis of specific types of salivary gland tumors. Cancer 1984;54:1620–1624. [DOI] [PubMed] [Google Scholar]
- 15. Ferlito A, Shaha AR, Silver CE, Rinaldo A, Mondin V. Incidence and sites of distant metastases from head and neck cancer. ORL J Otorhinolaryngol Relat Spec 2001;63:202–207. [DOI] [PubMed] [Google Scholar]
- 16. Bradley PJ. Adenoid cystic carcinoma of the head and neck: a review. Curr Opin Otolaryngol Head Neck Surg. 2004;12:127–132. [DOI] [PubMed] [Google Scholar]
- 17. Prasad ML, Barbacioru CC, Rawal YB, Husein O, Wen P. Hierarchical cluster analysis of myoepithelial/basal cell markers in adenoid cystic carcinoma and polymorphous low‐grade adenocarcinoma. Mod Pathol 2008;21:105–114. doi: 10.1038/modpathol.3800983. [DOI] [PubMed] [Google Scholar]
- 18. Chen JC, Gnepp DR, Bedrossian CW. Adenoid cystic carcinoma of the salivary glands: an immunohistochemical analysis. Oral Surg Oral Med Oral Pathol 1988;65:316–326. [DOI] [PubMed] [Google Scholar]
- 19. Prasad AR, Savera AT, Gown AM, Zarbo RJ. The myoepithelial immunophenotype in 135 benign and malignant salivary gland tumors other than pleomorphic adenoma. Arch Pathol Lab Med 1999;123:801–806. [DOI] [PubMed] [Google Scholar]
- 20. Szanto PA, Luna MA, Tortoledo ME, White RA. Histologic grading of adenoid cystic carcinoma of the salivary glands. Cancer 1984;54(6):1062–1069. [DOI] [PubMed] [Google Scholar]
- 21. Perzin KH, Gullane P, Clairmont AC. Adenoid cystic carcinomas arising in salivary glands: a correlation of histologic features and clinical course. Cancer 1978;42:265–282. [DOI] [PubMed] [Google Scholar]
- 22.Westra WH. The surgical pathology of salivary gland neoplasms. Otolaryngol Clin North Am 1999;32:919–943. [DOI] [PubMed] [Google Scholar]
- 23. Spiro RH, Huvos AG. Stage means more than grade in adenoid cystic carcinoma. Am J Surg 1992;164:623–628. [DOI] [PubMed] [Google Scholar]
- 24. Seethala RR. An update on grading of salivary gland carcinomas. Head Neck Pathol 2009;3:69–77. doi: 10.1007/s12105-009-0102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang M, Ma D, Sun K, Yu G, Guo C, Gao F. Factors influencing survival rate in adenoid cystic carcinoma of the salivary glands. Int J Oral Maxillofac Surg 1997;26:435–439. [DOI] [PubMed] [Google Scholar]
- 26. Sequeiros Santiago G, Rodrigo Tapia JP, Llorente Pendas JL, Suarez Nieto C. [Prognostic factors in adenoid cystic carcinoma of salivary glands]. Acta Otorrinolaringol Esp 2005;56:361–367. [DOI] [PubMed] [Google Scholar]
- 27. Chen AM, Weinberg V, Garcia J, et al. Adenoid cystic carcinoma of the head and neck treated by surgery with or without postoperative radiation therapy: prognostic features of recurrence. Int J Radiat Oncol Biol Phys. 2006;66:152–159. [DOI] [PubMed] [Google Scholar]
- 28. Garden AS, Weber RS, Morrison WH, Ang KK, Peters LJ. The influence of positive margins and nerve invasion in adenoid cystic carcinoma of the head and neck treated with surgery and radiation. Int J Radiat Oncol Biol Phys 1995;32:619–625. [DOI] [PubMed] [Google Scholar]
- 29. Jensen AD, Poulakis M, Nikoghosyan AV, et al. Re‐irradiation of adenoid cystic carcinoma: analysis and evaluation of outcome in 52 consecutive patients treated with raster‐scanned carbon ion therapy. Radiother Oncol 2015;114:182–188. doi: 10.1016/j.radonc.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 30. Laurie SA, Ho AL, Fury MG, Sherman E, Pfister DG. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: a systematic review. Lancet 2011;12:815–824. [DOI] [PubMed] [Google Scholar]
- 31. van Herpen CM, Buter J, Thomas J, et al. Phase II study on gemcitabine in recurrent and/or metastatic adenoid cystic carcinoma of the head and neck. Eur J Cancer 2008;44:2542–2545. [DOI] [PubMed] [Google Scholar]
- 32. Gilbert J, Pinto HA, Jennings T, et al. Phase II trial of taxol in salivary gland malignancies (E1394): a trial of the Eastern Cooperative Oncology Group. Head Neck 2006;28:197–204. [DOI] [PubMed] [Google Scholar]
- 33. Schramm VL Jr, Srodes C, Myers EN. Cisplatin therapy for adenoid cystic carcinoma. Arch Otolaryngol 1981;107:739–741. [DOI] [PubMed] [Google Scholar]
- 34. Licitra L, Spinazze S, Rossi A, et al. Cisplatin in advanced salivary gland carcinoma. A phase II study of 25 patients. Cancer 1991;68:1874–1877. [DOI] [PubMed] [Google Scholar]
- 35. Mattox DE, Von Hoff DD, Balcerzak SP. Southwest Oncology Group study of mitoxantrone for treatment of patients with advanced adenoid cystic carcinoma of the head and neck. Invest New Drugs 1990;8:105–107. [DOI] [PubMed] [Google Scholar]
- 36. Verweij J, de Graeff A, Vermorken JB, et al. Phase II study on mitoxantrone in adenoid cystic carcinomas of the head and neck. EORTC Head and Neck Cancer Cooperative Group. Ann Oncol 1996;7:867–869. [DOI] [PubMed] [Google Scholar]
- 37. Vermorken JB, de Mulder PH, Cognetti F, et al. Epirubicin in patients with advanced or recurrent adenoid cystic carcinoma of the head and neck: a phase II study of the EORTC Head and Neck Cancer Cooperative Group. Ann Oncol 1993;4:785–788. [DOI] [PubMed] [Google Scholar]
- 38. Airoldi M, Succo G, Gabriele AM, et al. Phase II randomized trial comparing vinorelbine versus vinorelbine plus cisplatin in patients with recurrent salivary gland malignancies. Cancer 2001;91:541–547. [DOI] [PubMed] [Google Scholar]
- 39. Dreyfuss AI, Fallon BG, Posner MR, et al. Cyclophosphamide, doxorubicin, and cisplatin combination chemotherapy for advanced carcinomas of salivary gland origin. Cancer 1987;60:2869–2872. [DOI] [PubMed] [Google Scholar]
- 40. Belani CP, Eisenberger MA, Gray WC. Preliminary experience with chemotherapy in advanced salivary gland neoplasms. Med Pediatr Oncol 1988;16:197–202. [DOI] [PubMed] [Google Scholar]
- 41. Creagan ET, Woods JE, Rubin J, Schaid DJ. Cisplatin‐based chemotherapy for neoplasms arising from salivary glands and contiguous structures in the head and neck. Cancer 1988;62:2313–2319. [DOI] [PubMed] [Google Scholar]
- 42. Licitra L, Grandi C, Palma SD, et al. Cisplatin, doxorubicin and cyclophosphamide in advanced salivary gland carcinoma: a phase II trial of 22 patients. Ann Oncol 1996;7:640–642. [DOI] [PubMed] [Google Scholar]
- 43. Lin CH, Jeng YM, Tzen CY, et al. Unexpected rapid progression of metastatic adenoid cystic carcinoma during treatment with imatinib mesylate. Head Neck 2005;27:1022–1027. [DOI] [PubMed] [Google Scholar]
- 44. Pfeffer MR, Catane R, Symon Z, et al. A phase II study of Imatinib for advanced adenoid cystic carcinoma of head and neck salivary glands. Oral Oncol 2007;43:33–36. [DOI] [PubMed] [Google Scholar]
- 45. Hotte SJ, Lamont E, MacKenzie M, et al. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c‐kit: a Princess Margaret Hospital phase II consortium study. J Clin Oncol 2005;23:585–590. [DOI] [PubMed] [Google Scholar]
- 46. Guigay JM, Temam S, Janot F, et al. Antitumor activity of imatinib in progressive, highly expressing KIT adenoid cystic carcinoma of the salivary glands: a phase II study. Proc Am Soc Clin Oncol 2007;25:6086. [Google Scholar]
- 47. Ochel HJ, Gademann G, Rocken C, Wordehoff H. Effects of imatinib mesylate on adenoid cystic carcinomas. Anticancer Res 2005;25:3659–3664. [PubMed] [Google Scholar]
- 48. Slevin NJ, Mais KL, Bruce I. Imatinib with cisplatin in recurrent and/or metastatic adenoidcystic carcinoma‐preliminary results of a phase II study of 18 patients with response assessed by morphological and functional imaging. Eur J Cancer 2005;3:292–293. [Google Scholar]
- 49. Vered M, Braunstein E, Buchner A. Immunohistochemical study of epidermal growth factor receptor in adenoid cystic carcinoma of salivary gland origin. Head Neck 2002;24:632–636. [DOI] [PubMed] [Google Scholar]
- 50. Jakob JA, Glisson BS, Kupferman ME, et al. Phase II study of gefitinib in patients with advanced salivary gland cancers. Head Neck 2015;37:644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Agulnik M, Cohen RB, Chen EX, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol 2007;25:3978–3984. [DOI] [PubMed] [Google Scholar]
- 52. Locati LD, Perrone F, Potepan P, et al. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: a phase II study. Oral Oncol 2009;45:574–578. [DOI] [PubMed] [Google Scholar]
- 53. Hitre E, Takacsi‐Nagy Z, Rubovszky G, et al. Cetuximab and platinum‐based chemoradio‐ or chemotherapy of patients with epidermal growth factor receptor expressing adenoid cystic carcinoma: a phase II trial. Br J Cancer 2013;109:1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stephens PJ, Davies HR, Mitani Y, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest 2013;123:2965–2968. doi: 10.1172/JCI67201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ho AS, Kannan K, Roy DM, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet 2013;45:791–798. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Keam B, Shin SH, Cho BC, Lee KW, et al. Phase 2 study of dovitinib in patients with metastatic or unresectable adenoid cystic carcinoma. Cancer 2015;121:2612–2617. [DOI] [PubMed] [Google Scholar]
- 57. Chau NG, Chen EX, Chin SF, et al. A phase II study of sunitinib in recurrent and/or metastatic adenoid cystic carcinoma (ACC) of the salivary glands: current progress and challenges in evaluating molecularly targeted agents in ACC. Ann Oncol 2012;23:1562–1570. [DOI] [PubMed] [Google Scholar]
- 58. Argiris A, Burtness B, Axelrod RS, et al. A phase 2 trial of bortezomib followed by the addition of doxorubicin at progression in patients with recurrent or metastatic adenoid cystic carcinoma of the head and neck: a trial of the Eastern Cooperative Oncology Group (E1303). Cancer 2011;117:3374–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim DW, Shin SH, Kang JH, et al. A multicenter phase II study of everolimus in patients with progressive unresectable adenoid cystic carcinoma. BMC Cancer 2014;14:795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frierson HF Jr, Moskaluk CA. Mutation signature of adenoid cystic carcinoma: evidence for transcriptional and epigenetic reprogramming. J Clin Invest 2013;123:2783–2785. doi: 10.1172/JCI69070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Persson M, Andren Y, Moskaluk CA, et al. Clinically significant copy number alterations and complex rearrangements of MYB and NFIB in head and neck adenoid cystic carcinoma. Genes Chromosomes Cancer 2012;51:805–817. doi: 10.1002/gcc.21965. [DOI] [PubMed] [Google Scholar]
- 62. Rao PH, Roberts D, Zhao YJ, et al. Deletion of 1p32‐p36 is the most frequent genetic change and poor prognostic marker in adenoid cystic carcinoma of the salivary glands. Clin Cancer Res 2008;14:5181–5187. doi: 10.1158/1078-0432.CCR-08-0158. [DOI] [PubMed] [Google Scholar]
- 63. Sandros J, Mark J, Happonen RP, Stenman G. Specificity of 6q‐ markers and other recurrent deviations in human malignant salivary gland tumors. Anticancer Res 1988;8:637–643. [PubMed] [Google Scholar]
- 64. Stenman G, Sandros J, Dahlenfors R, Juberg‐Ode M, Mark J. 6q‐ and loss of the Y chromosome—two common deviations in malignant human salivary gland tumors. Cancer Genet Cytogenet 1986;22:283–293. [DOI] [PubMed] [Google Scholar]
- 65. Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci U S A 2009;106(44):18740–18744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Enlund F, Persson F, Stenman G. Molecular analyses of the candidate tumor suppressor gene, PLAGL1, in benign and malignant salivary gland tumors. Eur J Oral Sci 2004;112:545–547. doi: 10.1111/j.1600-0722.2004.00174.x. [DOI] [PubMed] [Google Scholar]
- 67. Mitani Y, Li J, Rao PH, et al. Comprehensive analysis of the MYB‐NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clin Cancer Res 2010;16:4722–4731. doi: 10.1158/1078-0432.CCR-10-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Brill LB 2nd, Kanner WA, Fehr A, et al. Analysis of MYB expression and MYB‐NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod Pathol 2011;24:1169–1176. doi: 10.1038/modpathol.2011.86. [DOI] [PubMed] [Google Scholar]
- 69. Mitani Y, Rao PH, Futreal PA, et al. Novel chromosomal rearrangements and break points at the t(6;9) in salivary adenoid cystic carcinoma: association with MYB‐NFIB chimeric fusion, MYB expression, and clinical outcome. Clin Cancer Res 2011;17:7003–7014. doi: 10.1158/1078-0432.CCR-11-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. West RB, Kong C, Clarke N, et al. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol 2011;35:92–99. doi: 10.1097/PAS.0b013e3182002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Drier Y, Cotton MJ, Williamson KE, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet 2016;48:265–272. doi: 10.1038/ng.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stenman G, Andersson MK, Andren Y. New tricks from an old oncogene. Cell Cycle 2010;9:3058–3067. doi: 10.4161/cc.9.15.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bandopadhayay P, Ramkissoon LA, Jain P, et al. MYB‐QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 2016;48:273–282. doi: 10.1038/ng.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dash AB, Orrico FC, Ness SA. The EVES motif mediates both intermolecular and intramolecular regulation of c‐Myb. Genes Dev 1996;10:1858–1869. [DOI] [PubMed] [Google Scholar]
- 75. George OL, Ness SA. Situational awareness: regulation of the myb transcription factor in differentiation, the cell cycle and oncogenesis. Cancers (Basel) 2014;6:2049–2071. doi: 10.3390/cancers6042049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhou Y, Ness SA. Myb proteins: angels and demons in normal and transformed cells. Front Biosci (Landmark Ed) 2011;16:1109–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bell D, Roberts D, Karpowicz M, Hanna EY, Weber RS, El‐Naggar AK. Clinical significance of Myb protein and downstream target genes in salivary adenoid cystic carcinoma. Cancer Biol Ther 2011;12:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gao R, Cao C, Zhang M, et al. A unifying gene signature for adenoid cystic cancer identifies parallel MYB‐dependent and MYB‐independent therapeutic targets. Oncotarget 2014;5:12528–12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Phuchareon J, Overdevest JB, McCormick F, Eisele DW, van Zante A, Tetsu O. Fatty acid binding protein 7 is a molecular marker in adenoid cystic carcinoma of the salivary glands: implications for clinical significance. Transl Oncol 2014;7:780–787. doi: 10.1016/j.tranon.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cimino‐Mathews A, Subhawong AP, Elwood H, et al. Neural crest transcription factor Sox10 is preferentially expressed in triple‐negative and metaplastic breast carcinomas. Hum Pathol 2013;44:959–965. doi: 10.1016/j.humpath.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ramkissoon LA, Horowitz PM, Craig JM, et al. Genomic analysis of diffuse pediatric low‐grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A 2013;110:8188–8193. doi: 10.1073/pnas.1300252110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Mitani Y, Liu B, Rao P, et al. Novel MYBL1 gene rearrangements with recurrent MYBL1‐NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clin Cancer Res 2015. 2016;22:725–733. doi: 10.1158/1078-0432.CCR-15-2867-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ross JS, Wang K, Rand JV, et al. Comprehensive genomic profiling of relapsed and metastatic adenoid cystic carcinomas by next‐generation sequencing reveals potential new routes to targeted therapies. Am J Surg Pathol 2014;38:235–238. doi: 10.1097/PAS.0000000000000102. [DOI] [PubMed] [Google Scholar]
- 84. Watson IR, Takahashi K, Futreal PA, Chin L. Emerging patterns of somatic mutations in cancer. Nat Rev Genet 2013;14:703–718. doi: 10.1038/nrg3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Spivakov M, Fisher AG. Epigenetic signatures of stem‐cell identity. Nat Rev Genet 2007;8:263–271. [DOI] [PubMed] [Google Scholar]
- 86. Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington's canal. Nat Rev Mol Cell Biol 2009;10:526–537. [DOI] [PubMed] [Google Scholar]
- 87. Avgustinova A, Benitah SA. The epigenetics of tumour initiation: cancer stem cells and their chromatin. Curr Opin Genet Dev 2016;36:8–15. doi: 10.1016/j.gde.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 88. Gao YB, Chen ZL, Li JG, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 89. Nickerson ML, Dancik GM, Im KM, et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer. Clin Cancer Res 2014;20:4935–4948. doi: 10.1158/1078-0432.CCR-14-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Roy DM, Walsh LA, Chan TA. Driver mutations of cancer epigenomes. Protein Cell 2014;5:265–296. doi: 10.1007/s13238-014-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yap KL, Kiyotani K, Tamura K, et al. Whole‐exome sequencing of muscle‐invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clin Cancer Res 2014;20:6605–6617. doi: 10.1158/1078-0432.CCR-14-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Cojoc M, Mabert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol 2015;31:16–27. doi: 10.1016/j.semcancer.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 94. Sun S, Wang Z. ALDH high adenoid cystic carcinoma cells display cancer stem cell properties and are responsible for mediating metastasis. Biochem Biophys Res Commun 2010;396:843–848. doi: 10.1016/j.bbrc.2010.04.170. [DOI] [PubMed] [Google Scholar]
- 95. Zhou JH, Hanna EY, Roberts D, Weber RS, Bell D. ALDH1 immunohistochemical expression and its significance in salivary adenoid cystic carcinoma. Head Neck 2013;35:575–578. doi: 10.1002/hed.23003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ivanov SV, Panaccione A, Brown B, et al. TrkC signaling is activated in adenoid cystic carcinoma and requires NT‐3 to stimulate invasive behavior. Oncogene 2013;32:3698–3710. doi: 10.1038/onc.2012.377. [DOI] [PubMed] [Google Scholar]
- 97. Ivanov SV, Panaccione A, Nonaka D, et al. Diagnostic SOX10 gene signatures in salivary adenoid cystic and breast basal‐like carcinomas. Br J Cancer 2013;109:444–451. doi: 10.1038/bjc.2013.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Achilleos A, Trainor PA. Neural crest stem cells: discovery, properties and potential for therapy. Cell Res 2012;22:288–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Le Douarin NM, Dupin E. The pluripotency of neural crest cells and their role in brain development. Curr Top Dev Biol 2016;116:659–678. doi: 10.1016/bs.ctdb.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 100. Kaufman CK, Mosimann C, Fan ZP, et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016;351:aad2197. doi: 10.1126/science.aad2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Simoes‐Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development 2015;142:242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 2013;12:15–30. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nonaka D, Chiriboga L, Rubin BP. Sox10: a pan‐schwannian and melanocytic marker. Am J Surg Pathol 2008;32:1291–1298. doi: 10.1097/PAS.0b013e3181658c14. [DOI] [PubMed] [Google Scholar]
- 104. Pozniak CD, Langseth AJ, Dijkgraaf GJ, Choe Y, Werb Z, Pleasure SJ. Sox10 directs neural stem cells toward the oligodendrocyte lineage by decreasing suppressor of fused expression. Proc Natl Acad Sci U S A 2010;107:21795–21800. doi: 10.1073/pnas.1016485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Shakhova O, Zingg D, Schaefer SM, et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat Cell Biol 2012;14:882–890. doi: 10.1038/ncb2535. [DOI] [PubMed] [Google Scholar]
- 106. Ohtomo R, Mori T, Shibata S, et al. SOX10 is a novel marker of acinus and intercalated duct differentiation in salivary gland tumors: a clue to the histogenesis for tumor diagnosis. Mod Pathol 2013;26:1041–1050. doi: 10.1038/modpathol.2013.54. [DOI] [PubMed] [Google Scholar]
- 107. Panaccione A, Chang MT, Carbone BE, et al. NOTCH1 and SOX10 are essential for proliferation and radiation resistance of cancer stem‐like cells in adenoid cystic carcinoma. Clin Cancer Res 2016;22:2083–2095. doi: 10.1158/1078-0432.CCR-15-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer 2008;8:523–534. [DOI] [PubMed] [Google Scholar]
- 109. Westin EH, Gallo RC, Arya SK, et al. Differential expression of the amv gene in human hematopoietic cells. Proc Natl Acad Sci U S A 1982;79:2194–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Gonda TJ, Metcalf D. Expression of myb, myc and fos proto‐oncogenes during the differentiation of a murine myeloid leukaemia. Nature 1984;310:249–251. [DOI] [PubMed] [Google Scholar]
- 111. Malaterre J, Carpinelli M, Ernst M, et al. c‐Myb is required for progenitor cell homeostasis in colonic crypts. Proc Natl Acad Sci U S A 2007;104:3829–3834. doi: 10.1073/pnas.0610055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Malaterre J, Pereira L, Putoczki T, et al. Intestinal‐specific activatable Myb initiates colon tumorigenesis in mice. Oncogene 2016;35:2475–2484. doi: 10.1038/onc.2015.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Malaterre J, Mantamadiotis T, Dworkin S, et al. c‐Myb is required for neural progenitor cell proliferation and maintenance of the neural stem cell niche in adult brain. Stem Cells 2008;26:173–181. [DOI] [PubMed] [Google Scholar]
- 114. Trauth K, Mutschler B, Jenkins NA, Gilbert DJ, Copeland NG, Klempnauer KH. Mouse A‐myb encodes a trans‐activator and is expressed in mitotically active cells of the developing central nervous system, adult testis and B lymphocytes. EMBO J 1994;13:5994–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Betancur P, Simoes‐Costa M, Sauka‐Spengler T, Bronner ME. Expression and function of transcription factor cMyb during cranial neural crest development. Mech Dev 2014;132:38–43. doi: 10.1016/j.mod.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pattabiraman DR, Gonda TJ. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia 2013;27:269–277. doi: 10.1038/leu.2012.225. [DOI] [PubMed] [Google Scholar]
- 117. Lahortiga I, De Keersmaecker K, Van Vlierberghe P, et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet 2007;39:593–595. [DOI] [PubMed] [Google Scholar]
- 118. Liu J, Sato C, Cerletti M, Wagers A. Notch signaling in the regulation of stem cell self‐renewal and differentiation. Curr Top Dev Biol 2010;92:367–409. doi: 10.1016/S0070-2153(10)92012-7. [DOI] [PubMed] [Google Scholar]
- 119. Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci 2005;8:709–715. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]
- 120. Koch U, Lehal R, Radtke F. Stem cells living with a Notch. Development 2013;140:689–704. doi: 10.1242/dev.080614. [DOI] [PubMed] [Google Scholar]
- 121. Coffman CR, Skoglund P, Harris WA, Kintner CR. Expression of an extracellular deletion of Xotch diverts cell fate in Xenopus embryos. Cell 1993;73:659–671. [DOI] [PubMed] [Google Scholar]
- 122. Jimenez‐Rojo L, Granchi Z, Graf D, Mitsiadis TA. Stem cell fate determination during development and regeneration of ectodermal organs. Front Physiol 2012;3:107. doi: 10.3389/fphys.2012.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hartenstein AY, Rugendorff A, Tepass U, Hartenstein V. The function of the neurogenic genes during epithelial development in the drosophila embryo. Development 1992;116:1203–1220. [DOI] [PubMed] [Google Scholar]
- 124. Dang H, Lin AL, Zhang B, Zhang HM, Katz MS, Yeh CK. Role for Notch signaling in salivary acinar cell growth and differentiation. Dev Dyn 2009;238:724–731. doi: 10.1002/dvdy.21875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Stockhausen MT, Kristoffersen K, Poulsen HS. Notch signaling and brain tumors. Adv Exp Med Biol 2012;727:289–304. doi: 10.1007/978-1-4614-0899-4_22. [DOI] [PubMed] [Google Scholar]
- 126. Teodorczyk M, Schmidt MH. Notching on cancer's door: Notch signaling in brain tumors. Front Oncol 2014;4:341. doi: 10.3389/fonc.2014.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood 2006;107:2223–2233. doi: 10.1182/blood-2005-08-3329. [DOI] [PubMed] [Google Scholar]
- 128. Abel EV, Kim EJ, Wu J, et al. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PloS One 2014;9:e91983. doi: 10.1371/journal.pone.0091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Reedijk M. Notch signaling and breast cancer. Adv Exp Med Biol 2012;727:241–257. doi: 10.1007/978-1-4614-0899-4_18. [DOI] [PubMed] [Google Scholar]
- 130. Jhappan C, Gallahan D, Stahle C, et al. Expression of an activated Notch‐related int‐3 transgene interferes with cell differentiation and induces neoplastic transformation in mammary and salivary glands. Genes Dev 1992;6:345–355. [DOI] [PubMed] [Google Scholar]
- 131. Su BH, Qu J, Song M, et al. NOTCH1 signaling contributes to cell growth, anti‐apoptosis and metastasis in salivary adenoid cystic carcinoma. Oncotarget 2014;5:6885–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang K, Zhang Q, Li D, et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple‐negative breast cancer sensitive to a gamma‐secretase inhibitor. Clin Cancer Res 2015;21:1487–1496. doi: 10.1158/1078-0432.CCR-14-1348. [DOI] [PubMed] [Google Scholar]
- 133. Stoeck A, Lejnine S, Truong A, et al. Discovery of biomarkers predictive of GSI response in triple‐negative breast cancer and adenoid cystic carcinoma. Cancer Discov 2014;4:1154–1167. doi: 10.1158/2159-8290.CD-13-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Park EY, Chang E, Lee EJ, et al. Targeting of miR34a‐NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res 2014;74:7573–7582. doi: 10.1158/0008-5472.CAN-14-1140. [DOI] [PubMed] [Google Scholar]
- 135. Feng L, Hatten ME, Heintz N. Brain lipid‐binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron 1994;12:895–908. [DOI] [PubMed] [Google Scholar]
- 136. Anthony TE, Mason HA, Gridley T, Fishell G, Heintz N. Brain lipid‐binding protein is a direct target of Notch signaling in radial glial cells. Genes Dev 2005;19:1028–1033. doi: 10.1101/gad.1302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Keilani S, Sugaya K. Reelin induces a radial glial phenotype in human neural progenitor cells by activation of Notch‐1. BMC Dev Biol 2008;8:69. doi: 10.1186/1471-213X-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Li H, Chang YW, Mohan K, et al. Activated Notch1 maintains the phenotype of radial glial cells and promotes their adhesion to laminin by upregulating nidogen. Glia 2008;56:646–658. doi: 10.1002/glia.20643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Patten BA, Peyrin JM, Weinmaster G, Corfas G. Sequential signaling through Notch1 and erbB receptors mediates radial glia differentiation. J Neurosci 2003;23:6132–6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Patten BA, Sardi SP, Koirala S, Nakafuku M, Corfas G. Notch1 signaling regulates radial glia differentiation through multiple transcriptional mechanisms. J Neurosci 2006;26:3102–3108. doi: 10.1523/JNEUROSCI.4829-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Jacob C, Lotscher P, Engler S, et al. HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. J Neurosci 2014;34:6112–6122. doi: 10.1523/JNEUROSCI.5212-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Giachino C, Basak O, Lugert S, et al. Molecular diversity subdivides the adult forebrain neural stem cell population. Stem Cells 2014;32:70–84. doi: 10.1002/stem.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Morihiro Y, Yasumoto Y, Vaidyan LK, et al. Fatty acid binding protein 7 as a marker of glioma stem cells. Pathol Int 2013;63:546–553. doi: 10.1111/pin.12109. [DOI] [PubMed] [Google Scholar]
- 144. Yun SW, Leong C, Zhai D, et al. Neural stem cell specific fluorescent chemical probe binding to FABP7. Proc Natl Acad Sci U S A 2012;109:10214–10217. doi: 10.1073/pnas.1200817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Liu RZ, Graham K, Glubrecht DD, Lai R, Mackey JR, Godbout R. A fatty acid‐binding protein 7/RXRbeta pathway enhances survival and proliferation in triple‐negative breast cancer. J Pathol 2012;228:310–321. doi: 10.1002/path.4001. [DOI] [PubMed] [Google Scholar]
- 146. Bensaad K, Favaro E, Lewis CA, et al. Fatty acid uptake and lipid storage induced by HIF‐1alpha contribute to cell growth and survival after hypoxia‐reoxygenation. Cell Rep 2014;9:349–365. doi: 10.1016/j.celrep.2014.08.056. [DOI] [PubMed] [Google Scholar]
- 147. Wang C, Chang JYF, Yang C, et al. Type 1 fibroblast growth factor receptor in cranial neural crest cell‐derived mesenchyme is required for palatogenesis. J Biol Chem 2013;288:22174–22183. doi: 10.1074/jbc.M113.463620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wehrle‐Haller B, Weston JA. Receptor tyrosine kinase‐dependent neural crest migration in response to differentially localized growth factors. Bioessays 1997;19:337–345. doi: 10.1002/bies.950190411. [DOI] [PubMed] [Google Scholar]
- 149. Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chao MV. Neurotrophins and their receptors: a convergence point for many signaling pathways. Nat Rev Neurosci 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 151. Pyle AD, Lock LF, Donovan PJ. Neurotrophins mediate human embryonic stem cell survival. Nat Biotechnol 2006;24:344–350. [DOI] [PubMed] [Google Scholar]
- 152. Castellanos DA, Tsoulfas P, Frydel BR, Gajavelli S, Bes JC, Sagen J. TrkC overexpression enhances survival and migration of neural stem cell transplants in the rat spinal cord. Cell Transplant 2002;11:297–307. [PubMed] [Google Scholar]
- 153. Youn YH, Feng J, Tessarollo L, Ito K, Sieber‐Blum M. Neural crest stem cell and cardiac endothelium defects in the TrkC null mouse. Mol Cell Neurosci 2003;24:160–170. [DOI] [PubMed] [Google Scholar]
- 154. Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov 2015;5:25–34. doi: 10.1158/2159-8290.CD-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Edsjo A, Lavenius E, Nilsson H, et al. Expression of trkB in human neuroblastoma in relation to MYCN expression and retinoic acid treatment. Lab Invest 2003;83:813–823. [DOI] [PubMed] [Google Scholar]
- 156. Okamura K, Harada T, Wang S, et al. Expression of TrkB and BDNF is associated with poor prognosis in non‐small cell lung cancer. Lung Cancer 2012;78:100–106. doi: 10.1016/j.lungcan.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 157. Lam CT, Yang ZF, Lau CK, Tam KH, Fan ST, Poon RT. Brain‐derived neurotrophic factor promotes tumorigenesis via induction of neovascularization: implication in hepatocellular carcinoma. Clin Cancer Res 2011;17:3123–3133. doi: 10.1158/1078-0432.CCR-10-2802. [DOI] [PubMed] [Google Scholar]
- 158. Kupferman ME, Jiffar T, El‐Naggar A, et al. TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene 2010;29:2047–2059. doi: 10.1038/onc.2009.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Bouzas‐Rodriguez J, Cabrera JR, Delloye‐Bourgeois C, et al. Neurotrophin‐3 production promotes human neuroblastoma cell survival by inhibiting TrkC‐induced apoptosis. J Clin Invest 2010;120:850–858. doi: 10.1172/JCI41013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett 2001;169:107–114. [DOI] [PubMed] [Google Scholar]
- 161. Kengaku M, Okamoto H. Basic fibroblast growth factor induces differentiation of neural tube and neural crest lineages of cultured ectoderm cells from Xenopus gastrula. Development 1993;119:1067–1078. [DOI] [PubMed] [Google Scholar]
- 162. Monsoro‐Burq AH, Fletcher RB, Harland RM. Neural crest induction by paraxial mesoderm in Xenopus embryos requires FGF signals. Development 2003;130:3111–3124. [DOI] [PubMed] [Google Scholar]
- 163. Dienstmann R, Rodon J, Prat A, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol 2014;25:552–563. doi: 10.1093/annonc/mdt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Myoken Y, Myoken Y, Okamoto T, et al. Immunohistochemical study of overexpression of fibroblast growth factor‐1 (FGF‐1), FGF‐2, and FGF receptor‐1 in human malignant salivary gland tumours. J Pathol 1996;178:429–436. [DOI] [PubMed] [Google Scholar]
- 165. Vekony H, Ylstra B, Wilting SM, et al. DNA copy number gains at loci of growth factors and their receptors in salivary gland adenoid cystic carcinoma. Clin Cancer Res 2007;13:3133–3139. doi: 10.1158/1078-0432.CCR-06-2555. [DOI] [PubMed] [Google Scholar]
- 166. Xerri L, Battyani Z, Grob JJ, et al. Expression of FGF1 and FGFR1 in human melanoma tissues. Melanoma Res 1996;6:223–230. [DOI] [PubMed] [Google Scholar]
- 167. Blanckaert VD, Hebbar M, Louchez MM, Vilain MO, Schelling ME, Peyrat JP. Basic fibroblast growth factor receptors and their prognostic value in human breast cancer. Clin Cancer Res 1998;4:2939–2947. [PubMed] [Google Scholar]
- 168. Zhao D, Lu Y, Yang C, Zhou X, Xu Z. Activation of FGF receptor signaling promotes invasion of non‐small‐cell lung cancer. Tumour Biol 2015;36:3637–3642. doi: 10.1007/s13277-014-3001-y. [DOI] [PubMed] [Google Scholar]
- 169. Rosini P, Bonaccorsi L, Baldi E, et al. Androgen receptor expression induces FGF2, FGF‐binding protein production, and FGF2 release in prostate carcinoma cells: role of FGF2 in growth, survival, and androgen receptor down‐modulation. Prostate 2002;53:310–321. doi: 10.1002/pros.10164. [DOI] [PubMed] [Google Scholar]
- 170. Tomlinson DC, Knowles MA. Altered splicing of FGFR1 is associated with high tumor grade and stage and leads to increased sensitivity to FGF1 in bladder cancer. Am J Pathol 2010;177:2379–2386. doi: 10.2353/ajpath.2010.100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Sato T, Oshima T, Yoshihara K, et al. Overexpression of the fibroblast growth factor receptor‐1 gene correlates with liver metastasis in colorectal cancer. Oncol Rep 2009;21:211–216. [PubMed] [Google Scholar]
- 172. Miglarese MR, Halaban R, Gibson NW. Regulation of fibroblast growth factor 2 expression in melanoma cells by the c‐MYB proto‐oncoprotein. Cell Growth Differ 1997;8:1199–1210. [PubMed] [Google Scholar]
- 173. Dutt A, Salvesen HB, Chen TH, et al. Drug‐sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A 2008;105:8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Pollock PM, Gartside MG, Dejeza LC, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007;26:7158–7162. doi: 10.1038/sj.onc.1210529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Park WJ, Theda C, Maestri NE, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet 1995;57:321–328. [PMC free article] [PubMed] [Google Scholar]
- 176. Gartside MG, Chen H, Ibrahimi OA, et al. Loss‐of‐function fibroblast growth factor receptor‐2 mutations in melanoma. Mol Cancer Res 2009;7:41–54. doi: 10.1158/1541-7786.MCR-08-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Grose R, Fantl V, Werner S, et al. The role of fibroblast growth factor receptor 2b in skin homeostasis and cancer development. EMBO J 2007;26:1268–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Turner N, Grose R. Fibroblast growth factor signaling: from development to cancer. Nat Rev Cancer 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 179. Chang J, Liu X, Wang S, et al. Prognostic value of FGFR gene amplification in patients with different types of cancer: a systematic review and meta‐analysis. PLoS One 2014;9:e105524. doi: 10.1371/journal.pone.0105524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene 2004;23:7267–7273. doi: 10.1038/sj.onc.1207946. [DOI] [PubMed] [Google Scholar]
- 181. Fujita S, Ikeda T. Cancer stem‐like cells in adenoid cystic carcinoma of salivary glands: relationship with morphogenesis of histological variants. J Oral Pathol Med 2012;41:207–213. doi: 10.1111/j.1600-0714.2011.01096.x. [DOI] [PubMed] [Google Scholar]
- 182. Karbanova J, Laco J, Marzesco AM, et al. Human prominin‐1 (CD133) is detected in both neoplastic and non‐neoplastic salivary gland diseases and released into saliva in a ubiquitinated form. PLoS One 2014;9:e98927. doi: 10.1371/journal.pone.0098927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Naka H, Nakamura S, Shimazaki T, Okano H. Requirement for COUP‐TFI and II in the temporal specification of neural stem cells in CNS development. Nat Neurosci 2008;11:1014–1023. doi: 10.1038/nn.2168. [DOI] [PubMed] [Google Scholar]
- 184. Doetsch F, Verdugo JM, Caille I, Alvarez‐Buylla A, Chao MV, Casaccia‐Bonnefil P. Lack of the cell‐cycle inhibitor p27Kip1 results in selective increase of transit‐amplifying cells for adult neurogenesis. J Neurosci 2002;22:2255–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Nguyen L, Besson A, Heng JI, et al. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev 2006;20:1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Nguyen L, Besson A, Roberts JM, Guillemot F. Coupling cell cycle exit, neuronal differentiation and migration in cortical neurogenesis. Cell Cycle 2006;5:2314–2318. [DOI] [PubMed] [Google Scholar]
- 187. Ben‐Izhak O, Akrish S, Gan S, Nagler RM. Skp2 and salivary cancer. Cancer Biol Ther 2009;8:153–158. [DOI] [PubMed] [Google Scholar]
- 188. Nagler RM, Ben‐Izhak O, Ostrovsky D, Golz A, Hershko DD. The expression and prognostic significance of Cks1 in salivary cancer. Cancer Invest 2009;27:512–520. doi: 10.1080/07357900802239116. [DOI] [PubMed] [Google Scholar]
- 189. Hao Z, Huang S. E3 ubiquitin ligase Skp2 as an attractive target in cancer therapy. Front Biosci (Landmark Ed) 2015;20:474–490. [DOI] [PubMed] [Google Scholar]
- 190. Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron 2003;38:17–31. [DOI] [PubMed] [Google Scholar]
- 191. Okamura Y, Saga Y. Notch signaling is required for the maintenance of enteric neural crest progenitors. Development 2008;135:3555–3565. doi: 10.1242/dev.022319. [DOI] [PubMed] [Google Scholar]
- 192. Moskaluk CA, Baras AS, Mancuso SA, et al. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Lab Invest 2011;91:1480–1490. doi: 10.1038/labinvest.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ruggeri BA, Camp F, Miknyoczki S. Animal models of disease: pre‐clinical animal models of cancer and their applications and utility in drug discovery. Biochem Pharmacol 2014;87:150–161. doi: 10.1016/j.bcp.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 194. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 195. Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu O. Genetic profiling reveals cross‐contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLoS One 2009;4:e6040. doi: 10.1371/journal.pone.0006040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Hu K, Li SL, Gan YH, Wang CY, Yu GY. Epiregulin promotes migration and invasion of salivary adenoid cystic carcinoma cell line SACC‐83 through activation of ERK and Akt. Oral Oncol 2009;45:156–163. doi: 10.1016/j.oraloncology.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 197. Li SL. [Establishment of a human cancer cell line from adenoid cystic carcinoma of the minor salivary gland]. Zhonghua Kou Qiang Yi Xue Za Zhi 1990;25:29–31, 62. [PubMed] [Google Scholar]
- 198. Wang L, Wang Y, Bian H, Pu Y, Guo C. Molecular characteristics of homologous salivary adenoid cystic carcinoma cell lines with different lung metastasis ability. Oncol Rep 2013;30:207–212. doi: 10.3892/or.2013.2460. [DOI] [PubMed] [Google Scholar]
- 199. Valentin MD, da Silva SD, Privat M, Alaoui‐Jamali M, Bignon YJ. Molecular insights on basal‐like breast cancer. Breast Cancer Res Treat 2012;134:21–30. doi: 10.1007/s10549-011-1934-z. [DOI] [PubMed] [Google Scholar]
- 200. Davis FM, Stewart TA, Thompson EW, Monteith GR. Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci 2014;35:479–488. doi: 10.1016/j.tips.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 201. Wallrapp C, Muller‐Pillasch F, Solinas‐Toldo S, et al. Characterization of a high copy number amplification at 6q24 in pancreatic cancer identifies c‐myb as a candidate oncogene. Cancer Res 1997;57:3135–3139. [PubMed] [Google Scholar]
- 202. Kauraniemi P, Hedenfalk I, Persson K, et al. MYB oncogene amplification in hereditary BRCA1 breast cancer. Cancer Res 2000;60:5323–5328. [PubMed] [Google Scholar]
- 203. Kasper LH, Boussouar F, Ney PA, et al. A transcription‐factor‐binding surface of coactivator p300 is required for haematopoiesis. Nature 2002;419:738–743. [DOI] [PubMed] [Google Scholar]
- 204. Pattabiraman DR, Gonda TJ. Role and potential for therapeutic targeting of MYB in leukemia. Leukemia 2013;27:269–277. [DOI] [PubMed] [Google Scholar]
- 205. Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett 2008;18:5904–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Toure M, Crews CM. Small‐molecule PROTACS: new approaches to protein degradation. Angew Chem Int Ed Engl 2016;55:1966–1973. doi: 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- 207. Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov 2014;13:357–378. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 208. Schott AF, Landis MD, Dontu G, et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res 2013;19:1512–1524. doi: 10.1158/1078-0432.CCR-11-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Adenoid Cystic Carcinoma Research Foundation . Selected phase i clinical trials of NOTCH inhibitors. 2014; updated May 2014. Available at: http://www.accrf.org/wp-content/uploads/Clinical-Trials-Phase-I-2014-May.pdf. Accessed December 25, 2015.
- 210. Xiao N, Lin Y, Cao H, et al. Neurotrophic factor GDNF promotes survival of salivary stem cells. J Clin Invest 2014;124:3364–3377. doi: 10.1172/JCI74096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Forthmann B, Aletta JM, Lee YW, et al. Coalition of nuclear receptors in the nervous system. J Cell Physiol 2015;230:2875–2880. doi: 10.1002/jcp.25036. [DOI] [PubMed] [Google Scholar]
- 212. Gu Y, Xue C, Zhu J, et al. Basic fibroblast growth factor (bFGF) facilitates differentiation of adult dorsal root ganglia‐derived neural stem cells toward Schwann cells by binding to FGFR‐1 through MAPK/ERK activation. J Mol Neurosci 2014;52:538–551. doi: 10.1007/s12031-013-0109-2. [DOI] [PubMed] [Google Scholar]
- 213. Lee YW, Stachowiak EK, Birkaya B, et al. NGF‐induced cell differentiation and gene activation is mediated by integrative nuclear FGFR1 signaling (INFS). PLoS One 2013;8:e68931. doi: 10.1371/journal.pone.0068931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Terranova C, Narla ST, Lee YW, et al. Global developmental gene programmingi a nuclear form of fibroblast growth factor receptor‐1 (FGFR1). PLoS One 2015;10:e0123380. doi: 10.1371/journal.pone.0123380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Hoffman MP, Kidder BL, Steinberg ZL, et al. Gene expression profiles of mouse submandibular gland development: FGFR1 regulates branching morphogenesis in vitro through BMP‐ and FGF‐dependent mechanisms. Development 2002;129:5767–5778. [DOI] [PubMed] [Google Scholar]
- 216. Jaskoll T Melnick M. Salivary Gland Branching Morphogenesis. Available at: http://www.ncbi.nlm.nih.gov/books/NBK6103/. Austin, TX: Landes Bioscience; 2000. −2013. [Google Scholar]
- 217. Jaskoll T, Abichaker G, Witcher D, et al. FGF10/FGFR2b signaling plays essential roles during in vivo embryonic submandibular salivary gland morphogenesis. BMC Dev Biol 2005;5:11. doi: 10.1186/1471-213X-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Jaskoll T, Zhou YM, Chai Y, et al. Embryonic submandibular gland morphogenesis: stage‐specific protein localization of FGFs, BMPs, Pax6 and Pax9 in normal mice and abnormal SMG phenotypes in FgfR2‐IIIc(+/Delta), BMP7(‐/‐) and Pax6(‐/‐) mice. Cells Tissues Organs 2002;170:83–98. [DOI] [PubMed] [Google Scholar]
- 219. Ohuchi H, Hori Y, Yamasaki M, et al. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi‐organ development. Biochem Biophys Res Commun 2000;277:643–649. doi: 10.1006/bbrc.2000.3721. [DOI] [PubMed] [Google Scholar]
- 220. Chopin V, Lagadec C, Toillon RA, Le Bourhis X. Neurotrophin signaling in cancer stem cells. Cell Mol Life Sci 2016;73:1859–1870. doi: 10.1007/s00018-016-2156-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev 2014;34:280–300. doi: 10.1002/med.21288. [DOI] [PubMed] [Google Scholar]
- 222. Steelman LS, Fitzgerald T, Lertpiriyapong K, et al. Critical roles of EGFR family members in breast cancer and breast cancer stem cells: targets for therapy. Curr Pharm Des 2016;22:2358–2388. [DOI] [PubMed] [Google Scholar]
- 223. Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: what are the cancer relevant targets? Cancer Lett 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 224. Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 225. Goncalves P, Kummar S, Siu L, et al. A phase II study of suberoylanilide hydroxamic acid (SAHA) in subjects with locally advanced, recurrent, or metastatic adenoid cystic carcinoma. J Clin Oncol 2013;31:suppl. [Google Scholar]
- 226. Junwei S, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Molecular Cell 2014;54:728–736. doi: 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010;468:1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Mansour MR, Abraham BJ, Anders L, et al. Oncogene regulation. An oncogenic super‐enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Wu S‐Y, Lee AY, Lai H‐T, Zhang H, Chiang C‐M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene‐specific targeting. Molecular Cell 2013;49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Kwiatkowski N, Zhang T, Rahl PB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Sridharan V, Liao X, Chau NG, et al. Immunologic profiling of adenoid cystic carcinoma (ACC). J Immunother Cancer 2015;3(suppl 2):105–106. [Google Scholar]
- 234. Yi KH, Chen L. Fine tuning the immune response through B7‐H3 and B7‐H4. Immunol Rev 2009;229:145–151. doi: 10.1111/j.1600-065X.2009.00768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]