

Abstract
Metformin improves obesity-associated metabolic dysregulation, but has controversial effects on adipose tissue inflammation.
Objective
To examine the direct effect of metformin on adipocyte inflammatory responses, and elucidate the underlying mechanisms.
Methods
Adipocytes were differentiated from 3T3-L1 cells and treated with metformin at various doses and for different time periods. The treated cells were examined for the proinflammatory responses, as well as the phosphorylation states of AMPK and the expression of PFKFB3/iPFK2. In addition, PFKFB3/iPFK2-knockdown adipocytes were treated with metformin and examined for changes in the proinflammatory responses.
Results
Treatment of adipocytes with metformin decreased the effects of lipopolysaccharide on inducing the phosphorylation states of JNK p46 and on increasing the mRNA levels of IL-1β and TNFα. In addition, treatment with metformin increased the expression of PFKFB3/iPFK2, but failed to significantly alter the phosphorylation states of AMPK. In PFKFB3/iPFK2-knockdown adipocytes, treatment with metformin did not suppress the proinflammatory responses as did it in control adipocytes.
Conclusion
Metformin has a direct effect on suppressing adipocyte proinflammatory responses in an AMPK-independent manner. Also, metformin increases adipocyte expression of PFKFB3/iPFK2, which is involved in the anti-inflammatory effect of metformin.
Keywords: Inducible 6-phosphofructo-2-kinase, metformin, adipocyte, inflammatory responses, obesity
Introduction
Chronic low-grade adipose tissue inflammation is a characteristic of obesity (Lumeng et al. 2007, Weisberg et al. 2003, Xu et al. 2003) and critically contributes to the pathogenesis of type 2 diabetes (Greenberg and Obin 2006, Shoelson et al. 2007). As shown in rodent models of obesity, adipose tissue-specific overexpression of monocyte chemoattractant protein-1 causes adipose tissue macrophage accumulation, which is accompanied with increased severity of systemic insulin resistance and metabolic dysregulation (Kamei et al. 2006). Similarly, adipocyte-derived Th2 cytokines critically determine the status of adipose tissue inflammation by promoting macrophage alternative activation, which in turn contributes to improvement of systemic insulin sensitivity (Kang et al. 2008). These results, together with many other results from both human and rodent studies (Boyle et al. 2011, Huo et al. 2010, Huo et al. 2012, Kamei et al. 2006, Ye et al. 2007), validate the importance of adipose tissue inflammation in the development of insulin resistance and glucose and fat metabolic dysregulation. Specifically, inflammatory adipose tissue produces a number of pro-hyperglycemic factors that impair insulin signaling in insulin-sensitive tissues including the liver and skeletal muscle, leading to systemic insulin resistance (Berg et al. 2001, Cheung et al. 2000, Hotamisligil et al. 1996, Kabir et al. 2005, Trujillo and Scherer 2006). The latter results in increased hepatic glucose production and decreased muscle glucose disposal (Kim et al. 2004, Steinberg et al. 2006, Wu et al. 2005b), which are two essential metabolic events responsible for the development of hyperglycemia. Because of this, targeting adipose tissue inflammation to correct obesity-associated insulin resistance is of particular importance in the treatment of type 2 diabetes.
Since it effectively corrects hyperglycemia, metformin has been used as the first-line medicine for the treatment of type 2 diabetes for over several decades (Zheng et al. 2015). Mechanistically, metformin acts mainly through suppressing hepatic glucose production to lower plasma levels of glucose. In addition, metformin has been shown to improve fat metabolism in both the liver and muscle, which also contributes to the glucose-lowering effect of metformin (Heishi et al. 2006, Perriello et al. 1994, Song et al. 2001, Stumvoll et al. 1995, Woo et al. 2014). Following the validation of adipose tissue inflammation as a causal factor of the development of insulin resistance and hyperglycemia, a number of studies also have explored the effects of metformin on adipose tissue inflammation and metabolic functions. Unlike its effect on the liver, the effect of metformin on adipose tissue remains controversial. Specifically, metformin is shown to decrease adipose tissue inflammation in both human subjects and rodent models of obesity and type 2 diabetes (Shin et al. 2013, Zhao et al. 2016, Zulian et al. 2011), evidenced by decreased proinflammatory signaling and cytokine expression. In contrast, there are studies in which metformin does not alter body weight and/or adipose tissue inflammation, in particular in rodents fed an high-fat diet (HFD) (Anthony et al. 2013, Shin et al. 2013, Song et al. 2001, Woo et al. 2014). Given this discrepancy, there is a need to gain mechanistic insights of whether metformin has direct effects on adipocyte inflammatory and metabolic responses.
PFKFB3 is the gene that encodes for the inducible 6-phosphofructo-2-kinase (iPFK2). In adipose tissue, the expression of PFKFB3/iPFK2 is at high abundance (Huo et al. 2010). As a regulatory enzyme, iPFK2 generates fructose-2,6-bisphosphate, which in turn activates 6-phosphofructo-1-kinase to enhance glycolysis (Okar et al. 2004, Rider et al. 2004). PFKFB3/iPFK2 has been previously shown to critically regulate adipose tissue functions and systemic insulin sensitivity (Huo et al. 2010). For example, PFKFB3/iPFK2 disruption exacerbates diet-induced adipose tissue inflammation and systemic insulin resistance whereas adipose tissue-specific PFKFB3/iPFK2 overexpression decreases adipose tissue inflammation and improves systemic insulin sensitivity (Huo et al. 2010, Huo et al. 2012). Limited research has suggested that the expression of PFKFB3/iPFK2 is increased in DB-1 melanoma cells upon metformin treatment, likely through a mechanism involving AMP-activated protein kinase (AMPK) (Mendoza et al. 2012). However, it is not clear whether metformin alters adipocyte expression of PFKFB3/iPFK2 in the context of adipose tissue inflammatory responses. The present study provides the evidence for the first time to support that metformin stimulates adipocyte expression of PFKFB3/iPFK2. The latter, in turn, is involved in the effect of metformin on suppressing adipocyte inflammatory responses.
Materials and methods
Adipocyte differentiation and treatment
3T3-L1 cells were cultured in high glucose DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin as previously described (Huo et al. 2010). To differentiate 3T3-L1 cells, the 2 d post-confluent cells were incubated in DMEM supplemented with 0.5 mM 3-isobutyl-1-methyl-xanthine, 1 μM dexamethasone, and 10 μg/mL insulin for 48 h. Thereafter, the cells were incubated in DMEM supplemented with 10 μg/mL insulin for an additional 8 d. At 10 days post differentiation, adipocytes were used for a dose-response study or a time-course study to examine the anti-inflammatory effect of metformin. In the dose-response study, the differentiated adipocytes were treated with metformin at a dose of 5, 50, or 500 μM (dissolved in phosphate-buffered saline, PBS) or PBS for 24 h in the absence or presence of lipopolysaccharide (LPS, 100 ng/ml) for the last 30 min to harvest protein lysates or LPS (20 ng/mL) for the last 6 h to harvest total RNA samples. In the time-course study, the differentiated adipocytes were treated with metformin (50 μM) for 0, 1, 6, 24, or 48 h in the presence of LPS (100 ng/mL) for the last 30 min to harvest protein lysates or LPS (20 ng/mL) for the last 6 h to harvest total RNA samples. To analyze the proinflammatory signaling, adipocyte lysates were examined for total amount and phosphorylation states of Jun N-terminal kinase (JNK) p46 and nuclear factor kappa B (NF-κB) p65. To analyze the expression of PFKFB3/iPFK2, adipocyte RNA was subjected to reverse transcription and real-time PCR to quantify PFKFB3 mRNA levels whereas adipocyte lysates were subjected to Western blot analysis to examine iPFK2 amount. In addition, adipocyte RNA was used to examine the mRNA levels of proinflammatory cytokines including IL-1β, IL-6, and TNFα. Details were described in the pertinent assays.
Adipocyte PFKFB3/iPFK2 knockdown and metformin treatment
Stable PFKFB3/iPFK2 knockdown (iPFK2-KD) and control (iPFK2-Ctrl) 3T3-L1 cells were previously established (Huo et al. 2010). To examine the involvement of PFKFB3/iPFK2 in metformin actions, iPFK2-KD cells and iPFK2-Ctrl cells were differentiated in the same way as described above. After differentiation, the cells were treated with metformin (50 μM) or PBS for 24 h in the absence or presence of LPS (100 ng/mL) for the last 30 min to harvest protein lysates or LPS (20 ng/mL) for the last 6 h to harvest total RNA samples. Some differentiated iPFK2-KD cells and iPFK2-Ctrl cells were treated with metformin (50 μM) or PBS for 24 h in the absence or presence of insulin (100 nM) for the last 30 min. Protein lysates were harvested and used to examine insulin signaling using Western blot analysis.
Western blot analysis
Adipocyte lysates were prepared in a lysis buffer containing 50 mM HEPES (pH 7.4), 10 mM EDTA, 50 mM sodium pyrophosphate, 0.1 M sodium fluoride, 10 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 2 mM benzamidine, and 1% Triton X-100. After protein electrophoresis and transfer, immunoblots were performed using rabbit anti-serum as primary antibody at a 1:1,000 dilution. This dilution was used for each of the primary antibodies used for the present study. After washing, the blot was incubated with a 1:10,000 dilution of goat anti-rabbit horseradish peroxidase-conjugated secondary antibody and followed by a chemiluminescent kit (Immobilon Western; EMD Millipore, Billerica, MA, USA) as previously described (Wu et al. 2005a). GAPDH was used as a loading control. The maximum intensity of each band was quantified using ImageJ software. Ratios of P-p46/p46, Pp65/65, P-AMPK/AMPK, or P-Akt/Akt were normalized to GAPDH and adjusted relative to the average of PBS-treated control, which was arbitrarily set as 1 (AU). Also, the amount of iPFK2 was normalized to GAPDH and adjusted similarly. Antibodies against Pp46, p46, Pp65, p65, P-AMPK, and/or AMPK were products of Cell Signaling (Danvers, MA, USA). Antibodies against P-Akt and Akt and anti-rabbit IgG antisera were products of Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
RNA isolation, reverse transcription, and real-time PCR
The total RNA was isolated from the adipocytes described above. Reverse transcription was performed using the GoScript Reverse Transcription System (Promega) and real-time PCR analysis was performed using SYBR Green (LightCycler 480 system; Roche) (Guo et al. 2013, Guo et al. 2012, Wu et al. 2006). The mRNA levels were analyzed for IL-1β, IL-6, TNFα, and/or PFKFB3. A total of 0.1 μg RNA was used for the determination. Results were normalized to 18s ribosomal RNA as plotted as relative expression to the average of PBS-treated control, which was set as 1.
Statistical methods
Numeric data are presented as means ± se (standard error). Two-tailed ANOVA and/or Student’s t tests were used for statistical analyses. Differences were considered significant at the P < 0.05.
Results
Metformin suppresses adipocyte proinflammatory responses
The effect of metformin on obesity-associated adipose tissue inflammation remains controversial. We sought to examine the direct effect of metformin on adipocyte inflammatory responses. In differentiated 3T3-L1 adipocytes, treatment with LPS, a powerful proinflammatory stimulus, caused significant increases in the phosphorylation states of JNK p46 and NF-κB p65 and in the mRNA levels of IL-1β and TNFα (Fig. 1A–C). Upon treatment with metformin, the effect of LPS on inducing adipocyte proinflammatory responses was significantly weakened. Specifically, treatment with metformin at a dose of 50 or 500 μM brought about a significant decrease in LPS-induced JNK p46 phosphorylation compared with metformin treatment at a dose of 5 μM (Fig. 1A). At a fixed dose of 50 μM, treatment with metformin for 24 or 48 h caused a marked decrease in the phosphorylation states of JNK p46 compared with control treatment or metformin treatment for 6 h (Fig. 1B). Unlike its effects on JNK p46, metformin treatment did not significantly alter adipocyte phosphorylation states of NF-κB p65 in either dose-response or time-course study. When cytokine expression was examined, treatment with metformin at a dose of 50 μM for 24 h significantly blunted the effect of LPS on increasing adipocyte mRNA levels of IL-1β and TNFα (Fig. 1C). Of note, treatment of adipocytes with metformin at a relatively low dose, e.g., 5 μM, or for a relatively short time, e.g., 6 h, appeared to not suppress or even increase LPS-induced JNK p46 phosphorylation states (Fig. 1A, and B). These effects, however, occurred prior to an increase in iPFK2 amount and/or in the presence of a decrease in iPFK2 amount (below, Fig. 3). Considering that decreased iPFK2 results in an increase in LPS-induced JNK p46 phosphorylation (Huo et al. 2010), it is likely that prior to full induction of iPFK2 metformin acts through certain mechanisms to increase LPS-induced JNK p46 phosphorylation. Once iPFK2 is increased, the anti-inflammatory effect of iPFK2 starts to dominate over the proinflammatory effect, thereby decreasing LPS-induced JNK p46 phosphorylation. Regardless, these results suggest that metformin has a direct effect on suppressing adipocyte proinflammatory responses.
Figure 1. Metformin suppresses adipocyte proinflammatory responses.
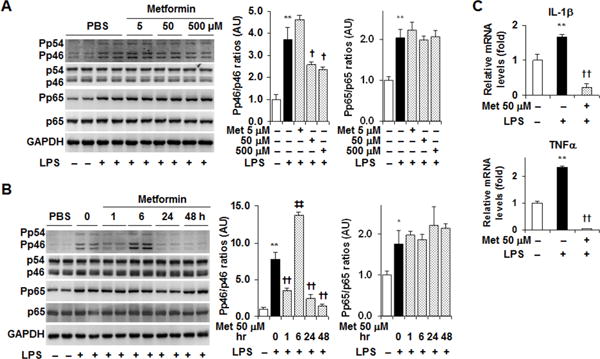
After differentiation, 3T3-L1 adipocytes were used to examine the effects of metformin on the proinflammatory responses. (A) Dose-response study. Adipocytes were treated with metformin (Met) at a dose of 5, 50, or 500 μM (dissolved in phosphate-buffered saline, PBS) or PBS for 24 hr. (B) Time-course study. Adipocytes were treated with metformin (50 μM) for 0, 1, 6, 24, or 48 hr. (C) Adipocyte cytokine expression. Adipocytes were treated with metformin (50 μM) for 24hr. For A – C, prior to harvest, the cells were also treated with lipopolysaccharide (LPS, 100 ng/ml) for 30 min to analyze inflammatory signaling or LPS (20 ng/ml) for 6 hr to analyze cytokine expression. For A and B, adipocyte lysates were subjected to Western blot analysis. Blots were quantified using densitometry. AU, arbitrary unit. For C, cytokine expression was examined using real-time PCR. For bar graphs (A – C), data are means ± S.E. n = 4. *, P < 0.05 and **, P < 0.01 LPS in the presence or absence of Met vs. PBS (empty bar); †, P < 0.05 and ††, P < 0.01 Met at 50 or 500 μM in the presence of LPS vs. LPS alone; ‡‡, P < 0.01 Met treatment for 6 hr vs. LPS alone (in B, left bar graph).
Figure 3. Metformin increases adipocyte expression of PFKFB3/iPFK2.
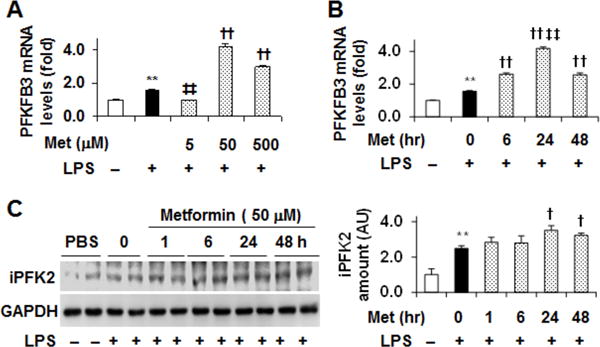
Adipocytes were differentiated and treated as described in Fig. 1. (A) PFKFB3 expression in the dose-response study. (B) PFKFB3 expression in the time-course study. For A and B, the mRNA levels of PFKFB3 were quantified using real-time PCR. (C) The amount of iPFK2 in time-course study was examined using Western blot analysis. Blots were quantified using densitometry. AU, arbitrary unit. Left panel, representative blots; right panel, quantification of blots. For bar graphs (A – C), data are means ± S.E. n = 4. **, P < 0.01 LPS alone vs. PBS (empty bar); ‡‡, P < 0.01 Met at 5 μM in the presence of LPS vs. LPS alone (in A) or Met for 24 hr vs. Met for any time period other than 24 hr (in B); †, P < 0.05 and ††, P < 0.01 Met at 50 or 500 μM in the presence of LPS vs. LPS alone (in A – C).
Metformin has limited effect on altering adipocyte AMPK phosphorylation
Metformin is shown to increase AMPK phosphorylation (activation) in various tissues/cells including the liver and muscle (Collier et al. 2006, Woo et al. 2014). However, the effect of metformin on adipose tissue AMPK phosphorylation remains controversial. Using adipocyte samples of the dose-response and time-course studies, we examined the direct effect of metformin on adipocyte AMPK phosphorylation. Consistent with our previous finding in adipose tissue of obese mice (Woo et al. 2014), treatment with metformin displayed limited effects on altering adipocyte AMPK phosphorylation (Fig. 2A, and B). This effect of metformin appeared to be different from that reported by Huypens et al. (Huypens et al. 2005). However, in the study by Huypens et al., adipocytes were treated with metformin at a high dose (1 mM) for 48 h. Based on the results of the present study, it appears that metformin, within a relatively low dose range, is not capable of increasing adipocyte AMPK phosphorylation.
Figure 2. Metformin does not significantly alter adipocyte AMPK phosphorylation.
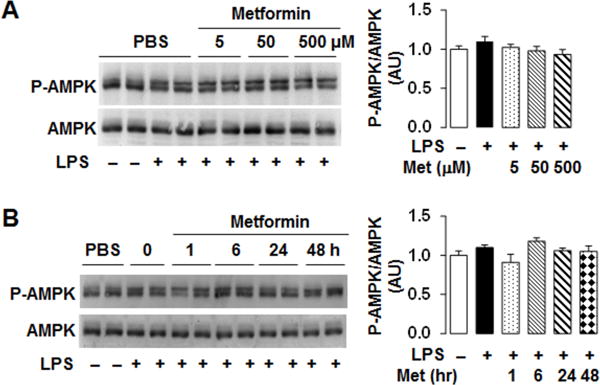
Adipocytes were differentiated and treated as described in Fig. 1. (A) AMPK phosphorylation in the dose-response study. (B) AMPK phosphorylation in the time-course study. For A and B, the amount and phosphorylation states of AMPK were examined using Western blot analysis. Blots were quantified using densitometry. AU, arbitrary unit.
Metformin stimulates adipocyte PFKFB3/iPFK2 expression
In adipocytes, PFKFB3/iPFK2 has a role in protecting against overnutrition-induced proinflammatory responses (Huo et al. 2010, Huo et al. 2012). Therefore, we examined the effect of metformin on adipocyte PFKFB3/iPFK2 expression. In the dose-response study detailed above, PFKFB3 expression was increased by LPS stimulation, indicating a defensive response. Upon treatment with metformin at a dose of 5 μM for 24 h, the effect of LPS on increasing PFKFB3 expression was blunted. However, treatment with metformin at a higher dose, i.e., 50 or 500 μM, for 24 h caused a significant increase in the mRNA levels of PFKFB3 compared with control in the presence of LPS (Fig. 3A). Moreover, treatment with metformin at a dose of 50 μM appeared to cause the strongest induction of PFKFB3 mRNA levels. In the time-course study, treatment with metformin at a dose of 50 μM for 6 h, 24 h, or 48 h significantly increased the mRNA levels of PFKFB3. Within a period of time between 0 and 48 hr, treatment with metformin for 24 h displayed the strongest effect on stimulating PFKFB3 mRNA levels (Fig. 3B). Consistently, treatment with metformin at a dose of 50 μM for 24 h or 48 h significantly increased protein amount of iPFK2 compared with control treatment in the presence of LPS (Fig. 3C); although iPFK2 amount in adipocytes upon metformin treatment for 24 h is not statistically different from that upon metformin treatment for 6 or 48 h (Fig. 3C). Taken together, these results suggest that metformin has a direct effect on stimulating adipocyte expression of PFKFB3/iPFK2.
PFKFB3/iPFK2 knockdown blunts the effect of metformin on suppressing adipocyte proinflammatory responses
To elucidate a role for PFKFB3/iPFK2 in regulating metformin actions, we examined the effect of metformin on the proinflammatory responses in iPFK2-KD adipocytes. Consistent with the findings in differentiated 3T3-L1 adipocytes (Fig. 3), treatment with metformin increased iPFK2 amount in control cells (iPFK2-Ctrl) (Fig. 4A). However, this effect of metformin was nearly blunted in iPFK2-KD cells. Also, treatment with metformin did not significantly alter the phosphorylation states of AMPK in either iPFK2-KD or iPFK2-Ctrl cells. Although the phosphorylation states of AMPK in iPFK2-KD cells appeared to be increased in relative to those in iPFK2-Ctrl cells, the ratios of P-AMPK to total AMPK (normalized to GAPHD in iPFK2-KD cells were comparable with those in iPFK2-Ctrl cells in either the presence or absence of metformin treatment. Next, we examined the anti-inflammatory effect of metformin. In iPFK2-Ctrl cells, treatment with metformin caused a significant decrease in the phosphorylation states of JNK p46; although not significantly altering the phosphorylation states of NF-κB p65. However, in iPFK2-KD cells, the phosphorylation states of JNK p46 were significantly higher than those in iPFK2-Ctrl cells, and remained high upon treatment with metformin (Fig. 4B). When proinflammatory cytokine expression was analyzed, the mRNA levels of IL-1β, IL-6, and/or TNFα in iPFK2-Ctrl cells were significantly reduced by treatment with metformin under basal conditions (in the absence of LPS) compared with control (in the absence of metformin). In contrast, the mRNA levels of IL-1β, IL-6, and/or TNFα in iPFK2-KD cells were increased or remained high upon treatment with metformin under either basal or LPS-stimulated conditions (Fig. 4C). Indeed, the mRNA levels of IL-1β, IL-6, and/or TNFα were much more induced in iPFK2-KD cells upon LPS stimulation in the presence or absence of metformin compared with their respective levels in iPFK2-Ctrl cells (Fig. 4C). Taken together, these results suggest that intact PFKFB3/iPFK2 is required for metformin to fully suppress adipocyte proinflammatory responses.
Figure 4. PFKFB3/iPFK2 knockdown impair the effect of metformin on suppressing adipocyte proinflammatory responses.
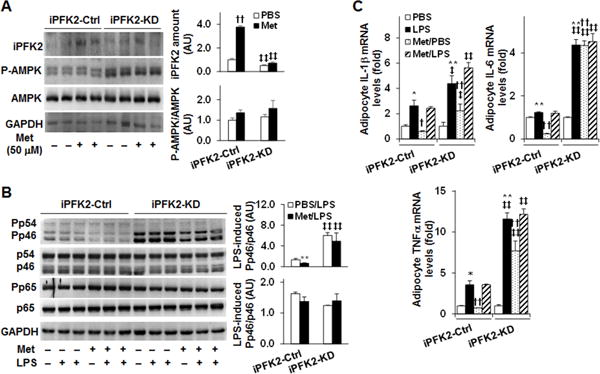
PFKFB3/iPFK2 knockdown (iPFK2-KD) adipocytes and control (iPFK2-Ctrl) adipocytes were generated as described in Methods. (A) Adipocyte iPFK2 amount and AMPK phosphorylation states. After differentiation, iPFK2-KD adipocytes and iPFK2-Ctrl adipocytes were treated with or without metformin (50 μM) for 24 hr. (B) Adipocyte proinflammatory signaling. Differentiated iPFK2-KD adipocytes and iPFK2-Ctrl adipocytes were treated with or without metformin (50 μM) for 24 hr in the absence or presence of LPS (100 ng/ml) for the last 30 min. For A and B, protein lysates were subjected to Western blot analysis. Blots were quantified using densitometry. AU, arbitrary unit. (C) Adipocyte expression of proinflammatory cytokines. Differentiated iPFK2-KD adipocytes and iPFK2-Ctrl adipocytes were treated with or without metformin (50 μM) for 24 hr in the absence or presence of LPS (20 ng/ml) for the last 6 hr. The mRNA levels of proinflammatory cytokines were quantified using real-time PCR. For C, data are means ± S.E. n = 4. For all bar graphs, *, P < 0.05 and **, P < 0.01 Met/LPS vs. PBS/LPS for iPFK2-Ctrl cells (in B) or LPS alone vs. PBS for the same type of cells (iPFK2-KD or iPFK2-Ctrl cells in C); ‡, P < 0.05 and ‡‡, P < 0.01 iPFK2-KD vs. iPFK2-Ctrl under the same condition (PBS or Met in A; PBS/LPS or Met/LPS in B; PBS, Met, PBS/LPS, or Met/LPS in C); †, P < 0.05 and ††, P < 0.01 Met vs. PBS for iPFK2-Ctrl cells (in A) or Met/PBS vs. PBS for the same type of cells (iPFK2-KD or iPFK2-Ctrl cells in C).
PFKFB3/iPFK2 knockdown blunts the effect of metformin on improving adipocyte insulin signaling
Adipocyte insulin signaling is reversely correlated with adipocyte proinflammatory status. We examined the involvement of PFKFB3/iPFK2 in metformin actions on improving adipocyte insulin signaling at the level of Akt phosphorylation. In iPFK2-Ctrl cells, treatment with metformin enhanced the effect of insulin on increasing the phosphorylation states of Akt compared with control treatment (Fig. 5A and B). However, this effect of metformin was blunted in iPFK2-KD cells (Fig. 5A and B). Therefore, intact PFKFB3/iPFK2 is needed for metformin to improve adipocyte insulin signaling.
Figure 5. PFKFB3/iPFK2 knockdown blunts the effect of metformin on improving adipocyte insulin signaling.
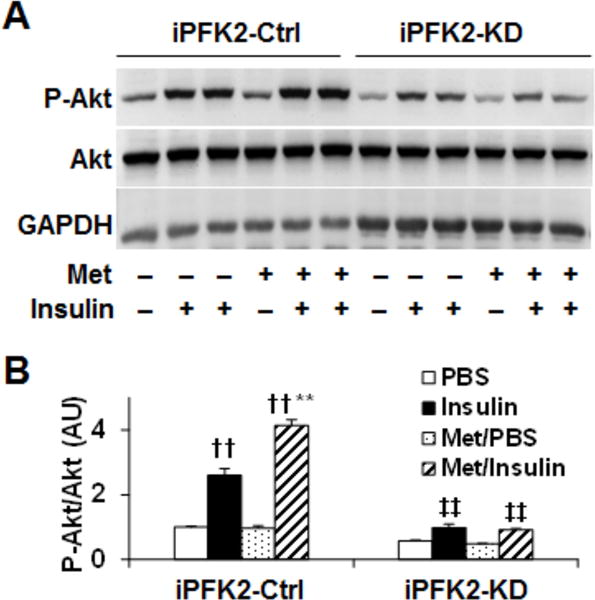
PFKFB3/iPFK2 knockdown (iPFK2-KD) adipocytes and control (iPFK2-Ctrl) adipocytes were generated as described in Methods. (A, B), After differentiation, iPFK2-KD adipocytes and iPFK2-Ctrl adipocytes were treated with or without metformin (50 μM) for 24 hr in the absence or presence of insulin (100 ng/ml) for the last 30 min. Adipocyte lysates were examined for the amount and phosphorylation states of Akt using Western blot analysis. Blots were quantified using densitometry. AU, arbitrary unit. A, representative bolts; B, quantification of blots. For B, data are means ± S.E. n = 4. **, P < 0.01 Met/Insulin vs. Insulin (iPFK2-Ctrl cells); ‡‡, P < 0.01 iPFK2-KD vs. iPFK2-Ctrl under the same condition (Insulin or Met/Insulin); ††, P < 0.01 Insulin vs. PBS or Met/Insulin vs. Met/PBS for iPFK2-Ctrl cells.
Discussion
The anti-diabetic effect of metformin has been well established. However, it remains controversial whether metformin suppresses adipose tissue inflammation, thereby contributing to the anti-diabetic effect of metformin (Shin et al. 2013, Woo et al. 2014, Zhao et al. 2016, Zulian et al. 2011). Because the discrepancy of metformin actions on adipose tissue inflammation appears to be associated with various factors including the strains of rodents used and the doses and paths of metformin delivery, the present study sought to determine the direct effect of metformin on adipocytes. Using in vitro systems involving differentiated 3T3-L1 adipocytes, the present study validated a direct effect of metformin on suppressing adipocyte proinflammatory responses. Furthermore, the present study revealed a stimulatory effect of metformin on adipocyte expression of PFKFB3/iPFK2, whose knockdown in adipocytes blunted the anti-inflammatory effect of metformin. Therefore, the present study provided the evidence to support metformin stimulation of adipocyte PFKFB3/iPFK2 as a novel mechanism by which metformin may suppress adipose tissue inflammation.
Metformin has a direct effect on suppressing adipocyte proinflammatory responses. In support of this, treatment with metformin at a dose of 50 or 500 μM caused a significant decrease in LPS-stimulated adipocyte phosphorylation states of JNK p46 although metformin treatment did not significantly alter LPS-stimulated phosphorylation states of NF-κB p65. In addition, metformin treatment at a fixed dose of 50 μM for 24 or 48 h caused significant decreases in LPS-stimulated adipocyte phosphorylation states of JNK p46. Consistently, treatment with metformin brought about a significant decrease in the mRNA levels of IL-1β and TNFα. Of note, metformin appeared to be more effective in adipocytes differentiated from un-transfected 3T3-L1 cells than in adipocytes differentiated from transfected and G418-selected 3T3-L1 cells while exhibiting anti-inflammatory effects. Specifically, metformin treatment caused a significant decrease in the mRNA levels of IL-1β and TNFα in un-transfected cells (Fig. 1C), but not in iPFK2-Ctrl cells (Fig. 4C) under LPS-stimulated conditions. This difference in metformin actions may be due to how adipocytes were prepared. Indeed, adipocytes differentiated from transfected cells exhibited a much stronger response to LPS than did adipocytes differentiated from un-transfected cells. Considering this, it is likely that there is a threshold of adipocyte proinflammatory status for metformin to exhibit anti-inflammatory effects. In other words, metformin may not be effective when LPS-induced proinflammatory responses are too strong. This appeared to be the case. In the present study, under basal conditions where the proinflammatory status was low or mild, metformin treatment significantly decreased the mRNA levels of IL-1β, IL-6, and TNFα, as well as the phosphorylation states of JNK p46. In contrast, in iPFK2-KD cells where the basal proinflammatory status was strong, metformin failed to decrease proinflammatory signaling and cytokine expression. The threshold view, however, needs to be further investigated. Nonetheless, the results argue in favor of a role for iPFK2 in mediating the anti-inflammatory effect of metformin (see below).
Metformin suppression of adipocyte proinflammatory responses does not involve AMPK. Previous results from both human and rodent studies indicate that treatment with metformin stimulates the phosphorylation of states of adipose tissue AMPK (Boyle et al. 2011, Lu et al. 2016). In contrast, there are studies in which treatment with metformin does not significantly alter the phosphorylation states of adipose tissue AMPK (Woo et al. 2014). Similarly, the direct effects of metformin on adipocyte AMPK phosphorylation vary among studies. In the present study, treatment with metformin did not alter adipocyte AMPK phosphorylation as this was supported by the results from both does-response and time course studies. These results were consistent with the findings of a previous study by Boyle et al. (Boyle et al. 2011), in which treatment with metformin at a dose of 1 mM for a time period of 0.5 to 24 h did not significantly alter adipocyte AMPK phosphorylation although treatment with metformin for 48 h increased adipocyte AMPK phosphorylation states. Of importance, in the present study, treatment with metformin significantly decreased the phosphorylation states of adipocyte JNK p46 and the mRNA levels of proinflammatory cytokines while exhibiting limited effect on increasing adipocyte AMPK phosphorylation states. Therefore, metformin appears to suppress adipocyte proinflammatory responses in an AMPK-independent manner. This is different from the effect of metformin on hepatocytes, in which treatment with metformin increases the phosphorylation states of AMPK and suppresses the proinflammatory responses (Woo et al. 2014). The mechanisms underlying the differential effects of metformin on AMPK phosphorylation states in hepatocytes versus in adipocytes are not clear, but may be due to the differences in the isoforms of the catalytic subunit of AMPK. In adipocytes, the α1 catalytic subunit is the predominant isoform and accounts for the major part of AMPK activity (Daval et al. 2005) whereas in hepatocytes α2-subunit is the main functional isoform of AMPK (Andreelli et al. 2006, Jelenik et al. 2010). As shown in muscles, treatment with metformin increases the activities of AMPKα2, but not AMPKα1 (Collier et al. 2006). Considering this, lacking responses in AMPKα1 to metformin likely explains the observation of the present study. Alternatively, a relatively low dose, e.g., 50 μM, is sufficient for metformin to exert anti-inflammatory effects whereas at a relatively high dose, e.g., 1 mM, is needed for metformin to increase AMPK phosphorylation as reported by Huypens et al (Huypens et al. 2005). In this case, metformin would still act to suppress adipocyte proinflammatory responses independent of AMPK phosphorylation.
It is a novel finding that treatment with metformin increased adipocyte expression of PFKFB3/iPFK2. Notably, treatment with metformin caused an increase in iPFK2 amount in both dose-response and time-course studies. In addition, treatment with metformin significantly increased the mRNA levels of PFKFB3 in the presence of LPS. Previously, AMPK activation upon lowering the pH values of the culture media is accompanied with increased PFKFB3/iPFK2 expression in glioblastoma cells (Mendoza et al. 2012). In the present study, metformin increased adipocyte PFKFB3/iPFK2 expression in the absence of stimulating AMPK phosphorylation. Considering this, metformin is likely capable of stimulating PFKFB3/iPFK2 expression in both AMPK-dependent and AMPK-independent manners. Regardless, increased amount of iPFK2 was correlated with the effect of metformin on decreasing adipocyte proinflammatory responses, suggesting the involvement of PFKFB3/iPFK2 in the anti-inflammatory effect of metformin. In adipocytes, PFKFB3/iPFK2 has been demonstrated as a metabolic regulator that links glucose and fatty acid metabolism and inflammatory responses. Specifically, PFKFB3/iPFK2, at an increase in its activity, is able to channel free fatty acids away from excessive oxidation to fat synthesis/storage, thereby reducing the generation of reactive oxygen species (Guo et al. 2010, Huo et al. 2012). Because of this, it is conceivable that metformin stimulation of PFKFB3/iPFK2 serves as a mechanism by which metformin suppresses adipocyte proinflammatory responses.
The involvement of PFKFB3/iPFK2 in the anti-inflammatory effect of metformin was further supported by the results from adipocytes upon PFKFB3/iPFK2 knockdown. In the present study, increased proinflammatory responses were confirmed in PFKFB3/iPFK2-knockdown adipocytes compared with those in control adipocytes. Of importance, metformin did not effectively suppress the proinflammatory responses in PFKFB3/iPFK2-knockdown adipocytes as did it in control adipocytes. As previously demonstrated by Guo et al., PFKFB3/iPFK2 knockdown increased adipocyte proinflammatory responses, which was due to, in large part, increased adipocyte oxidative stress (Guo et al. 2010). In contrast, PFKFB3/iPFK2 over-expression promoted adipocyte fat deposition, leading to decreased adipocyte proinflammatory responses (Huo et al. 2012). Based on this, it is very likely that intact PFKFB3/iPFK2 is needed for a metabolic environment in which metformin is able to effectively suppress the proinflammatory responses. As additional evidence, metformin treatment increased insulin singling, indicated by insulin-induced Akt phosphorylation, in control adipocytes, but not in PFKFB3/iPFK2-knockdown adipocytes.
In summary, the present study provided the evidence to support a direct effect of metformin on suppressing adipocyte proinflammatory responses. This effect of metformin appeared to be AMPK-independent. Moreover, the present study reported for the first time that metformin stimulated adipocyte expression of PFKFB3/iPFK2, which also was AMPK-independent. In PFKFB3/iPFK2-knockdown adipocytes, the proinflammatory responses were much exacerbated and were not suppressed upon metformin treatment compared with those in control adipocytes. Taken together, metformin appears to act through simulating PFKFB3/iPFK2 expression to suppress adipocyte proinflammatory responses.
Acknowledgments
This work was supported in part or in whole by grants from the National Institutes of Health (HL108922 and HL095556 to Y.H., and R01DK095828 and R01DK095862 to C.W.). Also, Y.H. is supported by National Natural Science Foundation of China (81400826), Guangdong Natural Science Foundation (2014A030312004), and Shenzhen Science and Technology Innovation Committee (JSGG20140717102922014, JCYJ20140903101709818, and the Shenzhen Peacock Program (KQCX2015032709315529). Y.C. is supported by the Science and Technology Plan projects of Guangdong province (2016A050502010); the 5010 Clinical Research Projects of Sun Yat-sen University (2015015); the Key Special Projects of Medical and Health Collaborative Innovation of Guangzhou city (201604020016); and the Special Scientific Research Project of Guangzhou city (2060404). C.W. is supported by the Hatch Program of the National Institutes of Food and Agriculture (NIFA).
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Authors’ contribution
C.W. Y.H. and Y.C. designed the research; T.Q., H.L., Y.P., S.L.W, X.G., and J.Z. conducted the research; T.Q., H.L., and C.W. analyzed the data and wrote the manuscript. Y.C., X.Q., and J.A. were involved in scientific discussion. C.W. and Y.H. had primary responsibility for the final content. All authors read and approved the final manuscript.
References
- Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, et al. Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology. 2006;147:2432–2441. doi: 10.1210/en.2005-0898. [DOI] [PubMed] [Google Scholar]
- Anthony J, Kelkar A, Wilankar C, Ranjith V, Bhumra SK, Mutt S, Deka N, Sivaramakrishnan H, Sharma S, Marita AR. Discovery of p1736, a novel antidiabetic compound that improves peripheral insulin sensitivity in mice models. PLoS ONE. 2013;8:e77946. doi: 10.1371/journal.pone.0077946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nature Medicine. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- Boyle JG, Logan PJ, Jones GC, Small M, Sattar N, Connell JMC, Cleland SJ, Salt IP. AMP-activated protein kinase is activated in adipose tissue of individuals with type 2 diabetes treated with metformin: a randomised glycaemia-controlled crossover study. Diabetologia. 2011;54:1799–1809. doi: 10.1007/s00125-011-2126-4. [DOI] [PubMed] [Google Scholar]
- Cheung AT, Wang J, Ree D, Kolls JK, Bryer-Ash M. Tumor necrosis factor-alpha induces hepatic insulin resistance in obese Zucker (fa/fa) rats via interaction of leukocyte antigen-related tyrosine phosphatase with focal adhesion kinase. Diabetes. 2000;49:810–819. doi: 10.2337/diabetes.49.5.810. [DOI] [PubMed] [Google Scholar]
- Collier CA, Bruce CR, Smith AC, Lopaschuk G, Dyck DJ. Metformin counters the insulin-induced suppression of fatty acid oxidation and stimulation of triacylglycerol storage in rodent skeletal muscle. American Journal of Physiology Endocrinology and Metabolism. 2006;291:E182–E189. doi: 10.1152/ajpendo.00272.2005. [DOI] [PubMed] [Google Scholar]
- Daval M, Diot-Dupuy F, Bazin R, Hainault I, Viollet Bt, Vaulont S, Hajduch E, Ferré P, Foufelle F. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. Journal of Biological Chemistry. 2005;280:25250–25257. doi: 10.1074/jbc.M414222200. [DOI] [PubMed] [Google Scholar]
- Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. American Journal of Clinical Nutrition. 2006;83:461S–465. doi: 10.1093/ajcn/83.2.461S. [DOI] [PubMed] [Google Scholar]
- Guo X, Li H, Xu H, Halim V, Thomas LN, Woo S-L, Huo Y, Chen YE, Sturino JM, Wu C. Disruption of inducible 6-phosphofructo-2-kinase impairs the suppressive effect of PPARγ activation on diet-induced intestine inflammatory response. Journal of Nutritional Biochemistry. 2013;24:770–775. doi: 10.1016/j.jnutbio.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Li H, Xu H, Halim V, Zhang W, Wang H, Ong KT, Woo SL, Walzem RL, Mashek DG, et al. Palmitoleate induces hepatic steatosis but suppresses liver inflammatory response in mice. PLoS ONE. 2012;7:e39286. doi: 10.1371/journal.pone.0039286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Xu K, Zhang J, Li H, Zhang W, Wang H, Lange AJ, Chen YE, Huo Y, Wu C. Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of PPARγ activation in mice. Journal of Biological Chemistry. 2010;285:23711–23720. doi: 10.1074/jbc.M110.123174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heishi M, Ichihara J, Teramoto R, Itakura Y, Hayashi K, Ishikawa H, Gomi H, Sakai J, Kanaoka M, Taiji M, et al. Global gene expression analysis in liver of obese diabetic db/db mice treated with metformin. Diabetologia. 2006;49:1647–1655. doi: 10.1007/s00125-006-0271-y. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- Huo Y, Guo X, Li H, Wang H, Zhang W, Wang Y, Zhou H, Gao Z, Telang S, Chesney J, et al. Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. Journal of Biological Chemistry. 2010;285:3713–3721. doi: 10.1074/jbc.M109.058446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Guo X, Li H, Xu H, Halim V, Zhang W, Wang H, Fan Y-Y, Ong KT, Woo S-L, et al. Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. Journal of Biological Chemistry. 2012;287:21492–21500. doi: 10.1074/jbc.M112.370379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huypens P, Quartier E, Pipeleers D, Van de Casteele M. Metformin reduces adiponectin protein expression and release in 3T3-L1 adipocytes involving activation of AMP activated protein kinase. European Journal of Pharmacology. 2005;518:90–95. doi: 10.1016/j.ejphar.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Jelenik T, Rossmeisl M, Kuda O, Jilkova ZM, Medrikova D, Kus V, Hensler M, Janovska P, Miksik I, Baranowski M, et al. AMP-activated protein kinase α2 subunit is required for the preservation of hepatic insulin sensitivity by n-3 polyunsaturated fatty acids. Diabetes. 2010;59:2737–2746. doi: 10.2337/db09-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir M, Catalano KJ, Ananthnarayan S, Kim SP, Van Citters GW, Dea MK, Bergman RN. Molecular evidence supporting the portal theory: a causative link between visceral adiposity and hepatic insulin resistance. American Journal of Physiology Endocrinology and Metabolism. 2005;288:E454–E461. doi: 10.1152/ajpendo.00203.2004. [DOI] [PubMed] [Google Scholar]
- Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. Journal of Biological Chemistry. 2006;281:26602–26614. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, Lee C-H. Adipocyte-derived Th2 cytokines and myeloid PPARδ regulate macrophage polarization and insulin sensitivity. Cell Metabolism. 2008;7:485–495. doi: 10.1016/j.cmet.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-J, Higashimori T, Park S-Y, Choi H, Dong J, Kim Y-J, Noh H-L, Cho Y-R, Cline G, Kim Y-B, et al. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes. 2004;53:1060–1067. doi: 10.2337/diabetes.53.4.1060. [DOI] [PubMed] [Google Scholar]
- Lu C-H, Hung Y-J, Hsieh P-S. Additional effect of metformin and celecoxib against lipid dysregulation and adipose tissue inflammation in high-fat fed rats with insulin resistance and fatty liver. European Journal of Pharmacology. 2016;789:60–67. doi: 10.1016/j.ejphar.2016.07.012. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, DeYoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- Mendoza EE, Pocceschi MG, Kong X, Leeper DB, Caro J, Limesand KH, Burd R. Control of glycolytic flux by AMP-activated protein kinase in tumor cells adapted to low pH. Translational Oncology. 2012;5:208–216. doi: 10.1593/tlo.11319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okar DA, Wu C, Lange AJ. Advances in Enzyme Regulation. Elsevier; 2004. Regulation of the regulatory enzyme, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphate; pp. 123–154. [DOI] [PubMed] [Google Scholar]
- Perriello G, Misericordia P, Volpi E, Santucci A, Santucci C, Ferrannini E, Ventura MM, Santeusanio F, Brunetti P, Bolli GB. Acute antihyperglycemic mechanisms of metformin in NIDDM. Evidence for suppression of lipid oxidation and hepatic glucose production. Diabetes. 1994;43:920–928. doi: 10.2337/diab.43.7.920. [DOI] [PubMed] [Google Scholar]
- Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochemical Journal. 2004;381:561–579. doi: 10.1042/BJ20040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin N-R, Lee J-C, Lee H-Y, Kim M-S, Whon TW, Lee M-S, Bae J-W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2013;63:727–735. doi: 10.1136/gutjnl-2012-303839. doi:710.1136/gutjnl-2012-303839. Epub 302013 Jun 303826. [DOI] [PubMed] [Google Scholar]
- Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132:2169–2180. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- Song S, Andrikopoulos S, Filippis C, Thorburn AW, Khan D, Proietto J. Mechanism of fat-induced hepatic gluconeogenesis: effect of metformin. American Journal of Physiology Endocrinology and Metabolism. 2001;281:E275–E282. doi: 10.1152/ajpendo.2001.281.2.E275. [DOI] [PubMed] [Google Scholar]
- Steinberg GR, Michell BJ, van Denderen BJW, Watt MJ, Carey AL, Fam BC, Andrikopoulos S, Proietto J, Görgün CZ, Carling D, et al. Tumor necrosis factor [alpha]-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metabolism. 2006;4:465–474. doi: 10.1016/j.cmet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin dependant diabetes mellitus. New England Journal of Medicine. 1995;333:550–554. doi: 10.1056/NEJM199508313330903. [DOI] [PubMed] [Google Scholar]
- Trujillo ME, Scherer PE. Adipose tissue-derived factors: impact on health and disease. Endocrine Reviews. 2006;27:762–778. doi: 10.1210/er.2006-0033. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AWJ. Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigtion. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo S-L, Xu H, Li H, Zhao Y, Hu X, Zhao J, Guo X, Guo T, Botchlett R, Qi T, et al. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS ONE. 2014;9:e91111. doi: 10.1371/journal.pone.0091111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Kang JE, Peng L, Li H, Khan SA, Hillard CJ, Okar DA, Lange AJ. Enhancing hepatic glycolysis reduces obesity: Differential effects on lipogenesis depend on site of glycolytic modulation. Cell Metabolism. 2005a;2:131–140. doi: 10.1016/j.cmet.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Wu C, Khan SA, Peng LJ, Li H, Camela S, Lange AJ. Perturbation of glucose flux in the liver by decreasing fructose-2,6-bisphosphate levels causes hepatic insulin resistance and hyperglycemia. American Journal of Physiology Endocrinology and Metabolism. 2006;291:E536–E543. doi: 10.1152/ajpendo.00126.2006. [DOI] [PubMed] [Google Scholar]
- Wu C, Okar DA, Kang J, Lange AJ. Reduction of hepatic glucose production as a therapeutic target in the treatment of diabetes. Current Drug Targets - Immune Endocrine & Metabolic Disorders. 2005b;5:51–59. doi: 10.2174/1568008053174769. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. Journal of Clinical Investigtion. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. American Journal of Physiology Endocrinology and Metabolism. 2007;293:E1118–E1128. doi: 10.1152/ajpendo.00435.2007. [DOI] [PubMed] [Google Scholar]
- Zhao W, Li A, Feng X, Hou T, Liu K, Liu B, Zhang N. Metformin and resveratrol ameliorate muscle insulin resistance through preventing lipolysis and inflammation in hypoxic adipose tissue. Cell Signal. 2016;28:1401–1411. doi: 10.1016/j.cellsig.2016.06.018. [DOI] [PubMed] [Google Scholar]
- Zheng J, Woo S-L, Hu X, Botchlett R, Chen L, Huo Y, Wu C. Metformin and metabolic diseases: a focus on hepatic aspects. Frontiers in Medicine. 2015:1–14. doi: 10.1007/s11684-015-0384-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulian A, Cancello R, Girola A, Gilardini L, Alberti L, Croci M, Micheletto G, Danelli P, Invitti C. In vitro and in vivo effects of metformin on human adipose tissue adiponectin. Obesity Facts. 2011;4:27–33. doi: 10.1159/000324582. [DOI] [PMC free article] [PubMed] [Google Scholar]