

Abstract
Spindle cell/sclerosing rhabdomyosarcoma (sRMS/scRMS), previously classified as a histologic variant of embryonal RMS (ERMS), has been recently defined as a distinct pathologic subtype of RMS. Despite a uniform histologic appearance, sRMS/scRMS is a heterogeneous group with different outcomes between children and adults and distinct genetic abnormalities with either NCOA2 or VGLL2 related fusions or MYOD1 mutations. As sRMS/scRMS show a predilection for the head and neck, we sought to investigate the clinicopathologic and molecular features of this patient population treated at our Institution in the past 2 decades. There were 13 patients (8 males and 5 females) with head and neck sRMS/scRMS with a mean age at diagnosis of 29 years, including 5 children and 8 adults. The common anatomic sites were buccal/masticator space, tongue and mandibular soft tissue. Three patients presented with distant metastases. Eight cases had spindle cell and five sclerosing morphology. All tumors tested showed diffuse reactivity for myoD1. None of the 7 cases tested showed PAX3/7-FOXO1 fusion. Three of the nine cases tested harbored MYOD1 with or without PIK3CA mutations, all having sclerosing histology and succumbing of disease with recurrences. All except one patient with sclerosing RMS died of disease, in contrast to only one of eight with spindle cell morphology. The only patient with infantile sRMS/scRMS showed an SRF-NCOA2 fusion. All patients except one were treated with multimodality chemotherapy, radiotherapy and/or surgery. Our results show that fatal outcome in head and neck sRMS/scRMS was associated with adult age, sclerosing morphology, and MYOD1 and PIK3CA mutations.
Keywords: Rhabdomyosarcoma, Spindle cell Rhabdomyosarcoma, Sclerosing rhabdomyosarcoma, MYOD1 mutations
INTRODUCTION
Spindle cell and sclerosing rhabdomyosarcoma (sRMS/scRMS) accounting for 5–10% of all RMS, was recently reclassified as a stand-alone pathologic entity in the latest WHO classification of soft tissue tumors [1]. Spindle cell RMS was first described by the German-Italian Cooperative Sarcoma Study on the basis of its distinct clinicopathologic features and favorable outcome, resulting in separation from the more common embryonal RMS (ERMS) [2]. Sclerosing RMS was first defined by Mentzel and Katenkamp as a ‘sclerosing pseudovascular RMS in adults’ [3]. As both spindle cell and sclerosing RMSs have similar clinical presentations in the paratesticular or head and neck region, affecting both pediatric and adult age groups with a male predilection [4, 5] and overlapping histologic features. It was suggested that they may represent a single pathologic entity sharing clinical, morphologic and genetic features [3, 6–8].
Our group has recently demonstrated that MYOD1 mutations are a recurrent finding in both pediatric and adult sRMS/scRMS patients[8], and that in pediatric patients MYOD1-mutant tumors are associated with a poor prognosis [9]. Furthermore, we have previously shown that congenital/infantile sRMS/scRMS have distinct genetic abnormalities and are characterized in most cases by the presence of NCOA2 and VGLL2 related fusions, being overwhelmingly associated with a favorable outcome, long-term survival and lack of distant metastases [9, 10]. In this study, we further investigate the clinicopathologic and molecular features of sRMS/scRMS presenting in the head and neck region at our Institution.
MATERIALS AND METHODS
The study was approved by the institutional review board of Memorial Sloan Kettering Cancer Center (MSKCC), New York. We have searched the MSKCC Department of Pathology patient charts for diagnosis of RMS involving the head and neck regions from 1996 to 2015. All cases with material available were re-reviewed and reclassified based on the 2013 WHO classification, applying strict morphologic criteria to identify cases of spindle cell/sclerosing RMS. The clinical features including: age, gender, anatomic site, size, pre-therapy stage, treatment, recurrence and follow-up information were obtained from the medical records in all cases. For each case, the hematoxylin-eosin stained sections and immunohistochemical profiles (myogenin, desmin) were reviewed. Unstained slides available in 12/13 cases were used for further molecular characterization, including FISH or RT-PCR for PAX3/7-FOXO1 fusions, PCR and targeted sequencing for hot-spot MYOD1 and PIK3CA mutations and FISH using custom BAC probes for NCOA2 and VGLL2 gene rearrangements, as previously described [8].
RESULTS
Clinical features
The clinical features are summarized in Table 1. Thirteen cases of spindle cell/sclerosing RMS were identified from our institutional and consultation archival files. Three cases were previously included in the study by Mosquera et al. [10], two of which were also studied in Agaram et al. [8] and the third case also studied by Alaggio et al. [9]. There were 8 males and 5 females with a wide age range at diagnosis 0.7–72 years (mean: 29), including 5 children (0.7–17 years, 1 infant) and 8 adults (27–72 years). The anatomic sites comprised various locations: buccal/masticator space (3), soft tissue of the mandible (2), tongue (2), neck (2), infratemporal fossa (1), soft tissue of skull (1), hypopharynx (1) and nasolabial/cheek (1). Three of the cases were considered as parameningeal sites, while the remaining were non-parameningeal, non-orbital sites. The tumor sizes ranged 0.8–10.5 cm with 7 cases being larger than 5 cm in greatest dimension. The images (MRI) showed large masses causing a mass-effect, erosion and destruction of nearby bony structures (Figure 1). Tumor staging was applied according to the Intergroup Rhabdomyosarcoma Study Group pretreatment classification: stage 1 (9), stage 3 (1) and stage 4 (3). The metastatic sites included lung (1), and spine and pelvis (2). None of the patients presented with bone marrow involvement on pre-therapy evaluation. All patients except one were treated with multimodality therapy with a combination of surgery and chemotherapy and/or radiotherapy. The patient (Case 7) treated with a unimodal therapy had a surgical resection for a 1.1 cm tongue lesion.
Table 1.
Clinical Features and Follow-Up Data of Spindle and Sclerosing RMS arising in the Head and Neck
| Case # | Age/Sex | Site | Size (cm) | Diagnosis | Stage | Therapy | Recurrence | Follow-up duration | Vital status |
|---|---|---|---|---|---|---|---|---|---|
| 1a,b | 34y/F | Buccal/masticator space | 2.4 | Sclerosing RMS | 1 | Surgery, CT and RT | DR | 43 mos | DOD |
| 2a,b,c | 14y/F | Infratemporal fossa | 7.3 | Sclerosing RMS | 3 | CT and RT | LR | 26 mos | DOD |
| 3 | 17y/F | Buccal/masticator space | 3.5 | Sclerosing RMS | 1 | Surgery, CT and RT | − | 31 mos | Alive (NED) |
| 4 | 28y/M | Buccal/masticator space | 10.5 | Sclerosing RMS | 4 | CT and RT | − | 12 mos | DOD |
| 5 | 33y/M | Soft tissue mandible | 5.8 | Sclerosing RMS | 1 | Surgery, CT and RT | LR (2), DR | 65 mos | DOD |
| 6 | 27y/F | Soft tissue skull | 5.1 | Spindle cell RMS | 4 | Surgery, CT and RT | |||
| 7 | 14y/M | Tongue | 1.1 | Spindle cell RMS | 1 | Surgery | |||
| 8a,c | 0.7y/M | Neck | 6.5 | Spindle cell RMS | 1 | Surgery, CT and RT | LR | 60 mos | Alive (NED) |
| 9 | 72y/M | Neck | 9.1 | Spindle cell RMS | 1 | Surgery, CT and RT | − | 29 mos | Alive (NED) |
| 10 | 41y/M | Soft tissue mandible | 4.5 | Spindle cell RMS | 1 | Surgery and RT | − | 4 mos | Alive (NED) |
| 11 | 60y/M | Hypopharynx | 8.3 | Spindle cell RMS | 4 | Surgery, CT and RT | − | 14 mos | DOD |
| 12 | 33y/M | Tongue | 0.8 | Spindle cell RMS | 1 | Surgery and CT | − | 94 mos | Alive (NED) |
| 13 | 1.75y/F | Nasolabial/cheek | 1.0 | Spindle cell RMS | 1 | Surgery and CT | − | 7 mos | Alive (NED) |
Cases included in Mosquera et al., Agaram et al. and Alaggio et al.
Stage, according to the Intergroup Rhabdomyosarcoma Study Group pretreatment staging classification
CT – chemotherapy, (−) – negative, RT – radiotherapy, LR – local recurrence, DR – distant recurrence, DOD – died of disease, NED – no evidence of disease
Figure 1. Radiographic findings of head and neck spindle cell/sclerosing rhabdomyosarcoma.
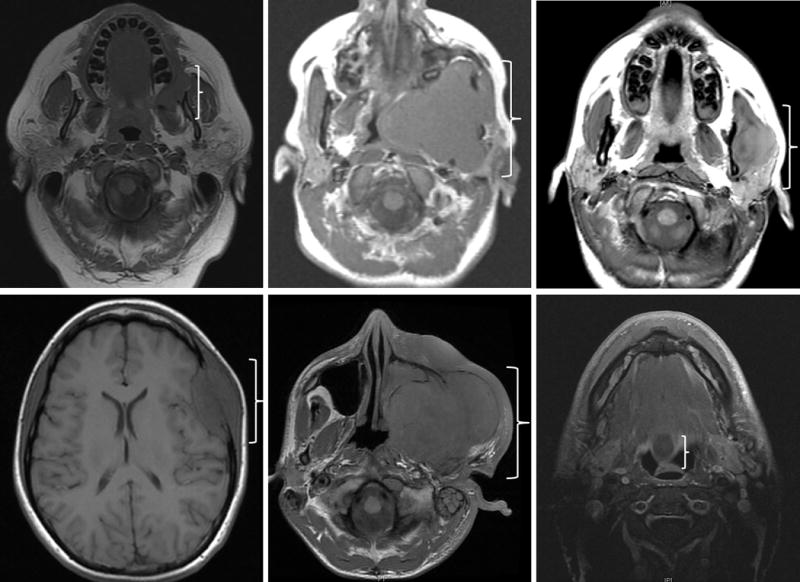
MRI axial views of 6 patients showing large tumor masses causing a mass-effect, erosion and destruction of nearby bony structures
Morphologic and Immunophenotypic features
Eight cases were classified as spindle cell and five cases as sclerosing RMS. The morphologic appearance of spindle cell RMS was that of compact spindle cells arranged in intersecting fascicles with a herring-bone, fascicular or fibromatosis-like growth pattern. The sclerosing RMS were defined as a proliferation of spindle to round cells embedded in an extensively hyalinized stroma showing a distinctive pseudoalveolar or pseudovascular pattern. Both histologic variants showed limited if any evidence of strap cells, rhabdomyoblastic differentiation, or nuclear pleomorphism. These features typically are associated with ERMS. Additionally, the typical myxoid stromal component seen in most ERMS was not present in either spindle or sclerosing RMS.
Immunohistochemical analysis showed reactivity for desmin and myogenin in all cases. The desmin expression was mostly diffuse and strong, while myogenin reactivity was focal to rare positive cells in 11/13 cases. Results are summarized in Table 2. Additionally, 7/7 case tested were positive for myoD1 in a diffuse pattern (Figures 2 and 3). Other stains performed in a subset of cases showed that 5/9 were positive for SMA (with 4/5 cases exhibiting only focal staining), 1/10 was positive for cytokeratin (AE1:AE3) and 1/3 was positive for CD99.
Table 2.
Immunohistochemical and molecular profile of spindle cell/sclerosing RMS
| Immunohistochemical stains | Molecular | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Case # | Myogenin | Desmin | MyoD1 | SMA | CK | CD99 | MYOD1 | PIK3CA | SRF-NCOA2 | PAX3/7-FOXO1 |
| 1a,b | +(f) | + | + | − | − | L122R (heterozygous) | E542V | − | − | |
| 2a,b,c | +(f) | + | − | − | + | L122R (homozygous) | E542K | − | − | |
| 3 | + | + | + | − | − | WT | WT | − | ||
| 4 | +(f) | + | + | − | ||||||
| 5 | + | + | +(f) | − | − | L122R (heterozygous) | WT | − | ||
| 6 | +(f) | + | + | +(f) | + | WT | WT | |||
| 7 | +(f) | +(f) | + | − | WT | WT | ||||
| 8a,c | +(f) | + | + | + | ||||||
| 9 | +(f) | + | +(f) | WT | WT | |||||
| 10 | +(f) | + | + | − | − | |||||
| 11 | +(f) | + | +(f) | − | WT | WT | ||||
| 12 | +(f) | + | + | − | − | − | ||||
| 13 | +(f) | + | − | WT | WT | − | − | |||
Cases included in Mosquera et al., Agaram et al. and Alaggio et al.
(+) – Positive, (−) – negative, f – focal, SMA – smooth muscle actin, CK - cytokeratin, WT – wild type
Figure 2. Pathologic features of SRF-NCOA2 fusion-positive congenital spindle cell RMS.
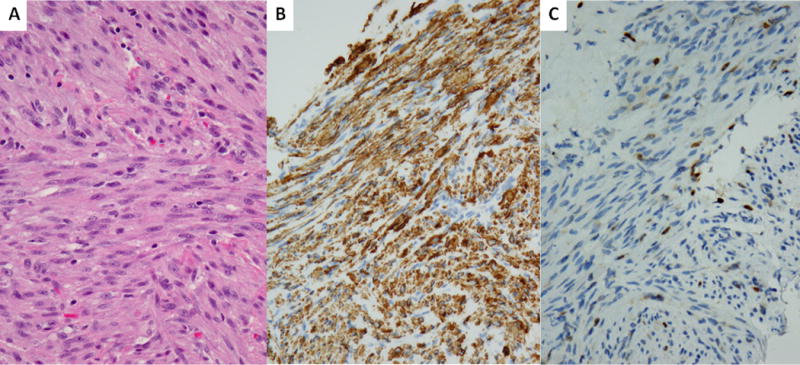
Spindle cell RMS occuring in a 7 month-old male in the neck soft tissue showing intersecting fascicles of relatively monomorphic fusiform cells with fibrillary eosinophilic cytoplasm, open chromatin and small nucleoli (A, 100×, case 8), which showed diffuse desmin immunoreactivity (B, 400×) and scattered nuclear myogenin (C, 400×).
Figure 3. Pathologic features of MYOD1-mutant positive sclerosing RMS.
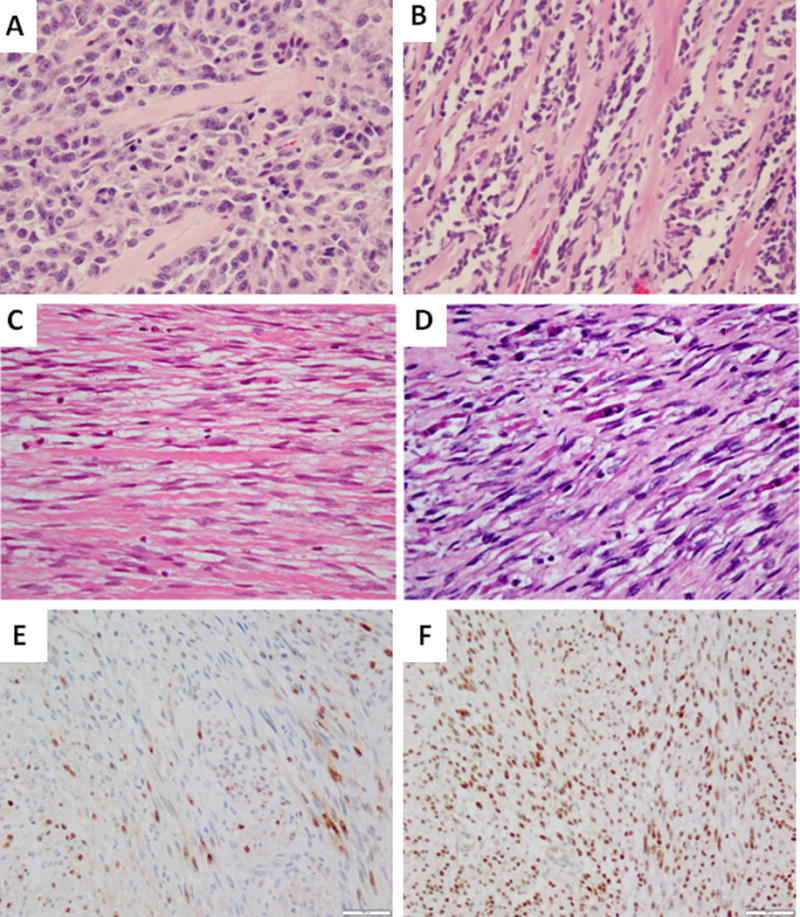
ScRMS arising in the buccal mucosa of a 34 year-old female showed predominant round cell phenotype, which was initially misdiagnosed as a solid ARMS (A, 200×, case 1). ScRMS in a 14 year-old female occurring in the infra-temporal fossa harboring co-existing MYOD1 L122R and PIK3CA E542 mutations showing a pathognomonic pseudovsacular growth pattern with parallel columns of hyalinization (B, case 2, 200×). Deceptively bland scRMS occurring in the mandibular soft tissue of a 33 year-old male showing spindle cells sepearated by thick collagen bundles in the primary tumor (C, 100×, case 5). Subsequent recurrence of the mandibular soft tissue tumor shows increased cellularity, hyperchromasia and strap cells (D, 100×). Immunohistochemically, the tumor shows scattered Myogenin (E, 100×) and diffuse MYOD1 nuclear staining (F, 100×)
Molecular Results
Results of molecular features are summarized in Table 2. Seven cases, including all 5 sclerosing RMS, were investigated for PAX3/7-FOXO1 gene fusions either by FISH or RT-PCR and were negative. Nine cases (5 spindle cell RMS and 4 sclerosing RMS) were also analyzed by PCR for hot spots mutations in MYOD1 exon 1 and PIK3CA exon 9 and 20 (Table 2). Three cases (Cases #1, 2, 5) showed mutations in MYOD1 exon 1 L122R (two heterozygous and one homozygous) and 2 cases showed co-existing PIK3CA exon 9 E542K/V mutations (cases #1, 2) [8]. As previously reported, the only one case of infantile head and neck sRMS showed the presence of an SRF-NCOA2 gene fusion [9].
Follow-up data
Follow-up information (Table 1) was available for 11 of the 13 cases, ranging from 4–94 months. Four patients developed recurrences at 3–24 months following treatment (3 sclerosing and 1 spindle cell RMS): 2 patients developed local recurrence (LR), 1 patient developed distant recurrence (DR) and 1 patient developed both LR (twice) and DR. The sites of DR included pleura and occiput. Six patients had no evidence of disease (NED) at an average follow-up time of 37.5 months (range: 4–94 months) and 5 patients died of the disease (DOD) at 12–65 months following diagnosis. Four of the 5 patients who DOD were adults, 2 of them presenting with distant metastases at initial diagnosis, 1 developed distant recurrence and 1 developed both local and distant recurrences. Four of the five patients (3 adults and 1 child) who DOD were classified as sclerosing RMS. All 3 patients with MYOD1-mutant RMS had sclerosing histology (Cases 1, 2 and 5), developed recurrences and DOD at 43, 26 and 65 months, respectively following diagnosis. All three patients received multiagent chemotherapy and radiotherapy to the primary tumor, two developed distant spread to the pleura and occiput and one patient also developed LR. The long-term survivor with sclerosing RMS (Case 3) was not found to have either MYOD1 or PIK3CA mutation.
The child with SRF-NCOA2 fusion positive sRMS/scRMS is alive with NED at 60 months following diagnosis.
DISCUSSION
Rhabdomyosarcoma (RMS), the most common soft tissue sarcoma in children, has been traditionally divided into 2 distinct pathologic entities based on their morphologic and genetic phenotype including the PAX-FOXO1 fusion-positive alveolar RMS and fusion negative ERMS. The spindle cell RMS (sRMS) is an uncommon variant characterized by monomorphic spindle cells arranged in intersecting fascicles, reminiscent of leiomyosarcoma or fibrosarcoma, and have predilection for head and neck and paratesticular location. Spindle cell RMS was initially grouped under ERMS, although they typically lack rhabdomyoblastic differentiation, nuclear pleomorphism or myxoid stroma. A subset of sRMS shows variable degrees of hyaline sclerosis and a pseudovascular growth pattern, and have overlapping features with the even less common sclerosing RMS (scRMS). ScRMS further shares with sRMS a very similar clinical presentation and biologic behavior [6–8]. ScRMS shows marked hyalinized matrix in a pseudovascular growth pattern associated with spindle to rounded cells mimicking entities like osteosarcoma, angiosarcoma or alveolar RMS (ARMS) [8, 11]. Subsequently, the latest WHO classification of soft tissue tumors recognized that sRMS and scRMS represent a morphologic continuum and should be grouped as a single pathologic entity distinct from embryonal RMS[1].
In this study, we sought to further investigate the clinicopathologic and molecular features of sRMS/scRMS occurring in the head and neck region. Among the 13 patients included, 8 had spindle cell features (sRMS) and 5 had prominent hyalinization defined as sclerosing subtype (scRMS). The patients had a wide age range at diagnosis as well as a variable distribution within the oral cavity, para-meningeal or soft tissue locations. Despite a relatively uniform morphology, the sRMS/scRMS arising in this location have diverse characteristics, depending on the age at presentation and genetic abnormalities, as previously reported in the pediatric age group or other locations [9]. First, tumors presenting within the first year of age show characteristic gene fusions either involving NCOA2 or VGLL2 genes, which are fused to critical transcription factors involved in skeletal muscle development or function [9]. The distinction of this clinical and genetic subset from other subtypes of RMS is critical since patients with congenital/infantile sRMS/scRMS are associated with a favorable outcome and lack distant metastases or death of disease. Indeed, the only infant patient in this study, a 7 month-old male with the tumor in the posterior neck harbored a SRF-NCOA2 fusion and had no evidence of disease at 60 months after diagnosis.
A second important genetic subgroup of sRMS/scRMS is characterized by the presence of MYOD1 mutations co-existing in most cases with PIK3CA mutations, and is seen often in older children or young adults [8, 9, 12]. Although these tumors show similar clinical presentations with MYOD1-negative tumors, they have a predilection for sclerosing histology and are associated with a highly aggressive outcome with early distant metastases and fatal course in most patients [8, 9]. The three head and neck cases (14 year-old female, infra-temporal fossa; 34 year-old female, buccal/masticator space; 33 year-old man, mandible soft tissue) in this series harbor MYOD1 L122R mutations and in two cases have co-existing PIK3CA helical domain mutations E542V/K. These cases presented with sclerosing histology and followed an aggressive course with recurrence and death of disease, despite multimodality therapy with chemotherapy and radiotherapy to the primary tumor. The long-term survivor with sclerosing RMS (17 year-old female, buccal/masticator space) was not found to have either MYOD1 or PIK3CA mutation.
Earlier studies have suggested that pediatric sRMS/scRMS has a more favorable prognosis compared with ERMS, while in adult patients has a poor prognosis [2, 13, 14]. In this study, among the 5 of 11 patients with follow-up information who succumbed of their disease, 4 were adults with 2 presenting with distant metastases at initial diagnosis, 1 with distant recurrence and 1 with both local and distant recurrences.
On the bases of morphologic and immunophenotypic similarities, the differential diagnosis of sRMS/scRMS is quite wide and includes a variety of other spindle cell sarcomas, such as leiomyosarcoma, fibrosarcoma and malignant peripheral nerve sheath tumor with RMS component (Triton tumor). Leiomyosarcoma shows similar intersecting long fascicles of spindle cells with fibrillary eosinophilic cytoplasm, but harbors more significant nuclear pleomorphism and by immunohistochemistry shows diffuse reactivity for SMA, and is negative for myogenin and myoD1. Fibrosarcoma, in particular congenital/infantile fibrosarcoma, shows overlapping features with sRMS in infantile age group due to its monomorphic spindle cells arranged in intersecting fascicles in a herringbone pattern. However, fibrosarcomas are negative for desmin, myogenin and myoD1. Triton tumors may mimic a sRMS as the S100 staining may be negative. However, the pattern of desmin is usually patchy rather than diffuse, highlighting mostly the strap cells, or well-differentiated rhabdomyoblastic cells. Furthermore, most Triton tumors occur in the setting of type I neurofibromatosis. An additional pitfall can occur in the setting of the so-called ‘pseudoalveolar architecture’ of scRMS, which may suggest an ARMS diagnosis. The correct diagnosis is usually suspected by the focal staining for myogenin, diffuse strong myoD1 expression and lack of FOXO1-gene abnormalities by FISH.
In conclusion, sRMS/scRMS shares similar clinical, histologic and molecular features. Diagnostic pitfalls from other look-alike sarcomatoid processes should be avoided with the use of appropriate immunohistochemical stains and relevant clinical information. Overall, sRMS/scRMS arising in adult patients showed a more aggressive clinical behavior compared to pediatric tumors. SRMS/scRMS lacking MYOD1 mutations carries a better prognosis compared to MYOD1-mutant tumors, irrespective of the patient’s age. We recommend that molecular evaluation of the MYOD1/PIK3CA mutations in children and adults with sRMS/scRMS should be performed for risk stratification. Additionally, in the setting of congenital or infantile sRMS/scRMS, FISH for NCOA2 and VGLL2 gene rearrangements should be performed to confirm these genetic abnormalities as this subset carries a favorable outcome and might be managed with less intensive chemotherapy.
HIGHLIGHTS.
Spindle cell rhabdomyosarcoma and sclerosing rhabdomyosarcoma share similar clinical, histologic and molecular features.
Diagnostic pitfalls from other look-alike sarcomatoid processes should be avoided with the use of appropriate immunohistochemical stains and relevant clinical information.
Our results show that fatal outcome in the head and neck spindle cell/sclerosing rhabdomyosarcoma was associated with adult age, sclerosing morphology and the presence of MYOD1 and PIK3CA mutations.
We recommend that molecular evaluation of the MYOD1/PIK3CA hot spot mutations in children and adults with spindle cell/sclerosing rhabdomyosarcoma should be performed for risk stratification.
Acknowledgments
Supported in part by: P50CA140146-01 (CRA), Cycle for Survival (LW, CRA) and Kristen Ann Carr Foundation (CRA)
Footnotes
Disclosure: All authors declare that there are no financial conflicts associated with this study and that the funding source has no role in conceiving and performing the study.
References
- 1.Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. WHO clasification of tumours of soft tissue and bone. Lyon, France: IARC Press; 2013. pp. 134–5. [Google Scholar]
- 2.Cavazzana AO, Schmidt D, Ninfo V, Harms D, Tollot M, Carli M, et al. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. The American journal of surgical pathology. 1992;16:229–35. doi: 10.1097/00000478-199203000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Archiv : an international journal of pathology. 2000;436:305–11. doi: 10.1007/s004280050451. [DOI] [PubMed] [Google Scholar]
- 4.Rekhi B, Singhvi T. Histopathological, immunohistochemical and molecular cytogenetic analysis of 21 spindle cell/sclerosing rhabdomyosarcomas. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2014;122:1144–52. doi: 10.1111/apm.12272. [DOI] [PubMed] [Google Scholar]
- 5.Yasui N, Yoshida A, Kawamoto H, Yonemori K, Hosono A, Kawai A. Clinicopathologic analysis of spindle cell/sclerosing rhabdomyosarcoma. Pediatric blood & cancer. 2015;62:1011–6. doi: 10.1002/pbc.25367. [DOI] [PubMed] [Google Scholar]
- 6.Mentzel T, Kuhnen C. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Archiv : an international journal of pathology. 2006;449:554–60. doi: 10.1007/s00428-006-0284-4. [DOI] [PubMed] [Google Scholar]
- 7.Mentzel T. [Spindle cell rhabdomyosarcoma in adults: a new entity in the spectrum of malignant mesenchymal tumors of soft tissues] Der Pathologe. 2010;31:91–6. doi: 10.1007/s00292-009-1249-6. [DOI] [PubMed] [Google Scholar]
- 8.Agaram NP, Chen CL, Zhang L, LaQuaglia MP, Wexler L, Antonescu CR. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes, chromosomes & cancer. 2014;53:779–87. doi: 10.1002/gcc.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alaggio R, Zhang L, Sung YS, Huang SC, Chen CL, Bisogno G, et al. A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. The American journal of surgical pathology. 2016;40:224–35. doi: 10.1097/PAS.0000000000000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosquera JM, Sboner A, Zhang L, Kitabayashi N, Chen CL, Sung YS, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes, chromosomes & cancer. 2013;52:538–50. doi: 10.1002/gcc.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. The American journal of surgical pathology. 2002;26:1175–83. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 12.Szuhai K, de Jong D, Leung WY, Fletcher CD, Hogendoorn PC. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. The Journal of pathology. 2014;232:300–7. doi: 10.1002/path.4307. [DOI] [PubMed] [Google Scholar]
- 13.Rubin BP, Hasserjian RP, Singer S, Janecka I, Fletcher JA, Fletcher CD. Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. The American journal of surgical pathology. 1998;22:459–64. doi: 10.1097/00000478-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Nascimento AF, Fletcher CD. Spindle cell rhabdomyosarcoma in adults. The American journal of surgical pathology. 2005;29:1106–13. [PubMed] [Google Scholar]