

Abstract
Crouzon syndrome (CS) is an genetic disorder with autosomal dominant inheritance caused by mutation of the gene for fibroblast growth factor receptor 2 (FGFR2) was described as one of the varieties of craniosynostosis. In this presented case, premature closure of the sutures had caused restricted skull growth and lack of space for the growing brain resulted to shallowed eyes and cranial and ophthalmic deformities and impairment in tooth development. Management of a patient of CS has two components. First is the release of prematurely fused sutures based on evidence of raised intracranial pressure. Surgery is mainly carried out early after 3–6 months. Second is the craniofacial reconstructive surgery including advancement of the maxilla and frontonasal complex; and other surgeries depending upon the deformities. An increased intracranial pressure impairs brain development and can lead to mental retardation. Because of the delayed diagnosis and treatment in this case, visual and hearing loses and decreased mental capacity and mild retardation.
Keywords: Crouzon syndrome, craniosynostosis, FGFR2 gene mutations
Introduction
Crouzon syndrome (CS) is a rare genetic disorder with autosomal dominant inheritance caused by mutation of the gene for fibroblast growth factor receptor 2 (FGFR2) has been mapped to the long arm of chromosome 10 and mutations in exon B of FGFR2 gene was described as one of the varieties of craniosynostosis caused by premature obliteration and ossification of two or more sutures.[1,2] Once the sutures become closed, growth potential to those sutures is restricted. However, multiple sutural synostoses frequently extend to premature fusion of skull base causing midfacial hypoplasia, shallow orbit, maxillary hypoplasia, and occasional upper airway obstruction. Intraoral manifestations include mandibular prognathism, overcrowding of upper teeth, and V-shaped maxillary dental arch. Narrow, high, or cleft palate and bifid uvula can also be seen. Occasional oligodontia, macrodontia, peg-shaped, and widely spaced teeth have been reported.[1,2]
A 13-year-old boy along with his parents admitted to our department of ophthalmology for treatment. The chief complaint being as presented by the mother was visual loss and headache. Review of medical history was unremarkable, specifically, the mother reported normal labor in and delivery. He is the fourth child of his family and there were no anomalies in any siblings or near relatives reported. The child was not on any medications and denied any medical allergies. He has never been to a dentist before and this was the first dental consultation. Head examination revealed elliptical-shaped head, with dolichofacial growth pattern, and convex facial profile. The presence of prominent eyeballs, which is the characteristics of the Crouzon's disease observed. Best corrected visual acuity was 5/10 for oculus dexter, count fingers at 2 m for oculus sinister with Snellen chart on ophthalmologic examination. Cycloplegic refraction was +1.75 for right eye, +5.75 (+0.75 × 58″) for left. Moreover, amblyopia was present at left eye. Bilateral exophthalmos was present. Anterior segment examination was normal for both eyes. Fundus examination showed that both of papillae were pale and atrophic. At visual evoked potential examination, there was 9 msn delay in P100 latency, bilaterally. Single sharp slow wave activity, which is related to central nervous system, was observed at electroencephalography examination. There was mild conductive hearing loss (30 dB) in the left ear in his examination. Dental examination made by dentists and showed that the patient is in early mixed dentition, with all primary teeth present in the maxillary and mandibular arch, the chronology of eruption and eruption status was normal for the child's age. The child also had a high arched palate. Since the child's appearance and head size was not normal, he was referred to the pediatrician and psychologist for their expertise. The pediatrician and psychologist did diagnose the child with CS associated with mild mental retardation.
The skull radiographs revealed the “scaphocephalic” skull shape, hypoplastic maxilla, and zygoma with shallow orbits. Prominent cranial markings of the inner surface of the cranial vault seen as multiple radiolucencies appearing as depressions resulting in the “hammered silver” (beaten metal/copper beaten) appearance indicating internal remodeling of the calvaria due to an increase in intracranial pressure as a result of premature cranial suture fusion. Three-dimensional computed tomographic (CT) scans of the skull showed fused sagittal and lambdoid sutures, moderate degree of hydrocephalus with diffuse indentation of inner table of skull. Other systemic examination was found to be normal.
Peripheral blood chromosomal analysis revealed 46, XX normal chromosomal count. Cardiovascular system, central nervous system, respiratory and abdominal examination was normal. Routine hematological and biochemical tests were within normal limits.
In this presented case, premature closure of the sutures had caused restricted skull growth and lack of space for the growing brain resulted to shallowed eyes and cranial and ophthalmic deformities and impairment in tooth development. Optic atrophy is frequently seen and has been reported in 30–80% of patients. Our patient had decrement in vision due to optic atrophy as seen in Figures 1–4. In addition, approximately one-third of patients with CS suffer from hearing loss due to middle ear deformities and upper airway obstruction occurs due to midfacial hypoplasia and narrow epipharynx and our patient had also suffer from hearing loss. Mental ability and psychomotor development is generally within normal limits. However, when the premature closure of the cranial suture lines impairs brain development due to increased intracranial pressure it can lead to mental retardation like this presented case.[3,4]
Figure 1.
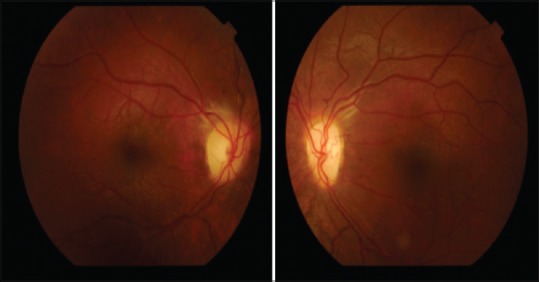
Pale and neuropathic pupilla in right eye (right picture) and left eye (left picture)
Figure 4.
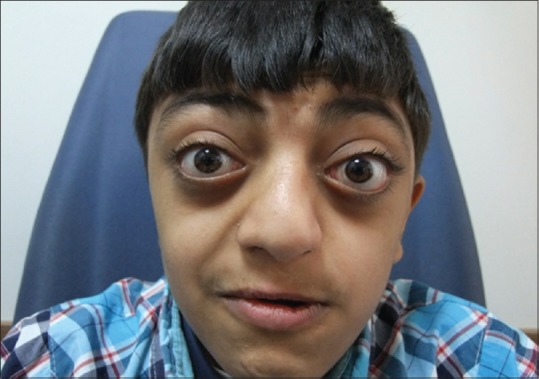
Characteristic face of Crouzon's syndrome
Figure 2.
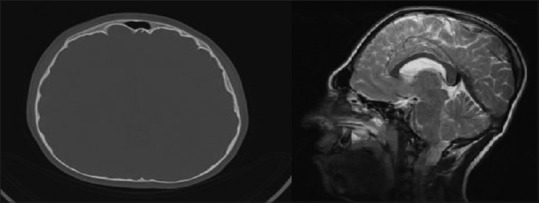
Synostosis (brachicephali) and diffuse indentation beaten Cooper appearance in axial computed tomography (left), elliptical shaped head in magnetic resonance imaging
Figure 3.
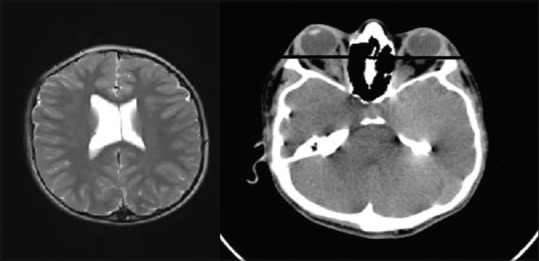
Axial magnetic resonance imaging revealed mild hydrocephaly in left and axial computed tomography revealed prominent exophtalmus in the right. Transverse line in the right picture is the interzygomatic line
In radiological examination, anteroposterior, lateral, and cephalometric views of the skull are taken. It shows the premature suture closure and provides information about maxillomandibular relation. “Paw marking” of the skull is seen due to raised intracranial pressure. CT scan confirms the standard radiographical findings and provides information on ventricular size.[4]
Early and accurate diagnosis of a patient of CS is essential in management. Management of a patient of CS has two components. First is the release of prematurely fused sutures based on evidence of raised intracranial pressure. Surgery is mainly carried out early after 3–6 months. Second is the craniofacial reconstructive surgery including advancement of the maxilla and frontonasal complex; and other surgeries depending upon the deformities in the patient such as rhinoplasty, oculoplasty, and cleft lip and cleft palate repair can be done. The management requires a multidisciplinary approach and the surgical treatment usually begins in the child's 1st year with cranial decompression. Early craniectomy of coronal sutures should be done soon to relieve increased intracranial pressure caused by premature multiple suture synostosis. An increased intracranial pressure impairs brain development and can lead to mental retardation. Because of the delayed diagnosis and treatment in this case, visual and hearing loses and decreased mental capacity and mild retardation and many oral, cranial, orbital, and maxillofacial dysmorphic features were found. The prognosis in the case of CS depends on the severity of malformation, and the patients usually have a normal lifespan. We referred our patient to neurosurgeons for possible surgical procedures to decrease the intracranial pressure to protect the visual and hearing capacity and maxillofacial surgeons to restore and improve palate and tooth.[1,5]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Padmanabhan V, Hegde AM, Rai K. Crouzon's syndrome: A review of literature and case report. Contemp Clin Dent. 2011;2:211–4. doi: 10.4103/0976-237X.86464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaur H, Singh Waraich H, Sharma CM. Crouzon syndrome: A case report and review of literature. Indian J Otolaryngol Head Neck Surg. 2006;58:381–2. doi: 10.1007/BF03049602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glaser RL, Jiang W, Boyadjiev SA, Tran AK, Zachary AA, Van Maldergem L, et al. Paternal origin of FGFR2 mutations in sporadic cases of Crouzon syndrome and Pfeiffer syndrome. Am J Hum Genet. 2000;66:768–77. doi: 10.1086/302831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen MM Jr, editor. Craniosynostosis: Diagnosis, Evaluation, and Management. 2nd ed. New York: Oxford University Press; 2000. Craniosynostosis: Diagnosis, evaluation, and management Crouzon syndrome; p. 361. [Google Scholar]
- 5.Golabi M. Radiographic abnormalities of Crouzon syndrome. A survey of 23 cases. Proc Greenwood Genet Ctr. 1984;3:102. [Google Scholar]