

Abstract
Yeast species are extremely diverse and not monophyletic. Because the majority of yeast research focuses on ascomycetes, the mutational determinants of genetic diversity across yeast species are not well understood. By combining mutation-accumulation techniques with whole-genome sequencing, we resolved the genomic mutation rate and spectrum of the oleaginous (oil-producing) ‘red yeast’ Rhodotorula toruloides, the first such study in the fungal phylum Basidiomycota. We find that the mutation spectrum is quite different from what has been observed in all other studied unicellular eukaryotes, but similar to that in most bacteria—a predominance of transitions relative to transversions. Rhodotorula toruloides has a significantly higher A:T→G:C transition rate—possibly elevated by the abundant flanking G/C nucleotides in the GC-rich genome, as well as a much lower G:C→T:A transversion rate. In spite of these striking differences, there are substantial consistencies between R. toruloides and the ascomycete model yeasts: a spontaneous base-substitution mutation rate of 1.90 × 10 −10 per site per cell division as well as an elevated mutation rate at non-methylated 5'CpG3' sites. These results imply the evolution of variable mutation spectra in the face of similar mutation rates in yeasts.
Keywords: spontaneous mutation, mutation accumulation, yeast evolution, evolutionary genomics
Introduction
In order to systematically understand the evolution of mutational features, such as the mutation rate and spectrum, investigations of organisms broadly sampled from the Tree of Life are required. In contrast to recent progress on bacterial mutation rates (Lee et al. 2012; Long et al. 2015a; Dillon et al. 2015; Sung et al. 2015; Kucukyildirim et al. 2016), studies on unicellular eukaryotes remain rare. Currently unbiased mutation spectra are available in only a handful of species, and the only two fungi are ascomycetes (Sung et al. 2012; Zhu et al. 2014; Behringer and Hall 2015; Farlow et al. 2015; Ness et al. 2015), which comprise less than 1% of all fungal species (Kurtzman and Piškur 2006). Most recently, studies of spontaneous mutation have primarily relied on mutation-accumulation techniques, repeatedly bottlenecking large numbers of parallel lineages through single individual/colony transfers (Bateman 1959; Mukai 1964; Kibota and Lynch 1996; Halligan and Keightley 2009). Such experimental design maximizes genetic drift, overwhelming the ability of selection to eliminate mutations prior to accumulation. Therefore, unless associated with extremely large fitness effects, most mutations have an equal chance of accumulating in the genome.
Rhodotorula toruloides, a yeast in the phylum Basidiomycota (∼62% GC content; ∼20 Mb genome size; synonym: Rhodosporidium toruloides) (Wang et al. 2015), has been given the nickname ‘red yeast’ due to the red color conferred by cellular carotenoids. This particular yeast is also oleaginous (oil-producing), with 70% of its cellular dry weight consisting of lipids. Previous spontaneous mutation studies on yeasts have been confined to the two AT-rich model species Saccharomyces cerevisiae (budding yeast, ∼38% GC content) and Schizosaccharomyces pombe (fission yeast, ∼36%), both of which belong to the phylum Ascomycota and are approximately 800 million years diverged from R. toruloides (Lynch et al. 2008; Parfrey et al. 2011; Zhu et al. 2014; Behringer and Hall 2015; Farlow et al. 2015). Recent comparison of spontaneous mutations in fission and budding yeasts showed that the per site mutation rate is fairly constant, while the mutation spectrum differs slightly with respect to G:C→T:A transversions (Behringer and Hall 2015; Farlow et al. 2015). By exploring spontaneous mutation in a GC-rich yeast of another phylum, we will gain a greater understanding of variation in the mutation rate and spectrum of yeasts. Thus, we have conducted a mutation-accumulation experiment on R. toruloides, resolved its mutation rate and spectrum, and contrasted these with the patterns in S. pombe and S. cerevisiae.
Materials and Methods
Strains and Transfers
The haploid R. toruloides strain was ordered from American Type Culture Collection (ATCC, catalogue number: 10788). From a single cell ancestor we created 60 MA lines. Single colonies were transferred by re-streaking onto fresh Yeast Mold agar plates (Becton Dickinson, catalogue number: 271210) at 25 °C every other day. Every month, single colonies from 10 randomly selected MA lines were cut from agar plates and serially diluted to count colony forming units (CFU), and the mean cell divisions passed from a single cell to a colony were estimated by log2(CFU). The total number of cell divisions of each MA line is the product of the grand mean (19.3) of all cell division estimates and the total transfer number for each line. On an average, each MA line experienced 181 transfers. The MA lines transferring took 362 days. We excluded two MA lines (RT6, RT10) from the final mutation analyses due to genome library construction failure.
DNA Extraction, Library Construction, and Genome Sequencing
DNA was extracted from the resulting MA lines using a phenol–chloroform protocol (Sambrook et al. 1989) and libraries constructed using the Nextera® DNA Library Preparation Kit (Illumina). Fragments were size-selected for an insert size of 300 bp and sequenced at the Hubbard Center for Genome Studies, University of New Hampshire on a HiSeq2500 2 × 150 bp rapid run. Across the 58 samples, a median depth of 70× coverage was achieved.
Mutation Analyses
We first trimmed library adaptors of paired-end reads with Trimmomatic 0.32 (Bolger et al. 2014), and then mapped reads to the reference genome of R. toruloides NP11 (GenBank genome accession number: GCA_000320785.2) using BWA mem -ver. 0.7.10 (Li and Durbin 2009). Duplicate reads were removed using picardtools-1.141 and reads mapping around indels were realigned across all 58 MA lines using GATK-3.5 before preforming SNP and indel discovery with standard hard filtering parameters described by GATK Best Practices recommendations (except that we set the Phred-scaled quality score QUAL > 100 and RMS mapping quality MQ > 59 for both variant and non-variant sites) (McKenna et al. 2010; DePristo et al. 2011; Van der Auwera et al. 2013). Base-pair substitutions and small indels were called using the UnifiedGenotyper in GATK. We also required greater than 99% of reads in a line to determine the line-specific consensus nucleotide at a candidate site—1% was set to allow for aberrant reads originated from sequencing errors, not absolutely pure indexes during library construction, or barcodes degeneracy during sequence demultiplexing. In order to confirm the presence of mutations that were identified bioinformatically, 96 base-substitution and 36 indel calls were randomly sampled and PCR primers were designed using BatchPrimer3 (You et al. 2008). We successfully PCR amplified and Sanger sequenced 89 base-substitution mutations and found no false positives (0 false positive rate) and validated 27 small indels out of 28 successfully sequenced candidates, i.e. 3.57% false positive rate for indels. These low error rates showed the high fidelity of our mutation calls. Mutation rate µ was calculated by µ, where m is the total number of mutations across all MA lines, n is the total number of lines, N is the analyzed sites in one line, and T is the number of cell divisions passed in the entire mutation accumulation process of the MA line. 95% Poisson confidence intervals of mutation rates were calculated by using an exact Poisson test in R (poisson.test). Equilibrium GC content under mutation pressure alone was calculated by p = (Lynch 2007), where u is the mutation rate from A/T to G/C nucleotides ; and v is , which is the sum of u and the mutation rate in the other direction; standard error of p was calculated by following Lynch and Walsh (1998):
where and are the sample variance of u and v across all MA lines, respectively, is the covariance of u and v—calculated by the mean value of across all lines and N is the total number of MA lines in the final mutation analysis, i.e. 56. Standard error of the transition/transversion (ts/tv) ratio was also calculated following this formula and 95% confidence intervals of the ratio using ts/tv ± 1.96 × SE.
Mutation rate outliers were determined by being greater than Q3 + 1.5 × IQR or Q1 − 1.5 × IQR, where Q3 is the third quartile, Q1 is the first quartile, IQR—interquartile range—is the distance between the first and the third quartile (Mcgill et al. 1978); mutation enrichment analysis of genes—mutations were limited to coding regions—was adapted from Long et al. (2016): briefly, for each gene, given the expected mutation number of each gene across all MA lines, we estimated the Poisson probability of finding greater than or equal to the observed number of mutations. The expected number of mutations per gene was calculated as the product of the mutation rate per site per generation multiplied by the gene length and the total generations of all lines. The cutoff P value was determined using Bonferroni correction, i.e. P = 0.05/(G − 1), where G equals 8,171—the total number of genes in the genome.
Using the BAM files for all MA lines from the above pipelines, we analyzed the presence of structural variants using the Delly software package (workflow available on GitHub, https://github.com/behrimg/R_toruloides) (Rausch et al. 2012). For structural variants present on the shorter contigs (contigs 49–94), variants were identified visually with the Integrated Genome Viewer (IGV v. 2.3.5) (Thorvaldsdottir et al. 2013). Potential large-scale insertions due to mobile genetic elements were additionally detected using RetroSeq (workflow available on GitHub, https://github.com/behrimg/R_toruloides) and a library of all reported fungal mobile elements, created using RepBase Update v. 20.12 (Jurka et al. 2005; Keane et al. 2013).
ts/tv Ratio of R. toruloides Natural Strains
We retrieved raw sequence reads of two geographically distant natural strains—ATCC10788 and JCM10049—with a genome sequence identity of 97.00% (ncbiblast+/2.2.31). Such high sequence identity could avoid potential species misidentification. For ATCC10788, the raw sequences of the MA ancestor cell of this study were used and DRR032558—NCBI SRA accession number—for the strain JCM10049. We mapped the raw reads to the NP11 reference genome and performed SNP calling using UnifiedGenotyper in GATK-3.5 and applied the same filters. We detected 147,722 bi-allelic sites, and the ts/tv ratio was then calculated using the two alleles of each site.
Results
In order to resolve the mutation rate and spectrum, we sequenced 58 MA lines, which were established from a single-cell ancestor and single-colony transferred every other day for 362 days. Each MA line experienced ∼181 single-cell bottlenecks and on average ∼3,486 cell divisions. The single-cell bottlenecks provide strong genetic drift, which dominates selection during clonal expansion, allowing mutations to accumulate almost neutrally (Bateman 1959; Mukai 1964; Kibota and Lynch 1996).
Base-Substitution Mutation Rate and Spectrum
We detected 789 base-substitution mutations from the 58 successfully sequenced MA lines. Two MA lines (RT17, RT30) were identified as outliers (supplementary fig. S1, Supplementary Material online), which have significantly higher mutation rate than other lines. Because specific genes enriched with mutations could bias the mutation rate and spectrum, we performed a mutation enrichment test for the coding region of each gene and found two genes with significantly higher numbers of mutations than others: gene4182 (coding one argonaute) and gene3886, a hypothetical protein without known significant homology with any other organisms in NCBI databases. Such enrichment could be from inadvertent selection on these genes, though the biological details are not clear due to the incomplete status of the genome and annotation. We thus removed the two outlier MA lines—56 MA lines were used in the final mutation analyses—and the mutations in the two enriched genes from the following analyses. The remaining 714 base-pair substitutions yield a mutation rate of 1.90 ×10−10 (95% Poisson confidence intervals: 1.76–2.04 ×10−10) (table 1 and fig. 1; supplementary tables S1–S5).
Table 1.
Mutation spectrum of R. toruloides. Mutation rates (µ) are in the unit of × 10 −10 per site per cell division; CI is 95% Poisson confidence interval of µ
| Substitutions | Count | µ | CI |
|---|---|---|---|
| Transitions | |||
| G:C→A:T | 288 | 1.23 | 1.10, 1.39 |
| A:T→G:C | 174 | 1.22 | 1.04, 1.41 |
| Transversions | |||
| A:T→T:A | 23 | 0.16 | 0.10, 0.24 |
| G:C→T:A | 75 | 0.32 | 0.25, 0.40 |
| A:T→C:G | 68 | 0.48 | 0.37, 0.60 |
| G:C→C:G | 86 | 0.37 | 0.29, 0.46 |
| Insertions | 33 | 0.09 | 0.06, 0.12 |
| Deletions | 50 | 0.13 | 0.10, 0.18 |
Fig. 1.—
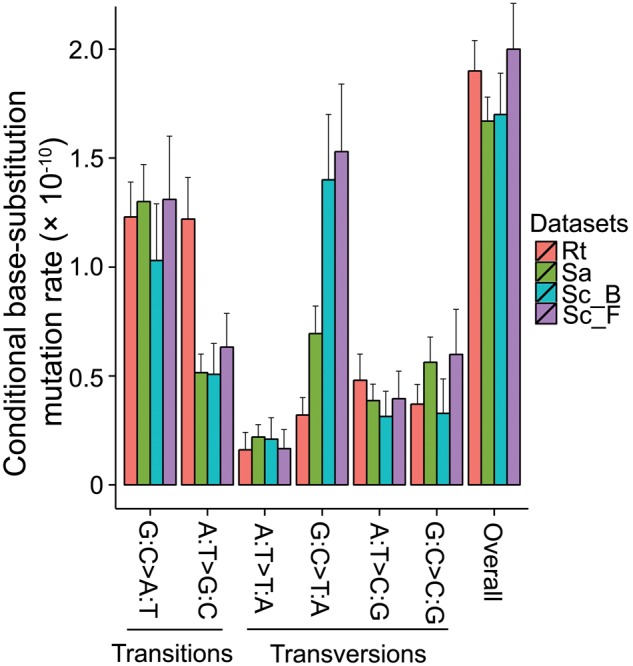
Mutation spectra of R. toruloides vs. fission and budding yeasts. G:C→A:T denotes G→A and C→T mutations, with similar designations for other x axis labels; Rt, Rhodotorula toruloides from this study; Sa, Saccharomyces cerevisiae (Zhu et al. 2014); Sc_B, Schizosaccharomyces pombe from Behringer and Hall (2015); Sc_F, S. pombe from Farlow et al. (2015); error bars are 95% Poisson confidence intervals.
We parsed all possible synonymous and non-synonymous changes in the coding region of the R. toruloides genome and calculated the expected non-synonymous:synonymous mutation ratio to be 2.76 in the absence of selection, which is not significantly different from the observed ratio of 2.53 (χ2 = 0.22, df = 1, P = 0.64). Thus, selection did not significantly bias the accumulation of mutations.
Among these mutations, transitions are the most abundant, with a ts/tv of 1.83 (95% confidence intervals: 1.54, 2.12). This ratio is ∼1.9× higher than that of S. cerevisiae (∼0.95; Zhu et al. 2014) and 2.4× that of S. pombe (∼0.75; Behringer and Hall 2015; Farlow et al. 2015). This ts/tv ratio is also higher than those of all other unicellular eukaryotes with resolved mutation spectra, but similar to the ts/tv ratios of bacteria (table 2).
Table 2.
Transition to transversion ratios (ts/tv) of bacteria and unicellular-eukaryote MA lines with whole genomes sequenced
| Organisms | ts/tv | GC content | References | |
|---|---|---|---|---|
| Unicellular eukaryotes | ||||
| Chlamydomonas reinhardtii | 0.76 | 0.64 | 0.40 | Ness et al. (2015)* |
| Rhodotorula toruloides | 1.83 | 0.62 | 0.99 | This study |
| Saccharomyces cerevisiae | 0.95 | 0.38 | 0.40 | Zhu et al. (2014) |
| Schizosaccharomyces pombe | 0.75a | 0.36 | 0.48a | Behringer and Hall (2015) |
| Farlow et al. (2015) | ||||
| Bacteria | ||||
| Bacillus subtilis | 2.46 | 0.43 | 0.76 | Sung et al. (2015) |
| Burkholderia cenocepacia | 1.21 | 0.67 | 0.86 | Dillon et al. (2015) |
| Deinococcus radiodurans | 1.71 | 0.67 | 1.14 | Long et al. (2015a) |
| Escherichia coli | 1.28 | 0.51 | 0.62 | Lee et al. (2012) |
| Mesoplasma florum | 1.26 | 0.27 | 0.08 | Sung et al. (2012) |
| Mycobacterium smegmatis | 1.48 | 0.66 | 1.43 | Kucukyildirim et al. (2016) |
Note.—The ts/tv ratio is calculated by , where n is the number of MA lines.
Mutations from strains CC2344 and CC2931 are not included due to presence of MA lines with affected DNA repair.
Average value of Behringer and Hall (2015) and Farlow et al. (2015).
We are also curious about if the ts/tv ratio in natural strains of R. toruloides is different from the above MA lines. We thus aligned raw genome sequences of ATCC10788 (the strain used in this study) and JCM10049 to the NP11 reference genome, analyzed SNPs (single nucleotide polymorphisms), and estimated the natural ts/tv ratio at replacement sites to be 1.72. This ratio is not significantly different from that of the MA lines, implying that natural selection may not heavily influence the segregating transitions.
The high ts/tv ratio arises because the mutation spectrum of R. toruloides is strikingly different from the ascomycete yeasts. The A:T→G:C transition mutation rate in R. toruloides is significantly higher than those in the ascomycete yeasts, while the G:C→T:A transversion mutation rate is significantly lower (see details below; fig. 1). Consistent with observations in fission and budding yeasts (Zhu et al. 2014; Behringer and Hall 2015; Farlow et al. 2015), mutations at 5'CpG3' sites are significantly elevated above those at cytosine sites without a flanking 3'G (χ2 = 132.84, P < 2.2 × 10−16). As in these other species, R. toruloides is not known to have a DNA C-5 methyltransferase. Therefore, it is unlikely that the mutational enrichment at 5'CpG3' sites is attributable to methylated cytosines.
The mutation rate in the AT direction (including G:C→A:T transitions and G:C→T:A transversions) is 1.56 × 10−10 (95% Poisson confidence interval: 1.40–1.72 × 10−10), while the mutation rate in the GC direction (A:T→G:C transitions and A:T→C:G transversions) is 1.69 × 10−10 (1.49–1.92 × 10−10). Thus, the mutation rate in this GC-rich organism is slightly biased to G/C, with the equilibrium GC content expected from mutation pressure alone being 52.13% (SE: 2.23%), lower than the 62% genome GC content, implying that evolutionary forces such as selection are operating to increase the genome GC content in this GC-rich organism, at least at some sites.
Unusual A:T→G:C Transition and G:C→T:A Transversion Mutation Rates
In R. toruloides, we find the A:T→G:C transition mutation rate to be 1.22 × 10−10 and the G:C→A:T transition rate to be 1.23 × 10−10 (95% Poisson confidence intervals overlap; table 1); with a ratio of 0.99 for A:T→G:C to G:C→A:T transition mutation rate. When examining unicellular organisms, we observed a significant correlation between GC content and the (A:T→G:C)/(G:C→A:T) transition mutation-rate ratios (Pearson’s correlation test, r = 0.76, P = 0.01; table 2), implying that organisms with higher GC content have a stronger transition bias in the GC direction.
We asked if the relatively high A:T→G:C transition mutation rate could be related to flanking nucleotide contexts. About 98.28% of the A/T sites with A:T→G:C transitions are flanked by at least one G or C nucleotide, and this proportion is significantly different from the proportion of A/T sites with at least one G or C nucleotide in the R. toruloides NP11 genome (92.54%) (χ2 test, P = 0.02). Flanking G:C pair(s) may elevate the mutation rate by orders of magnitude due to their stable anchoring and base-stacking power (Yakovchuk et al. 2006; Lee et al. 2012; Long et al. 2015b; Ness et al. 2015; Sung et al. 2015). Indeed, the transition mutation rate for A/T sites flanked by G or C is ∼3-fold higher than that without such flanking (F-test for equal variance—F = 1.06, P = 0.83; two-sample Student t test P = 0.04; fig. 2). Therefore, the nucleotide context is a significant factor accounting for the unusually high A:T→G:C transition mutation rate in this GC-rich yeast.
Fig. 2.—

Context-dependent mutation rate of R. toruloides. Analytical methods were modified from Long et al. (2015b), AAA denotes a focal A nucleotide in the middle with two flanking As in the same DNA strand, contexts in red have no flanking G or C, and error bars are 95% Poisson confidence intervals.
In contrast to the elevated A:T→G:C transition mutation rate found in R. toruloides, the G:C→T:A transversion mutation rate of 3.22 × 10−11 is much lower than that in the ascomycetous yeasts (∼1/5 of the S. pombe and ∼1/2 of the S. cerevisiae rates; fig. 2). This may be explained by the extremely high concentration of cellular triglycerides (70% of cellular dry weight) and carotenoids in R. toruloides. A large fraction of G:C→T:A transversions are thought to result from DNA oxidative damage (Grollman and Moriya 1993; Foster et al. 2015), and triglycerides and carotenoids are known to reduce oxidative mutation (Kobayashi et al. 2008; Rock 2009), for example, by scavenging reactive oxygen species.
Insertions and Deletions (Indels)
We detected 50 small deletions (total size of 267 bp; < 29 bp) and 33 small insertions (317 bp; < 31 bp) (supplementary tables S1 and S3, Supplementary Material online). These indels yield an indel mutation rate of 2.21 × 10−11 per site per cell division (95% Poisson confidence intervals: 1.76–2.74 × 10−11), or 11.51% of the base-substitution mutation rate. This small-indel rate is ∼4.4× higher than that of S. cerevisiae (Zhu et al. 2014), but the proportion of indels occurring next to simple sequence repeats is much lower than that of the budding yeast (25.30% vs. 61.54%). Such difference may reflect the high divergence in the genome content or the DNA replication and repair machineries of these two yeasts from different phyla.
Across all 56 MA lines, we detected 39 large deletions (> 50 bp) for a total loss of 40,183 bp (supplementary table S4, Supplementary Material online). The ends of chromosomes are particularly susceptible to deletion, most likely due to recombination between subtelomeric regions (McEachern 2008). This is particularly true for the chromosome represented by the scaffold KB722717.1, which experienced a deletion in 7 out of the 56 MA lines, with the nucleotide sequence affected being flanked by CCCTAA repeats in all the seven cases, characteristic of telomeres (Meyne et al. 1989; Guzman and Sanchez 1994; Underwood et al. 1996). The second most common hotspot for deletions is located on the chromosome represented by the scaffold KB722690.1, where 4 out of the 56 MA lines are affected. Again, the sequencing reads mapping around these deletions contain CCCTAA repeats.
We additionally attempted to identify novel mobile-element insertions, but none were detected. This may be caused by the incomplete status of the genome assembly and/or annotation as mobile elements are difficult to assemble, but another possibility is that fungi, and yeast species in particular, are extremely adept at inactivating mobile-genetic elements through various pathways (Hood et al. 2005; Hu et al. 2013). The latter possibility is further supported when comparing the genome size of R. toruloides (∼20 Mb) to that of other members of subphylum Pucciniomycotina such as Puccinia graminis (∼89 Mb) and Melampsora larici-populina (∼101 Mb), which have high numbers of mobile elements, while Sporobolomyces roseus (∼10 Mb), also a ‘red yeast’, has extremely low mobile-element content (Muszewska et al. 2011, 2013).
Discussion
Despite being separated by 800 million years of evolution, the total base-substitution mutation rates of the three yeasts are ∼ 2 × 10−10 per nucleotide site per cell division. This is particularly striking in light of their dramatic biological differences: such as gene structure (e.g., only 5% of S. cerevisiae genes have introns, whereas most genes have introns in S. pombe and R. toruloides), genome size, chromosome number, GC content, etc. (Spingola et al. 1999; Cherry et al. 2012; Wood et al. 2002; Zhu et al. 2012). This shared mutation rate implies limited variation of effective population sizes in yeasts, under the drift-barrier hypothesis (Sung et al. 2012; Farlow et al. 2015), where selection lowers the total genomic mutation rate until any further drop in the mutation rate cannot provide an advantage to overcome the power of genetic drift. Once the organism reaches the drift barrier, although the mutation rate may remain stable, the mutation spectrum may be free to vary (Lynch 2012).
The elevated mutation rate at 5'CpG3' sites remains to be explained. We speculate that different replication error rates of DNA polymerases at these sites or different pre-mutation repair biases may be involved. Given that oxidative damage is a very strong endogenous mutation determinant (Foster et al. 2015), and antioxidant triglycerides and carotenoids are extremely abundant in R. toruloides, the much lower G:C→T:A transversion mutation rate suggests that this oleaginous yeast may provide benefits as an antioxidant nutritional oil candidate. Based on this observation, we predict that organisms with high cellular antioxidants will generally have a lower G:C→T:A transversion mutation rate relative to organisms with low antioxidants or without. However, these differences may also be due to undetermined fundamental differences between ascomycete and basidiomycete yeasts. By investigating more fungal species, especially filamentous ones, it may be possible to determine the mechanisms underlying the variation in the fungal mutation spectrum.
Supplementary Material
Acknowledgments
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Acknowledgments
This research is funded by a Multidisciplinary University Research Initiative award (W911NF-09-1-0444) from the US Army Research Office and a NIH R01 grant (R01GM036827). We thank Thomas G. Doak and Samuel F. Miller for technical assistance and comments on the manuscript.
Literature Cited
- Bateman AJ. 1959. The viability of near-normal irradiated chromosomes. Int J Radiat Biol. 1:170–180. [Google Scholar]
- Behringer MG, Hall DW. 2015. Genome wide estimates of mutation rates and spectrum in Schizosaccharomyces pombe indicate CpG sites are highly mutagenic despite the absence of DNA methylation. G3 (Bethesda) 6:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, et al. 2012. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon MM, Sung W, Lynch M, Cooper VS. 2015. The rate and molecular spectrum of spontaneous mutations in the GC-rich multichromosome genome of Burkholderia cenocepacia. Genetics 200:935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow A, et al. 2015. The spontaneous mutation rate in the fission yeast Schizosaccharomyces pombe. Genetics 201:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Lee H, Popodi E, Townes JP, Tang H. 2015. Determinants of spontaneous mutation in the bacterium Escherichia coli as revealed by whole-genome sequencing. Proc Natl Acad Sci U S A. 112:E5990–E5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grollman AP, Moriya M. 1993. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 9:246–249. [DOI] [PubMed] [Google Scholar]
- Guzman PA, Sanchez JG. 1994. Characterization of telomeric regions from Ustilago maydis. Microbiology 140(Pt 3):551–557. [DOI] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD. 2009. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst. 40:151–172. [Google Scholar]
- Hood ME, Katawczik M, Giraud T. 2005. Repeat-induced point mutation and the population structure of transposable elements in Microbotryum violaceum. Genetics 170:1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Stenlid J, Elfstrand M, Olson A. 2013. Evolution of RNA interference proteins dicer and argonaute in Basidiomycota. Mycologia 105:1489–1498. [DOI] [PubMed] [Google Scholar]
- Jurka J, et al. 2005. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110:462–467. [DOI] [PubMed] [Google Scholar]
- Keane TM, Wong K, Adams DJ. 2013. RetroSeq: transposable element discovery from next-generation sequencing data. Bioinformatics 29:389–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibota T, Lynch M. 1996. Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature 381:694–696. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, et al. 2008. Direct assessments of the antioxidant effects of propofol medium chain triglyceride/long chain triglyceride on the brain of stroke-prone spontaneously hypertensive rats using electron spin resonance spectroscopy. Anesthesiology 109:426–435. [DOI] [PubMed] [Google Scholar]
- Kucukyildirim S, et al. 2016. The rate and spectrum of spontaneous mutations in Mycobacterium smegmatis, a bacterium naturally devoid of the postreplicative mismatch repair pathway. G3 (Bethesda) 6:2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzman CP, Piškur J. 2006. Taxonomy and phylogenetic diversity among the yeasts In: Sunnerhagen P, Piskur J, editors. Comparative genomics: using fungi as models. Berlin: Springer; p. 29–46. [Google Scholar]
- Lee H, Popodi E, Tang H, Foster PL. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A. 109:E2774–E2783. 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, et al. 2015a. Background mutational features of the radiation-resistant bacterium Deinococcus radiodurans. Mol Biol Evol. 32:2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, et al. 2015b. Mutation rate, spectrum, topology, and context-dependency in the DNA mismatch repair (MMR) deficient Pseudomonas fluorescens ATCC948. Genome Biol Evol. 7:262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, et al. 2016. Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc Natl Acad Sci U S A. 113:E2498–E2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2012. Evolutionary layering and the limits to cellular perfection. Proc Natl Acad Sci U S A. 109:18851–18856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2007. The origins of genome architecture. Sunderland, MA: Sinauer Associates, Inc. [Google Scholar]
- Lynch M, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 105:9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Walsh B. 1998. Genetics and analysis of quantitative traits. Sunderland, MA: Sinauer; p. 818. [Google Scholar]
- McEachern MJ. 2008. Telomeres: guardians of genomic integrity or double agents of evolution? In: Nosek J, Tomaska L, editors. Origin and evolution of telomeres. Austin, TX: Landes Bioscience; p. 100–113. [Google Scholar]
- Mcgill R, Tukey JW, Larsen WA. 1978. Variations of box plots. Am Stat. 32:12–16. [Google Scholar]
- McKenna A, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyne J, Ratliff RL, Moyzis RK. 1989. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci U S A. 86:7049–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T. 1964. The genetic structure of natural populations of Drosophila melanogaster. I. Spontaneous mutation rate of polygenes controlling viability. Genetics 50:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muszewska A, Hoffman-Sommer M, Grynberg M. 2011. LTR retrotransposons in fungi. PLoS One 6:e29425.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muszewska A, Steczkiewicz K, Ginalski K. 2013. DIRS and Ngaro retrotransposons in Fungi. PLoS One 8:e76319.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Vasanthakrishnan RB, Colegrave N, Keightley PD. 2015. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii. Genome Res. 25:1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Lahr DJ, Knoll AH, Katz LA. 2011. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc Natl Acad Sci U S A. 108:13624–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch T, et al. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28:i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock CL. 2009. Carotenoids and cancer. Basel, Switzerland: Birkhäuser. [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Spingola M, Grate L, Haussler D, Ares M., Jr. 1999. Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA 5:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, et al. 2015. Asymmetric context-dependent mutation patterns revealed through mutation-accumulation experiments. Mol Biol Evol. 32:1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M. 2012. Drift-barrier hypothesis and mutation rate evolution. Proc Natl Acad Sci U S A. 109:18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood AP, Louis EJ, Borts RH, Stringer JR, Wakefield AE. 1996. Pneumocystis carinii telomere repeats are composed of TTAGGG and the subtelomeric sequence contains a gene encoding the major surface glycoprotein. Mol Microbiol. 19:273–281. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, et al. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 11:11.10.11–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QM, et al. 2015. Phylogenetic classification of yeasts and related taxa within Pucciniomycotina. Stud Mycol. 81:149–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood V, et al. 2002. The genome sequence of Schizosaccharomyces pombe. Nature 415:871–880. [DOI] [PubMed] [Google Scholar]
- Yakovchuk P, Protozanova E, Frank-Kamenetskii MD. 2006. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 34:564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You FM, et al. 2008. BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 9:253.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 111:E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, et al. 2012. A multi-omic map of the lipid-producing yeast Rhodosporidium toruloides. Nat Commun. 3:1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.