

Abstract
Photoexcited arylketones catalyze the direct chlorination of C(sp3)–H groups by N-chlorosuccinimide. Acetophenone is the most effective catalyst for functionalization of unactivated C–H groups while benzophenone provides better yields for benzylic C–H functionalization. Activation of both acetophenone and benzophenone can be achieved by irradiation with a household compact fluorescent lamp. This light-dependent reaction provides a better control of the reaction as compared to the traditional chlorination methods that proceed through a free radical chain propagation mechanism.
Keywords: C–H functionalization, Chlorination, Arylketone, Photochemistry, Visible light
Graphical abstract
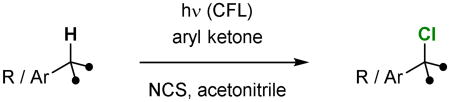
1. Introduction
Halogenation is a common strategy to enhance the potency or alter the physical properties of small molecule drugs.1-3 Naturally occurring halogenated molecules also often display medicinally useful activities.4,5 Catalysts that promote C–H halogenation are thus highly valuable. We report herein a catalytic, light-dependent method for C(sp3)–H chlorination (Fig. 1).
Figure 1.

A catalytic light-dependent method for C–H chlorination.
Whereas a variety of C–H fluorination methods6-18 have been reported, free radical chain reactions remain to be the most frequently used method for C–H chlorination. In contrast, biological C–H chlorination is catalyzed by α-ketoglutarate (αKG)-dependent non-heme iron (FeNH) halogenases.19-22 A good example is the sequential chlorination of the BarA-loaded l-leucine by BarB2 and BarB1 in the biosynthesis of barbamide (1) (Fig. 2).23,24 Mechanistically, this halogenase-catalyzed C–H chlorination reaction is similar to C–H hydroxylation reactions catalyzed by the corresponding oxygenases.25-27
Figure 2.

C–H chlorination in the biosynthesis of barbamide (1) and the mechanism of C–H chlorination catalyzed by FeNH-αKG halogenase.
The reaction of 2 has inspired Que and co-workers to develop biomimetic chlorinating complexes.28 They showed that [Fe(TPA)Cl2](ClO4) (8) promotes C(sp3)–H chlorination upon activation with tert-butyl hydroperoxide (Fig. 3) (TPA = tri-(2-pyridylmethyl)amine). However, mechanistic studies indicated that the Fenton-type free radical chain reaction is also operative and there is little or no turnover of the catalyst.29,30 Later, Groves and co-workers found that Mn(TMP)Cl (9) catalyzes C(sp3)–H chlorination by bleach in the presence of a catalytic amount of tetra-n-butylammonium chloride (TMP = tetramesitylporphyrin).31,32 However, heme mimetics could also promote aromatic C(sp2)–H oxidation leading to low selectivity for aromatic substrates. For example, Fuji and co-workers found that Fe(TPFP)(NO3) (10) catalyzed chlorination of electron-rich arenes by ozone and tetra-n-butylammonium chloride upon activation by a catalytic amount of trifluoroacetic acid (TPFP = tetrakis(pentafluorophenyl)porphyrin).33
Figure 3.

Examples of reported C–H chlorination complexes.
A variety of directing groups have been developed to better control the regioselectivity of C–H chlorination. For example, Sanford and co-workers showed that a pyridine group can direct and facilitate catalytic halogenation of benzylic C(sp3)–H groups by palladium.34,35 Yu and co-workers also demonstrated that an oxazoline group facilitates palladium-catalyzed C(sp3)–H halogenation,36,37 and 2-nitrobenzenesulfonamide is an excellent directing group for copper-catalyzed C(sp3)–H bromination.38 Additional directing groups for palladium-catalyzed C(sp3)–H halogenation include 2-pyridylsulfoximine,39 amide,40 and 8-aminoquinoline.41 However, C(sp3)–H halogenation without over-oxidation is still challenging. Notably, C(sp2)–H chlorination can be achieved more easily by using, for example, chlorobis(methoxycarbonyl)guanidine (CBMG)42 developed by Baran and co-workers as chlorinated arenes are less electron-rich and thus less reactive than their precursors toward aromatic substitution.
Baran and co-workers have demonstrated that site-selective halogenation can be realized by trifluoroethyl N-halocarbamate-mediated Hofmann–Löffler–Freytag reaction.43 Unlike the N-chloroamine-mediated method, the N-cyclization product is not formed with the carbamate group. Ball and co-workers also showed that a peroxide group can be used as an internal oxidant to achieve copper-catalyzed regioselective C(sp3)–H chlorination without over-oxidation.44 Intermolecular oxidative radical halogenation has also been achieved by Alexanian, Vanderwal, and co-workers by N-haloamides.45,46 They showed that N-chloro-N-(tert-butyl)-3,5-bis(trifluoromethyl)benzamide chlorinates sclareolide selectively and effectively. Addition of cesium carbonate helped suppress dichlorination. Thus, a large excess of substrates is not needed to prevent over-oxidation. However, one potential safety concern for performing large-scale free radical chain reactions is that the chain propagation process may lead to a runaway reaction. To date, the most practical method for introducing a chlorine atom onto an aliphic chain is arguably the silver-catalyzed decarboxylative chlorination reaction developed by Li and co-workers, although pre-installation of a carboxylic acid group is required.47
Walling discovered serendipitously in 1965 that photoreduction of triplte benzophenone by cyclohexane in the presence of internal standard Freon 112 (CFCl2CFCl2) led to the formation of cyclohexyl chloride.48 He subsequently found that carbon tetrachloride is a better chlorine atom donor. UV-irradiation of a mixture of cyclohexane and benzophenone in carbon tetrachloride gave cyclohexyl chloride and benzpinacol in good yields. However, there is no report of the development of a catalytic system for this C–H chlorination reaction. We have demonstrated that triplet arylketones are functionally similar to the metal-oxo species of 5 and can catalyze C(sp3)–H fluorination.6,7 We now show that catalytic C(sp3)–H chlorination can also be achieved through this photochemical reaction.
2. Results and discussion
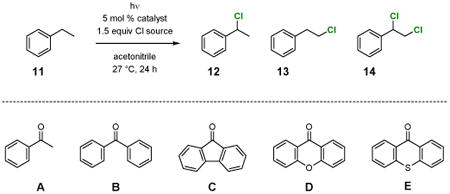
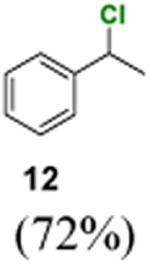
Our work started with optimization of the catalyst system for benzylic chlorination using ethylbenzene (11) as the standard substrate (Table 1). Because benzophenone ketyl radical is rather stable and susceptible to deactivation by dimerization, we first tested if acetophenone could offer a better catalyst turnover number. However, irradiation of 11 with UV light in carbon tetrachloride in the presence of 5 mol % of acetophenone gave only 12% yield of benzylic chloride 12 along with 7% of the homobenzylic chloride 13 (entry 1). Whereas there was nearly no reaction when irradiated with violet light for 24 h (entry 2), switching the chlorine atom donor to N-chlorosuccinimide (NCS) led to a quick reaction and 11 was consumed completely to give 12 together with 13 and the dichlorination product 14 (entry 3). The reaction proceeded well but slower when a household compact fluorescence lamp (CFL) was used as the light source (entry 4). Nonetheless, acetophenone, benzophenone, 9-fluorenone, xanthone, and thioxanthone can all catalyst C–H chlorination by NCS upon activation by CFL-irradiation (entries 4–8). Among these arylketones, benzophenone and 9-fluorenone provide the best reaction rates and selectivity (entries 5 and 6). We have also examined the effectiveness of a series of other chlorinating reagents, but did not observe improvement in reactivity or selectivity by using N-chlorophthalimide, trichloroisocyanuric acid, 1,3-dichloro-5,5-dimethylhydantoin, chloramine T, dichloramine T, and CBMG.
Table 1. Effects of the catalyst, chlorine donor, and light source on the benzylic chlorination of 11.
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Entry | Catalyst | Cl source | Light source | 11 | 12 | 13 | 14 |
| 1 | A | CCl4a | 350 nmb | 81% | 12% | 7% | – |
| 2 | A | CCl4a | 419 nmc | >95% | – | – | – |
| 3 | A | NCS | 419 nmc | 0% | 68% | 12% | 20% |
| 4 | A | NCS | CFLd | 15% | 70% | 13% | 2% |
| 5 | B | NCS | CFLd | 0% | 74% | 7% | 19% |
| 6 | C | NCS | CFLd | 0% | 72% | 7% | 21% |
| 7 | D | NCS | CFLd | 6% | 70% | 15% | 8% |
| 8 | E | NCS | CFLd | 21% | 67% | 10% | 2% |
No acetonitrile, carbon tetrachloride is used as the solvent.
16× RPR-3500Å lamps (24 W, 300–420 nm).
16× RPR-4190Å lamps (24 W, 375–465 nm).
1× compact fluorescence lamp (19 W).
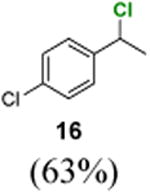
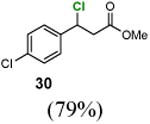
We next used benzophenone as the standard catalyst to explore the scope of this reaction (Table 2). Introduction of an electron-withdrawing group to the benzene ring at the ortho, meta, or para position did not affect the reaction significantly (entries 1–6). However, chlorination of ethylbenzene derivatives with an electron-donating group led to benzylic chlorides that are not stable under the reaction conditions. Primary and tertiary benzylic C–H groups could also be chlorinated smoothly (entries 7 and 8). Remarkably, an ester group at the β-position can be tolerated (entry 9). Chloride 30 did not undergo elimination under the reaction conditions.
Table 2. Scope of the benzophenone-catalyzed photochemical benzylic C–H chlorination.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Substrate | Time | Product (Isolated yield) | β-Clb | 1,2-Cl2b |
| 1 |
|
24 h |
|
7% | 19% |
| 2 |
|
24h |
|
17% | 19% |
| 3a |
|
9 h |
|
23% | ND |
| 4 |
|
24 h |
|
12% | 4% |
| 5 |
|
9 h |
|
27% | ND |
| 6 |
|
24 h |
|
24% | ND |
| 7a |
|
5 h |
|
– | – |
| 8a |
|
48 h |
|
3%c | ND |
| 9 |
|
48 h |
|
ND | ND |
Using 1.2 equiv NCS.
Determined by 1H NMR and ND denotes not detected.
Together with 9% γ-chlorination product.
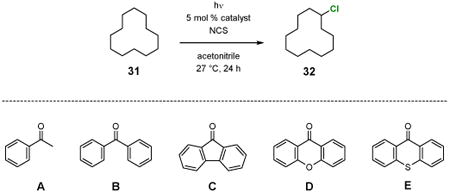
For non-benzylic chlorination, cyclododecane (31) was used as the standard substrate for catalyst screen (Table 3). Acetophenone, benzophenone, 9-fluorenone, xanthone, and thioxanthone all catalyzed the reaction well, giving good yields of cyclododecyl chloride (32) (entries 1–5). It is noteworthy that, unlike most innate C(sp3)–H chlorination reactions, this photochemical reaction does not require the use of a large excess of the substrate to suppress over-chlorination. Acetophenone, for example, catalyzed monochlorination of 31 in good yields even when a 1:1 or 1:1.2 ratio of substrate to oxidant was used (entries 6 and 7).
Table 3. Effects of the catalyst on the chlorination of 31.
| |||
|---|---|---|---|
|
| |||
| Entry | Catalyst | Cl equiv | NMR yield |
| 1 | A | 0.8 | 98%a |
| 2 | B | 0.8 | 93%a |
| 3 | C | 0.8 | 97%a |
| 4 | D | 0.8 | 91%a |
| 5 | E | 0.8 | 77%a |
| 6 | A | 1.0 | 85%b |
| 7 | A | 1.2 | 82%b |
Calculated based on NCS.
Calculated based on 31.
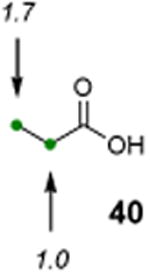
The utility of this photochemical reaction has also been briefly investigated (Table 4). Whereas only a slight excess of the substrate is needed to achieve monochlorination of simple hydrocarbons (entries 1 and 2), dichlorination of the tert-butyl group occurred even at low conversion, eroding the yields for monochlorination products (entries 3 and 4). Additionally, chlorination of propionic acid (39) and isovaleric acid (40) occurred at various positions (entries 5 and 6), and chlorination of sclareolide and cholesterol resulted in complex mixtures of products. The α-chlorination of 39 by NCS likely proceeded through an uncatalyzed, non-radical pathway. However, there was no significant amount of electrophilic chlorination product in the reaction of 40 with NCS possibly due to increased steric hindrance around the α-position.
Table 4. Scope of the benzophenone-catalyzed photochemical aliphatic C–H chlorination.

| ||||
|---|---|---|---|---|
|
| ||||
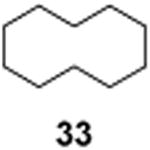
| Entry | Substrate | Product | Time | Isolated yielda or conversion |
| 1 |
|
|
24 h | 95% |
| 2 |
|
|
24 h | 95% |
| 3 |
|
|
9 h | 70% |
| 4 |
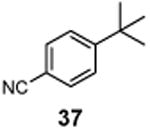
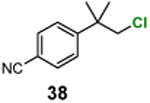
|
|
9 h | 82% |
| 5 |
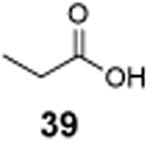
|
|
24 h | 86% conversion |
| 6 |
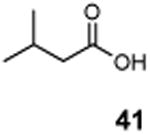
|
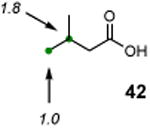
|
24 h | 76% conversion |
Calculated based on NCS.
Product distribution determined by 1H NMR.
The lower selectivity of triplet ketone-catalyzed C(sp3)–H chlorination comparing to the corresponding fluorination reaction suggests that the rate limiting step for chlorination is C–H abstraction. We suspect that the transfer of a chlorine atom from NSC to the alkyl radical resulted from C–H abstraction is a facile process, leading to kinetic C–H functionalization. In contrast, the transfer of a fluorine atom from Selectfluor to the alkyl radical49 is likely slower than C–H abstraction. Thermodynamic products were thus formed due to reversible C–H abstraction. Supportive to this hypothesis is the observation that dichlorination of ethylbenzene (11) gave (1,2-dichloroethyl)benzene (14) (Table 1) whereas difluorination of 11 provided (1,1-difluoroethyl)benzene.6 Based on the C–H bond strength, 1,1-dihalogenation should be favored in both cases. The C–H bond dissociation energies for H–CH3, H–CH2F, and H–CH2Cl, are 104.9, 101.3, and 100.1 kcal/mol, respectively.50 Finally, we have confirmed that benzophenone-catalyzed chlorination of 11 is not a free radical chain reaction (Fig. 4). The reaction stopped immediately after the light was turned off.
Figure 4.
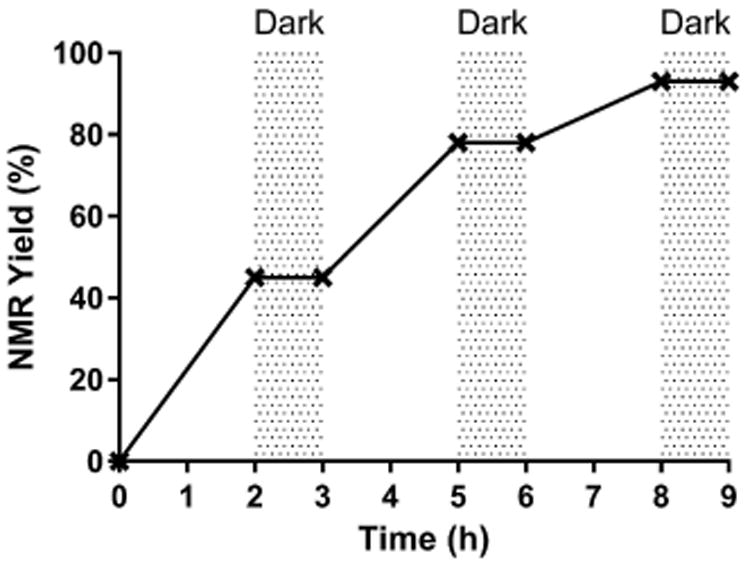
Benzophenone-catalyzed chlorination of 11 by NCS is a light-dependent process.
3. Conclusion
Triplet arylketones effectively catalyze kinetic C(sp3)–H chlorination by NCS. For simple substrates, good yields can be obtained without using a large excess of the substrates. Additionally, there is no competing aromatic chlorination. Unlike free radical chain reactions, this light-dependent reaction allows for control of degree of chlorination by irradiation time. However, the regioselectivity of this reaction is low, in particular for more complex substrates, limiting its utility to functionalization of simple organic compounds.
4. Experimental
General procedure for triplet ketone-catalyzed C(sp3)–H chlorination
To a 4 mL clear vial charged with the reaction substrate, N-chlorosuccinimide, ketone catalyst in anhydrous acetonitrile (0.2 M) was degassed and irradiated with a 19 W compact fluorescent lamp at room temperature for 24 h. The solvent was then removed and the residue was dissolved in diethyl ether, filtrated, concentrated and purified by preparative thin-layer chromatography.
(1-Chloroethyl)benzene (12)
1H NMR (400 MHz, CDCl3) δ 7.45 (d, J = 8.1 Hz, 2H), 7.41–7.30 (m, 3H), 5.12 (q, J = 6.8 Hz,1H), 1.88 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 128.8, 128.6, 128.3, 126.5, 58.8, 26.5; MS (EI) calcd for C8H9Cl [M+] 140.0, found 140.1.
1-Chloro-4-(1-chloroethyl)benzene (16)
1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 8.8 Hz, 2H), 5.06 (q, J = 6.8 Hz, 1H), 1.83 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 141.5, 134.1, 128.9, 128.1, 57.9, 26.6; MS (EI) calcd for C8H8Cl2 [M+] 174.0, found 174.0.
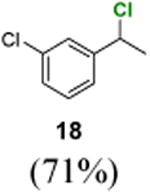
1-Chloro-3-(1-chloroethyl)benzene (18)
1H NMR (400 MHz, CDCl3) δ 7.43–7.40 (m, 1H), 7.32–7.26 (m, 3H), 5.03 (q, J = 6.8 Hz, 1H), 1.83 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 144.9, 134.6, 130.1, 128.5, 126.9, 124.9, 57.8, 26.6; MS (EI)calcd for C8H8Cl2 [M+] 174.0, found 174.0.
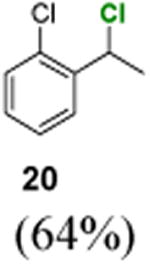
1-Chloro-2-(1-chloroethyl)benzene (20)
1H NMR (400 MHz, CDCl3) δ 7.53–7.07 (m, 4H), 5.59 (q, J = 6.8 Hz, 1H), 1.84 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 140.1, 132.5, 129.7, 129.4, 128.0, 127.5, 54.6, 25.8; MS (EI) calcd for C8H8Cl2 [M+] 174.0, found 174.0.
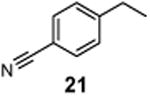
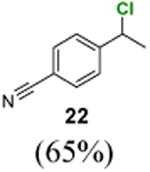
4-(1-Chloroethyl)benzonitrile (22)
1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 8.2 Hz, 2H), 7.53 (d, J = 8.2 Hz, 2H), 5.08 (q, J = 6.8 Hz, 1H), 1.83 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 147.9, 132.6, 127.5, 118.6, 112.2, 57.3, 26.4; MS (EI) calcd for C9H8ClN [M+] 165.0, found 165.0.
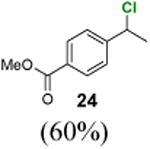
Methyl 4-(1-chloroethyl)benzoate (24)
1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.2 Hz, 2H), 7.49 (d, J = 8.2 Hz, 2H), 5.10 (q, J = 6.8 Hz, 1H), 3.92 (s, 3H), 1.85 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.7, 147.7, 130.1, 130.1, 126.7, 57.9, 52.4, 26.6; MS (EI) calcd for C9H11ClO2 [M+] 198.0, found 198.1.
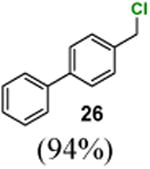
4-(Chloromethyl)-1,1′-biphenyl (26)
1H NMR (400 MHz, CDCl3) δ 7.62–7.57 (m, 4H), 7.50–7.41 (m, 4H), 7.42–7.34 (m, 1H), 4.65 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 141.5, 140.6, 136.6, 129.2, 129.0, 127.7, 127.6, 127.3, 46.2; MS (EI) calcd for C13H11Cl [M+] 202.1, found 202.1.
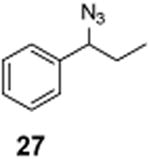
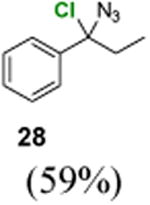
(1-Azido-1-chloropropyl)benzene (28)
1H NMR (500 MHz, CDCl3) δ 7.44–7.27 (m, 5H), 1.95–1.74 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 133.6, 131.2, 128.1, 128.1, 126.9, 67.9, 34.3, 10.8; MS (EI) calcd for C9H10ClN3 [M+] 195.1, found 195.0.
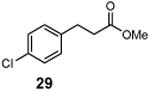
Methyl 3-chloro-3-(4-chlorophenyl)propanoate (30)
1H NMR (400 MHz, CDCl3) δ 7.42–7.27 (m, 4H), 5.31 (m, 1H), 3.70 (s), 3.09 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 169.9, 138.9, 134.7, 129.2, 128.5, 57.2, 52.3, 44.7; MS (EI) calcd for C10H10ClO2 [M+] 232.0, found 232.0.
Chlorocyclododecane (32)
1H NMR (400 MHz, CDCl3) δ 4.21–4.03 (m, 1H), 2.01–1.84 (m, 2H), 1.83–1.64 (m, 2H), 1.63– 1.46 (m, 2H), 1.44–1.18 (m, 16H); 13C NMR (100 MHz, CDCl3)δ 60.4, 34.0, 23.9, 23.8, 23.5, 23.5, 22.0; MS (EI) calcd for C12H23Cl [M+] 202.1, found 202.1.
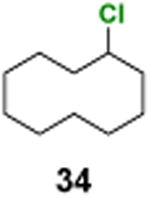
Chlorocyclodecane (34)
1H NMR (400 MHz, CDCl3) δ 4.47– 4.13 (m, 1H), 2.13–2.02 (m, 2H), 2.01–1.90 (m, 2H), 1.75–1.64 (m, 2H), 1.63–1.37 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 62.2, 34.3, 25.3, 24.9, 24.3, 23.0; MS (EI) calcd for C10H19Cl [M+] 174.1, found 174.1.
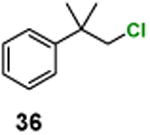
(1-Chloro-2-methylpropan-2-yl)benzene (36)
H NMR (400 MHz, CDCl3) δ 7.46–7.27 (m, 5H), 3.66 (s, 2H), 1.44 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 146.1, 128.5, 126.6, 126.0, 56.5, 39.9, 26.6; MS (EI) calcd for C10H13Cl [M+] 168.1, found 168.2.
4-(1-Chloro-2-methylpropan-2-yl)benzonitrile (38)
1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 3.65 (s, 2H), 1.45 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 151.5, 132.2, 127.0, 118.9, 110.5, 55.4, 40.5, 26.6.; MS (EI) calcd for C11H12ClN [M+] found 193.1.
Supplementary Material
Acknowledgments
We thank NIH/NIGMS (R01-GM079554), the Welch Foundation (I-1868), and UT Southwestern for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Hernandes MZ, Cavalcanti SMT, Moreira DRM, de Azevedo WF, Jr, Leite ACL. Curr Drug Targets. 2010;11:303–314. doi: 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y, Liu Y, Xu Z, Li H, Liu H, Zhu W. Expert Opin Drug Discov. 2012;7:375–383. doi: 10.1517/17460441.2012.678829. [DOI] [PubMed] [Google Scholar]
- 3.Gerebtzoff G, Li-Blatter X, Fischer H, Frentzel A, Seelig A. ChemBioChem. 2004;5:676–684. doi: 10.1002/cbic.200400017. [DOI] [PubMed] [Google Scholar]
- 4.Gribble GW. Mar Drugs. 2015;13:4044–4136. doi: 10.3390/md13074044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung Wj, Vanderwal CD. Angew Chem Int Ed. 2016;55:4396–4434. doi: 10.1002/anie.201506388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia JB, Zhu C, Chen C. J Am Chem Soc. 2013;135:17494–17500. doi: 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia JB, Zhu C, Chen C. Chem Commun. 2014;50:11701–11704. doi: 10.1039/c4cc05650g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia JB, Ma Y, Chen C. Org Chem Fornt. 2014;1:468–472. doi: 10.1039/C4QO00057A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hull KL, Anani WQ, Sanford MS. J Am Chem Soc. 2006;128:7134–7135. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Mei TS, Yu JQ. J Am Chem Soc. 2009;131:7520–7521. doi: 10.1021/ja901352k. [DOI] [PubMed] [Google Scholar]
- 11.Liu W, Huang X, Cheng MJ, Nielsen RJ, Goddard WA, III, Groves JT. Science. 2012;337:1322–1325. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- 12.Bloom S, Pitts CR, Miller DC, Haselton N, Holl MG, Urheim E, Lectka T. Angew Chem Int Ed. 2012;51:10580–10583. doi: 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]
- 13.Amaoka Y, Nagatomo M, Inoue M. Org Lett. 2013;15:2160–2163. doi: 10.1021/ol4006757. [DOI] [PubMed] [Google Scholar]
- 14.Braun MG, Doyle AG. J Am Chem Soc. 2013;135:12990–12993. doi: 10.1021/ja407223g. [DOI] [PubMed] [Google Scholar]
- 15.Fier PS, Hartwig JF. Science. 2013;342:956–960. doi: 10.1126/science.1243759. [DOI] [PubMed] [Google Scholar]
- 16.Xu P, Guo S, Wang L, Tang P. Angew Chem Int Ed. 2014 doi: 10.1002/anie.201400225. Early View. [DOI] [Google Scholar]
- 17.Halperin SD, Fan H, Chang S, Martin RE, Britton R. Angew Chem Int Ed. 2014;53:4690–4693. doi: 10.1002/anie.201400420. [DOI] [PubMed] [Google Scholar]
- 18.Kee CW, Chin KF, Wong MW, Tan CH. Chem Commun. 2014;50:8211–8214. doi: 10.1039/c4cc01848f. [DOI] [PubMed] [Google Scholar]
- 19.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 20.Neumann CS, Fujimori DG, Walsh CT. Chem Biol. 2008;15:99–109. doi: 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 21.Butler A, Sandy M. Nature. 2009;460:848–854. doi: 10.1038/nature08303. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal V, Miles ZD, Winter JM, Eustáquio AS, El Gamal AA, Moore BS. Chem Rev. 2017 doi: 10.1021/acs.chemrev.6b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang Z, Flatt P, Gerwick WH, Nguyen VA, Willis CL, Sherman DH. Gene. 2002;296:235–247. doi: 10.1016/s0378-1119(02)00860-0. [DOI] [PubMed] [Google Scholar]
- 24.Galonić DP, Vaillancourt FH, Walsh CT. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 25.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 26.Krebs C, Fujimori DG, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruijnincx PCA, van Koten G, Gebbink RJMK. Chem Soc Rev. 2008;37:2716–2744. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]
- 28.Leising RA, Zang Y, Que L., Jr J Am Chem Soc. 1991;113:8555–8557. [Google Scholar]
- 29.Kim J, Harrison RG, Kim C, Que L., Jr J Am Chem Soc. 1996;118:4373–4379. [Google Scholar]
- 30.MacFaul PA, Ingold KU, Wayner DDM, Que L., Jr J Am Chem Soc. 1997;119:10594–10598. [Google Scholar]
- 31.Liu W, Groves JT. J Am Chem Soc. 2010;132:12847–12849. doi: 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]
- 32.Lv XL, Wang K, Wang B, Su J, Zou X, Xie Y, Li JR, Zhou HC. J Am Chem Soc. 2017;139:211–217. doi: 10.1021/jacs.6b09463. [DOI] [PubMed] [Google Scholar]
- 33.Cong Z, Kurahashi T, Fujii H. J Am Chem Soc. 2012;134:4469–4472. doi: 10.1021/ja209985v. [DOI] [PubMed] [Google Scholar]
- 34.Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]
- 35.Kalyani D, Dick AR, Anani WQ, Sanford MS. Tetrahedron. 2006;62:11483–11498. [Google Scholar]
- 36.Giri R, Chen X, Yu JQ. Angew Chem Int Ed. 2005;44:2112–2115. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]
- 37.Wasa M, Yu JQ. J Am Chem Soc. 2008;130:14058–14059. doi: 10.1021/ja807129e. [DOI] [PubMed] [Google Scholar]
- 38.Liu T, Myers MC, Yu JQ. Angew Chem Int Ed. 2017;56:306–309. doi: 10.1002/anie.201608210. [DOI] [PubMed] [Google Scholar]
- 39.Rit RK, Yadav MR, Ghosh K, Shankar M, Sahoo AK. Org Lett. 2014;16:5258–5261. doi: 10.1021/ol502337b. [DOI] [PubMed] [Google Scholar]
- 40.Li B, Wang SQ, Liu B, Shi BF. Org Lett. 2015;17:1200–1203. doi: 10.1021/acs.orglett.5b00151. [DOI] [PubMed] [Google Scholar]
- 41.Xiong HY, Cahard D, Pannecoucke X, Besset T. Eur J Org Chem. 2016:3625–3630. [Google Scholar]
- 42.Rodriguez RA, Pan CM, Yabe Y, Kawamata Y, Eastgate MD, Baran PS. J Am Chem Soc. 2014;136:6908–6911. doi: 10.1021/ja5031744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen K, Richter JM, Baran PS. J Am Chem Soc. 2008;130:7247–7249. doi: 10.1021/ja802491q. [DOI] [PubMed] [Google Scholar]
- 44.Kundu R, Ball ZT. Org Lett. 2010;12:2460–2463. doi: 10.1021/ol100472t. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt VA, Quinn RK, Brusoe AT, Alexanian EJ. J Am Chem Soc. 2014;136:14389–14392. doi: 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]
- 46.Quinn RK, Könst ZA, Michalak SE, Schmidt Y, Szklarski AR, Flores AR, Nam S, Horne DA, Vanderwal CD, Alexanian EJ. J Am Chem Soc. 2016;138:696–702. doi: 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Zhu L, Yin F, Su Z, Li Z, Li C. J Am Chem Soc. 2012;134:4258–4263. doi: 10.1021/ja210361z. [DOI] [PubMed] [Google Scholar]
- 48.Walling C, Gibian MJ. J Am Chem Soc. 1965;87:3361–3364. [Google Scholar]
- 49.Rueda-Becerril M, Sazepin CC, Leung JCT, Okbinoglu T, Kennepohl P, Paquin JF, Sammis GM. J Am Chem Soc. 2012;134:4026–4029. doi: 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]
- 50.Further introduction of chlorine atoms decreases the bond dissociation energy of the adjacent C–H, whereas further introduction of fluorine atoms increases the bond dissociation energy of the adjacent C–H. The C–H bond dissociation energy for chlorinated methanes is 100.1, 96.2, and 93.8 kcal/mol for H–CH2Cl, H–CHCl2, H–CCl3, respectively, and the C–H bond dissociation energy for fluorinated methanes is 101.3, 103.2, 107.4 kcal/mol for H–CH2F, H–CHF2, H–CF3, respectively (CRC Handbook of Chemistry and Physics).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.