

Abstract
Background and Purpose
Mirabegron has been classified as a β3‐adrenoceptor agonist approved for overactive bladder syndrome. We investigated possible cardiac effects of mirabegron in the absence or presence of β‐adrenoceptor subtype antagonists. In view of its phenylethanolamine structure, we investigated whether mirabegron has indirect sympathomimetic activity by using neuronal uptake blockers.
Experimental Approach
Right atrial trabeculae, from non‐failing hearts, were paced and contractile force measured at 37°C. Single concentrations of mirabegron were added in the absence or presence of the phosphodiesterase inhibitor 3‐isobutyl‐1‐methylxanthine (IBMX), β3 (L‐748,337), β1 (CGP 20712A), β2 (ICI 118,551) ‐adrenoceptor antagonists, neuronal uptake inhibitors desipramine or phenoxybenzamine.
Key Results
Mirabegron significantly increased contractile force in human right atrium (1 μM, 7.6 ± 2.6%, n = 7; 10 μM, 10.2 ± 1.5%, n = 22 compared with (−)‐isoprenaline P < 0.05). In the presence of IBMX, mirabegron (10 μM) caused a greater contraction. L‐748,337 (100 nM) had no effect on the increase in contractile force caused by mirabegron (10 μM). In contrast, mirabegron (10 μM) reduced contractile force in the presence of CGP 20712A, which was not affected by L‐748,337 (100 nM) or ICI 118,551 (50 nM). Mirabegron (10 μM) also reduced contractile force in the presence of desipramine or phenoxybenzamine.
Conclusions and Implications
Mirabegron increases human atrial force through β1‐ but not β3‐adrenoceptors. Desipramine and phenoxybenzamine block neuronal uptake and conceivably prevent mirabegron from releasing noradrenaline. A non‐specific cardiodepressant effect is not mediated through β3 (or β2)‐adrenoceptors, consistent with lack of β3‐adrenoceptor function on human atrial contractility.
Abbreviations
- CGP 20712A
(2‐hydroxy‐5‐[2‐[[2‐hydroxy‐3‐[4‐[1‐methyl‐4‐(trifluorometyl)‐1H–imidazol‐2‐yl]phenoxy]propyl]amino]ethoxy]‐benzamide)
- IBMX
3‐isobutyl‐1‐methylxanthine
- ICI 118,551
(1‐[2,3‐dihydro‐7‐methyl‐1H‐inden‐4‐yl] oxy‐3‐[(1‐methylethyl)amino‐2‐butanol)]
- L‐748,337
N‐(3‐[3‐[2‐(4‐benzenesulphonylaminophenyl)ethylamino]‐2‐hydroxypropoxy]benzyl) acetamide
- NET
noradrenaline transporter
Introduction
Mirabegron has been classified as a β3‐adrenoceptor agonist (Takasu et al., 2007; Figure 1). It induces β3‐adrenoceptor‐mediated relaxation of detrusor smooth muscle and was approved as a ‘first‐in‐class’ medicine for the management of overactive bladder syndrome (Chapple et al., 2014). In view of its ability to activate β3‐adrenoceptors, we used it to determine the contribution of β3‐adrenoceptors to the contractility of human atria.
Figure 1.
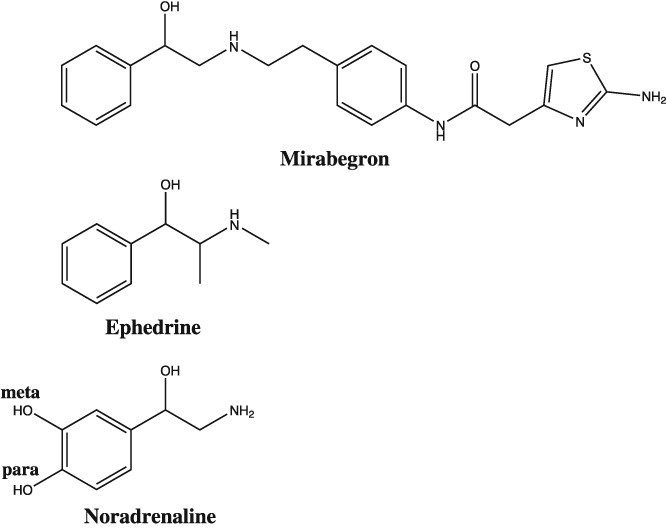
Chemical structures of mirabegron, ephedrine and noradrenaline, highlighting the absence (mirabegron, ephedrine) and presence (noradrenaline) of meta‐ and para‐hydroxyl groups on the phenyl group. Ephedrine causes β‐adrenoceptor‐mediated cardiostimulant effects in the heart by uptake into sympathetic nerve terminals and release of noradrenaline. The presence of meta‐ and para‐hydroxyl groups on noradrenaline is required for direct activation of β‐adrenoceptors.
In human isolated atrium, Skeberdis et al. (2008) proposed that activation of β3‐adrenoceptors caused an increase in contractile force and increased L‐type Ca2+ current in right atrial myocytes. However, Christ et al. (2011) demonstrated that the inotropic effects of the agonists used by Skeberdis et al. (2008) on human atrial trabeculae were not mediated through β3‐adrenoceptors. To further understand the cardiac effects of human atrial β3‐adrenoceptors, we investigated mirabegron directly on human atrial myocardium. Firstly, we studied the effects of mirabegron in the absence and presence of β‐adrenoceptor subtype antagonists. We utilized the PDE inhibitor 3‐isobutyl‐1‐methylxanthine (IBMX), a non‐selective inhibitor of PDE, to potentiate putative contractile effects of mirabegron, by limiting the metabolism of cAMP, as rationalized by Skeberdis et al. (2008) for β3‐adrenoceptors.
Interestingly, mirabegron has a phenylethanolamine structure, lacking meta‐ and para‐hydroxyl groups (Figure 1). Ephedrine is similarly a phenylethanolamine (Figure 1), which causes cardiostimulant effects by being taken up into sympathetic nerves and releasing noradrenaline (Waldeck and Widmark, 1985; Bönisch and Trendelenburg, 1988). This prompted us to investigate whether mirabegron is an indirect sympathomimetic amine. We therefore investigated the effects of mirabegron in the absence or presence of the neuronal blockers desipramine or phenoxybenzamine.
Methods
Human tissues
Right atrial appendages were obtained from patients undergoing coronary artery bypass surgery, aortic valve surgery, a combination of both or myomectomy surgery at The Prince Charles Hospital, Chermside. Patients provided written informed consent prior to surgery. Prospective patients were invited to participate in the study by the authors (W.M. and P.M.) who explained ethical considerations and provided patient information and revocation documents. The study, process for obtaining written informed consent, patient information, consent and revocation documents were approved by the Metro North Hospital and Health Services Human Ethics Committee, approval reference HREC/12/QPCH/275. Patient characteristics are outlined in Table 1.
Table 1.
Patient characteristics
| n | 64 |
| Gender, M/F | 57/7 |
| Age, years (mean±SD) | 60 ± 8 |
| CAD | 54 |
| AVD | 6 |
| CAD + AVD | 3 |
| HOCM | 1 |
| LVEF, % (mean±SEM) | 55 ± 10% |
| Sinus rhythm | 61 |
| Atrial fibrillation | 1 |
| Atrial rhythm information not available | 2 |
| Cardiovascular medication, n | |
| ACE inhibitors | 29 |
| AT1‐receptor blockers | 7 |
| β‐blockers | 47 |
| Atenolol | 4 |
| Metoprolol | 33 |
| Bisoprolol | 6 |
| Carvedilol | 2 |
| Propranolol | 2 |
| Calcium channel blockers | 22 |
| Diuretics | 4 |
| Lipid‐lowering drugs | 48 |
| Antidiabetics | 10 |
| Antidepressants | 4 |
CAD, coronary artery disease; AVD, aortic valve disease; HOCM, hypertrophic obstructive cardiomyopathy; LVEF, left ventricular ejection fraction; ACE, angiotensin‐converting enzyme.
Contractility studies
After excision, human right atrial appendages were immediately placed in modified oxygenated ice‐cold Krebs solution containing (mM): Na+ 125, K+ 5, Ca2+ 2.25, Mg2+ 0.5, Cl− 98.5, SO4 2−0.5, HCO3 −34, HPO4 2−1, EDTA 0.04, and equilibrated with 95% O2/5% CO2. Trabeculae from right atrial appendages were dissected and set up, on occasion, in pairs to contract at 1 Hz in an apparatus with a 50 mL tissue bath in the Krebs solution, supplemented with (mM): Na+ 15, fumarate 5, pyruvate 5, L‐glutamate 5, glucose 10 at 37°C, as described (Gille et al., 1985; Molenaar et al., 2007). The tissues were attached to Swema SG4‐45 strain gauge transducers, electrically stimulated with square‐wave pulses of 5 ms duration and just over threshold voltage and force recorded on Labchart 8 for Windows (ADInstruments, Bella Vista, Australia). After determination of a length–tension curve, the length of each atrial trabeculum was set to obtain 50% of the resting tension associated with maximum developed force.
Experimental protocols
Mirabegron was added to the tissue bath to obtain single concentrations of 0.1, 1 or 10 μM after tissues had been preincubated with or without the PDE inhibitor IBMX (10 μM), β3‐adrenoceptor antagonist L‐748,337 (100 nM; Candelore et al., 1999; Wuest et al., 2009), β1‐adrenoceptor antagonist CGP 20712A (300 nM; Kaumann, 1986; Buxton et al., 1987), β2‐adrenoceptor antagonist ICI 118,551 (50 nM; Lemoine et al., 1985; Buxton et al., 1987; Kaumann and Lemoine, 1987; Hall et al., 1990), neuronal uptake inhibitors desipramine (1 μM; Iversen, 1965) or phenoxybenzamine (5 μM; Gille et al., 1985; Iversen, 1965). Tissues were incubated for 20 min with IBMX and 60 min with L‐748,337, CGP 20712A, ICI 118,551 or desipramine before addition of mirabegron. Tissues were incubated for 90 min with phenoxybenzamine, which was then washed from the tissue bath (Gille et al., 1985) before mirabegron (10 μM) was added. Experiments were concluded by the addition of (−)‐isoprenaline (200 μM) to saturate β1‐, β2‐ and β3‐adrenoceptors (Hall et al., 1990; Gauthier et al., 1996).
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Patients presenting for heart surgery (coronary artery bypass surgery, aortic valve surgery or combination of both surgeries, myomectomy surgery) provided the samples of atrial tissue used. Group size numbers are indicated as numbers of (trabeculae/patients). Values of n shown refer to the number of patients (not replicates) for statistical analysis and n > 5 for statistical analysis. During the course of the study, two patients chronically treated with carvedilol were used, and the results presented as preliminary data. Trabeculae/patient numbers were not always the same because right atrial appendages are inconsistent in terms of size and numbers of trabeculae. If an experiment had four different conditions, as in the experiment shown in Figure 3, the priority was to allocate one trabeculae for each condition. However, some right atrial appendages did not have four trabeculae, and therefore, it was not always possible to complete the four conditions from tissues obtained from one patient. If there were >4 trabeculae from a patient, as for the example shown in Figure 3, a replicate was included. The availability of patients, numbers of trabeculae and other variables such as surgeon were completely out of the control of the researchers and therefore may be considered to contribute to randomization. The experimenter and analyst (W.M. and P.M.) were not blinded for practical reasons. The experimenter was required to carry out all parts of each experiment including dissection and preparation of trabeculae and addition of investigational drugs. Every experimental intervention was digitally recorded on the trace in real time. At the conclusion of the experiment, data were analysed by the same experimenter and maintained in one Excel file (W.M. and P.M.). Data were normalized to the reference agonist (−)‐isoprenaline (200 μM) and expressed as percentage for increases in contractility (Hall et al., 1990) to overcome variability in the magnitude of contractility (absolute values) due to differences in the size and other characteristics of trabeculae. Data were normalized to basal contractility for decreases in contractility and expressed as a percentage and not the (−)‐isoprenaline response since it was considered more meaningful to express cardiodepression against basal contractility. Increases or decreases of force caused by mirabegron and increases by (−)‐isoprenaline were measured as absolute values in units of mN prior to normalization. Only the increases or decreases in force were analysed statistically and not baseline values. Absolute values of basal and maximal forces for trabeculae of each group are given. Reductions in force of contraction were measured at 30 min (or minimum contractile force before 30 min, e.g. Figure 3). When comparing groups, the level of probability (P) was set at 0.05. Data were compared by one‐way ANOVA with post hoc tests if P < 0.05. Non‐paired t‐tests were used in experiments with two groups. Statistical analysis was carried out with the computer program GraphPad Instat® Version 3.10.
Figure 3.
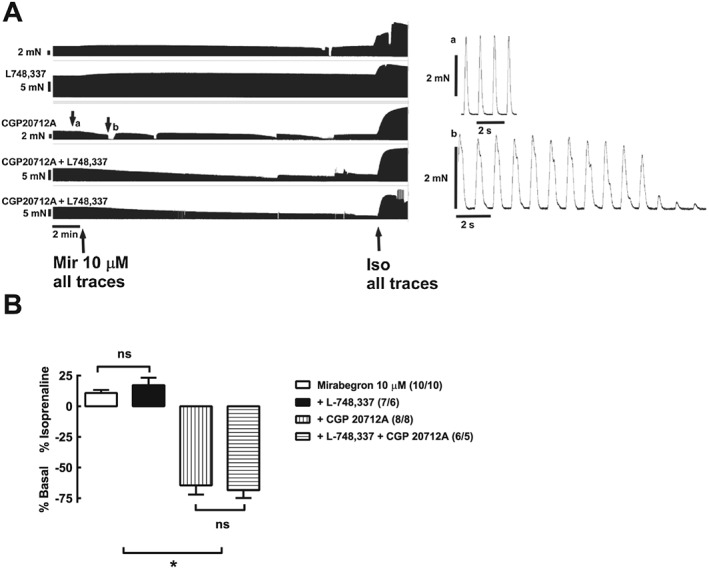
Critical requirement of β1‐adrenoceptors for cardiostimulant effects of mirabegron in human right atrium. The β3‐adrenoceptor antagonist L‐748,337 did not affect the mirabegron‐induced increase in contractile force. Representative recordings are shown of the effects of mirabegron (10 μM), with or without L748,337 (100 nM), CGP 20712A (300 nM) or CGP 20712A (300 nM) + L748,337 (100 nM), in trabeculae from a 67 year male patient with coronary artery disease. The right‐hand panels show expanded timescale recordings of individual contractions before (a) and after (b) the addition of mirabegron in the presence of CGP 20712A. Bar graph shows summary of data; numbers in brackets are trabeculae/patients. P < 0.05, *significantly different as indicated; ns, not significant; one‐way ANOVA, Please notice evidence for ‘contractile dysfunction’ shown after (b), consisting of sudden long‐lasting interruptions of contraction, prolonged contractions compared with contractions before CGP 20712A at subpanel (a) (please compare the fast‐speed right‐side panels a and b), fade of contraction or arrhythmic contractions.
Materials
Mirabegron was purchased from Sapphire Bioscience (Redfern, Australia); L‐748,337 from Tocris (Bristol, UK); and IBMX, (−)‐isoprenaline, desipramine, ICI 118,551 and CGP 20712A were from Sigma Aldrich (Castle Hill, Australia). Phenoxybenzamine was a gift from GlaxoSmithKline (Stevenage, UK). Mirabegron and desipramine solutions were made fresh each day. Mirabegron (0.1 M) stock was prepared in DMSO. The maximal quantity of DMSO added to the 50 mL tissue bath was 5 μL (1–10 000 dilution), which did not modify atrial contractions (data not shown).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Cardiostimulant effects of mirabegron
Mirabegron (1 and 10 μM) caused significant concentration‐dependent increases in contractile force in human right atrium (1 μM, 7.6 ± 2.6 %, n = 7; 10 μM, 10.2 ± 1.5%, n = 22, Figure 2). At a lower concentration (0.1 μM), mirabegron did not affect contraction (n = 8).
Figure 2.
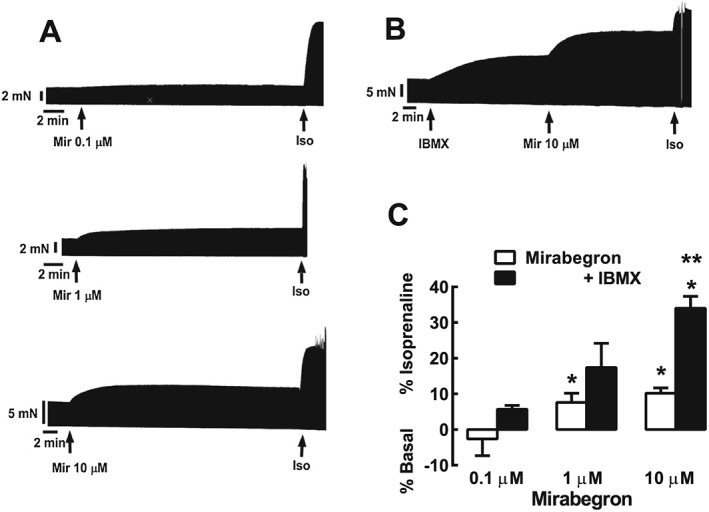
Cardiostimulant effects of mirabegron in human right atrium and potentiation by IBMX. (A) Representative recordings showing the effect of mirabegron (Mir) 0.1, 1 and 10 μM, followed by (−)‐isoprenaline (Iso) 200 μM in three separate male patients aged 65, 66 and 70 years respectively with coronary artery disease. Arrows indicate addition of mirabegron and (−)‐isoprenaline. (B) Representative recording of the effect of mirabegron 10 μM in the presence of the PDE inhibitor IBMX in a female patient aged 58 years with coronary artery disease. (C) Summary of data indicating that mirabegron (1 and 10 μM) significantly increased the contractile force (mirabegron 0.1 μM, 6/6; 1 μM, 7/6 and 10 μM, 22/22, as trabeculae/patients). Increases in contractile force are represented as a percentage of the response to 200 μM (−)‐isoprenaline (% Isoprenaline). Reductions in contractile force are represented as a percentage of basal force (% Basal). *P < 0.05. significant increase in force of contraction. The absolute mean values for contractile force (as mN) before and after mirabegron are given in Table 2. Responses to mirabegron in the presence of IBMX (0.1 μM, 6/5; 1 μM, 7/6; 10 μM, 15/13, as trabeculae/patients) showed that IBMX potentiated responses to mirabegron (10 μM). **P < 0.05, significant effect of IBMX.
Table 2.
Effect of mirabegron (0.1, 1 and 10 μM) on force of contraction (mN) in human right atrium. Data correspond to Figure 2A. n is number of trabeculae
| Force (mN) | |||
|---|---|---|---|
| Mirabegron concentration (μM) | Pre‐mirabegron | Post‐mirabegron | n |
| 0.1 | 8.9 ± 2.3 | 8.7 ± 2.2 | 6 |
| 1 | 8.5 ± 0.7 | 10.3 ± 1.2 * | 7 |
| 10 | 8.4 ± 1.1 | 10.0 ± 1.1 * | 22 |
Values shown are means ± SEM.
P < 0.05, significantly different from pre‐mirabegron; paired t‐test.
Enhancement of cardiostimulant effects of mirabegron by IBMX
IBMX (10 μM) caused a 32± 2% (n = 19, % (−)‐isoprenaline effect) increase in contractile force. In the presence of IBMX, mirabegron (0.1–10 μM) caused a further increase in force (Figure 2). The magnitude of the increase in contractile force caused by mirabegron (10 μM) was greater in the presence of IBMX than in its absence (Figure 2).
Mirabegron effects in the presence of β1‐, β2‐ and β3‐adrenoceptor antagonists
The β3‐adrenoceptor antagonist L‐748,337 (100 nM) had little effect on the increase in contractile force caused by mirabegron (10 μM) (Figure 3). In contrast, mirabegron (10 μM) caused a marked reduction in contractile force in the presence of the β1‐adrenoceptor antagonist CGP 20712A (300 nM), which was not affected by concomitant presence of L‐748,337 (Figure 3) or the β2‐adrenoceptor antagonist ICI 118,551 (Figure 4). In the presence of CGP 20712A, mirabegron often caused ‘contractile dysfunction’ (Figures 3 and 4), which could be overcome either by increasing the stimulus voltage or by (−)‐isoprenaline (Figures 3 and 4). L‐748,337 (100 nM) had no effect on ‘contractile dysfunction’ caused by mirabegron (10 μM) in the presence of CGP 20712A (Figure 3).
Figure 4.
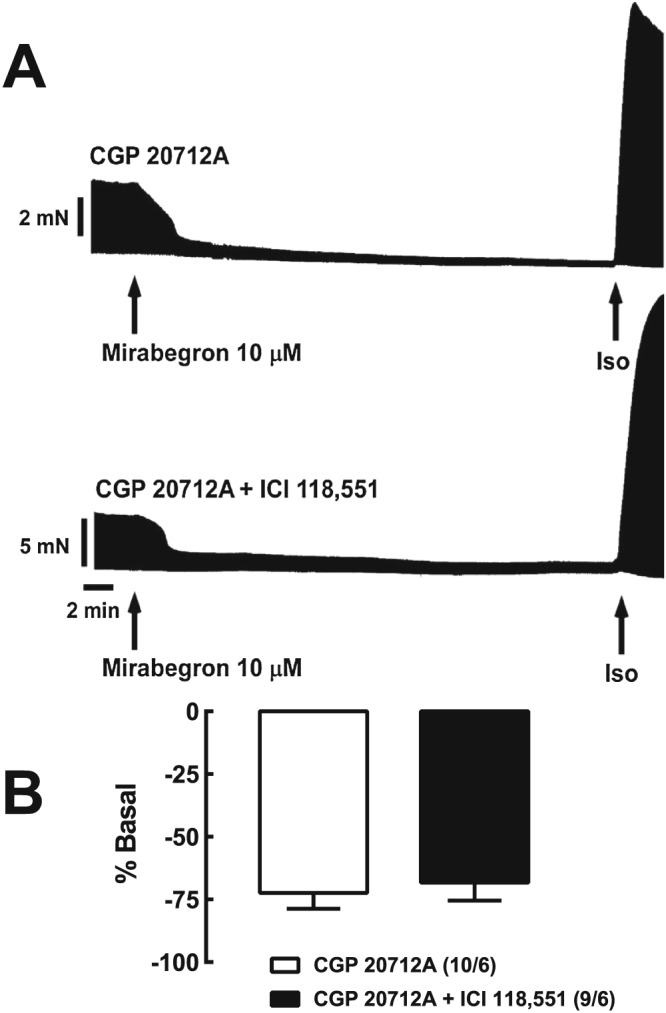
Lack of effect of the β2‐adrenoceptor antagonist ICI 118,551 on the cardiodepressant effects of mirabegron in the presence of the β1‐adrenoceptor antagonist CGP 20712A. (A) Representative recordings of mirabegron (10 μM) in the presence of CGP 20712A 300 nM (top recording) and CGP 20712A 300 nM + ICI 118,551 50 nM (bottom recording) in trabeculae from a 55 year female patient with aortic valve disease. (B) Bar graph shows summary of data; numbers in brackets are trabeculae/patients.
During the course of these studies, we obtained atrial tissues from two patients with coronary artery disease who were chronically treated with the β‐blocker carvedilol until heart surgery. Two trabeculae were obtained from each patient, equilibrated with IBMX (10 μM) and then mirabegron (10 μM). Mirabegron did not cause an increase in contractile force but, in contrast, reduced force in each trabeculum (individual values −24, −14, −16, − 90% of basal force). As carvedilol persistently blocks both β1‐ and β2‐adrenoceptors (Molenaar et al., 2006), these data also suggest that β1‐ and β2‐adrenoceptors are not responsible for the cardiodepressive effects of mirabegron.
Mirabegron decreases contractile force in the presence of desipramine or phenoxybenzamine
We considered further the mechanism through which mirabegron causes increases in contractile force through the β1‐adrenoceptor. The absence of meta‐ and para‐hydroxyl groups on the phenyl ring of mirabegron (Figure 1) suggests that this compound would not be an effective agonist at β1‐adrenoceptors. Rather, this β3‐adrenoceptor agonist may at least in part cause increases in force through an indirect sympathomimetic effect, as previously observed in isolated heart preparations with other sympathomimetic amines that also lack meta‐ and para‐hydroxyl groups (Pesce and Adler‐Graschinsky, 1983; Waldeck and Widmark, 1985). Therefore, we tested the effects of mirabegron (10 μM) in the presence of desipramine or phenoxybenzamine (after washout), compounds reported to inhibit noradrenaline uptake into sympathetic nerves (Iversen, 1965). Mirabegron (10 μM) reduced contractile force and caused ‘contractile dysfunction’ in the presence of desipramine or after phenoxybenzamine (5 μM), followed by washout of unbound phenoxybenzamine (Figure 5).
Figure 5.
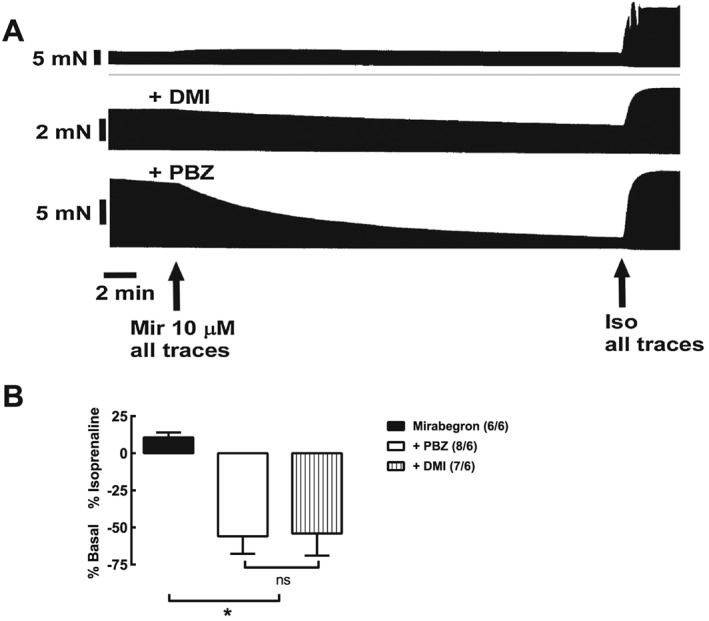
Possible dependance on neuronal uptake for cardiostimulant effects of mirabegron (Mir) in human right atrium. Reduction of contractile force of mirabegron by desipramine (DMI) or phenoxybenzamine (PBZ). Shown are recordings of the effects of mirabegron 10 μM in the absence or presence of DMI (1 μM) or phenoxybenzamine (5 μM; 90 min incubation followed by washout) in trabeculae from a 42 year old patient with aortic valve disease. Bar graphs show summary of data; numbers in brackets are trabeculae/patients. *P < 0.05, significantly different; ns, not significant as indicated; one‐way ANOVA,
Discussion
We investigated the effects of mirabegron on contractility in isolated human right atrium to further clarify the role of β3‐adrenoceptors. It was reported that β3‐adrenoceptors mediate increases in contractile force in human right atrium (Skeberdis et al., 2008) but not by Christ et al. (2011). The availability of mirabegron, which causes β3‐adrenoceptor meditated relaxation of human detrusor muscle, provided the opportunity to re‐address the question whether β3‐adrenoceptors modify contractility in human atrium. We found that mirabegron at concentrations of 1 and 10 μM caused small increases in contractile force in human right atrium. In the presence of the β1‐adrenoceptor selective antagonist CGP 20712A, desipramine or phenoxybenzamine (after washout), mirabegron caused a large reduction in contractile force and ‘contractile dysfunction’. Both cardiostimulant and cardiodepressant effects were not affected by the β3‐adrenoceptor antagonist L‐748,337 (100 nM), a concentration estimated to occupy 83% of β3‐adrenoceptors in human detrusor muscle (Wuest et al., 2009). Taken together, these findings are compatible with with a lack of functional β3‐adrenoceptors affecting contractile force in human atrium, in agreement with Christ et al. (2011), but suggest that mirabegron may increase contractile force through release of noradrenaline.
Cardiostimulant effect of mirabegron
A potency (pEC50) of mirabegron for relaxation of human‐isolated detrusor muscle of 6.1 (Takasu et al., 2007) and 6.2 (Svalǿ et al., 2013) has been reported. To determine whether mirabegron has direct effects on human heart, we therefore exposed isolated human right atrium to concentrations of 0.1, 1 and 10 μM. Concentration‐dependent increases in atrial contractile force were observed at 1 and 10 μM. The positive inotropic effects at 10 μM were, however, unaffected by the β3‐adrenoceptor antagonist L‐748,337 100 nM. The results of this study, which was carried out at 37°C, are consistent with the study of Christ et al. (2011), who also did not observe β3‐adrenoceptor‐mediated increases in human atrial contractile force at both 37 and 24°C.
The increase in contractile force of mirabegron 10 μM was completely prevented by the β1‐adrenoceptor antagonist CGP 20712A. The concentration of CGP 20712A used in the present study, 300 nM, occupies 99.7% β1‐adrenoceptors (Kaumann, 1986; Buxton et al., 1987; Kaumann and Lemoine, 1987), 9% β2‐adrenoceptors (Buxton et al., 1987; Kaumann and Lemoine, 1987) but only 0.015% β3‐adrenoceptors (Baker, 2010). These data indicate a cardiostimulant mechanism mediated through β1‐adrenoceptors but not β3‐adrenoceptors. Increases in contractile force were enhanced by the non‐selective PDE inhibitor IBMX, consistent with a β1‐adrenoceptor‐Gαs‐protein‐cAMP mechanism.
Cardiodepressant effects of mirabegron
Mirabegron caused a reduction in contractile force in the presence of the β1‐adrenoceptor antagonist CGP 20712A. Furthermore, the initial voltage applied across the electrodes to maintain contractility often became insufficient to maintain normal contractions (compare Figure 3A, B), possibly due to conduction block. An increase in voltage or the addition of (−)‐isoprenaline overcame ‘contractile dysfunction’ and increased force (Figures 3 and 4). The reduction in contractile force in the presence of CGP 20712A was not prevented by the β3‐adrenoceptor antagonist L‐748,337 or β2‐adrenoceptor antagonist ICI 118,551 and is therefore not likely to be due to a β3‐ or β2‐adrenoceptor mechanism.
Mirabegron (in the presence of IBMX) also caused a reduction in contractile force in the right atrium of two patients with evidence of heart failure that were chronically treated with carvedilol, in preliminary studies. Previously, we showed that chronic carvedilol treatment of patients with non‐terminal heart failure reduced the inotropic sensitivity of atrial trabeculae to noradrenaline at β1‐adenoceptors and adrenaline at β2‐adrenoceptors ~ 6‐fold and ~90‐fold, respectively, but the potency of 5HT at 5HT4 receptors was not reduced (Molenaar et al., 2006). It is likely that the persistent blockade of β‐adrenoceptors in right atrial trabeculae from patients chronically treated with carvedilol (Molenaar et al., 2006) prevented the observation of cardiostimulant effects but not cardiodepressant effects of mirabegron.
The mechanism of contractile dysfunction revealed in the presence of β1‐adrenoceptor antagonism, desipramine or phenoxybenzamine was not identified in this study. However, the observation that contractility was restored by increasing stimulus voltage or by administering a saturating concentration of isoprenaline (200 μM) provides a starting point for understanding cardiodepressant mechanisms. Excitation‐contraction coupling is complex, depending on precisely coordinated ion fluxes across sarcolemmal and intracellular membranes. Over 120 ion channels were identified and measured in human right atrium, each of which contribute to the action potential and therefore to the intracellular, time–dependent, concentrations of Ca2+ (Chandler et al., 2009). Systolic contractile force is Ca2+‐dependent and therefore changes to ion channel fluxes that cause decreased intracellular Ca2+ concentrations reduce systolic contractile force. Mirabegron may have membrane‐stabilizing properties. Membrane‐stabilizing properties of β‐adrenoceptor antagonists (and partial agonists) have been reported previously in human (Nayler et al., 1969) and animal cardiac tissues Nayler et al., 1969; Van Zwieten, 1969; Langslet, 1970; Fitzgerald and O'Donnell, 1971; Fitzgerald et al., 1972). ‘Membrane‐stabilizing properties’ are used to describe ‘local anaesthetic’ and ‘quinidine‐like’ effects of drugs (Davis, 1970). For example, propranolol, at concentrations as low as 0.3 μM [but higher than those required to block β‐adrenoceptors (Nayler et al., 1969; Gille et al., 1985)], caused negative inotropic responses in isolated human right atrial and ventricular preparations (Nayler et al., 1969). Similarly, high (micromolar) concentrations of propranolol and other β‐blockers caused cardiodepressant effects including negative inotropic effects (Nayler et al., 1969; van Zwieten, 1969; Langslet, 1970) and increased the stimulus threshold voltage for contraction (Langslet, 1970) in animal‐isolated cardiac preparations, effects not dissimilar to that observed for mirabegron. Additionally, Langslet (1970) observed ECG changes characterized by increased PQ, QRS and QT intervals in rat isolated perfused hearts, indicating changes in action potential generation and propagation. Activation of β‐adrenoceptors with isoprenaline increases I CaL in human right atrium (Christ et al., 2011) and was sufficient to overcome cardiodepression caused by mirabegron.
Desipramine and phenoxybenzamine prevent the cardiostimulant effects of mirabegron
Mirabegron is a phenylethanolamine derivative (Figure 1). A feature of its structure is the absence of meta‐ and para‐hydroxyl groups. An informative comparison can be made with other compounds with known sympathomimetic pharmacology that lack these groups, such as amphetamine, tyramine, mephentermine, ephedrine and β‐phenylethylamine. These compounds cause cardiostimulant effects in isolated cardiac tisssues, which are reduced or completely abolished in tissues taken from animals pretreated with reserpine (Pesce and Adler‐Graschinsky, 1983; Waldeck and Widmark, 1985), which depletes cardiac stores of noradrenaline (Häggendal and Lindqvist, 1963). Furthermore, pretreatment of tissues with cocaine, a sympathetic neuronal uptake inhibitor (Bönisch and Trendelenburg, 1988), reduces the potency of amphetamine and β‐phenylethylamine (Pesce and Adler‐Graschinsky, 1983). The action of compounds like ephedrine (Waldeck and Widmark, 1985) and amphetamine are mediated through release of noradrenaline. It was concluded that indirectly acting sympathomimetic amines are substrates of neuronal uptake and release vesicular noradrenaline (Bönisch and Trendelenburg, 1988). To determine whether the cardiostimulant effect of mirabegron is possibly mediated through an indirect mechanism, we investigated the effects of mirabegron in the absence and presence of desipramine or phenoxybenzamine, which inhibit neuronal uptake (Iversen, 1965).
Mirabegron reduced contractile force in the presence of desipramine or phenoxybenzamine. Both desipramine and phenoxybenzamine have mixed pharmacological properties. Desipramine (1 μM) causes 92% inhibition of noradrenaline uptake into sympathetic nerve terminals (Iversen, 1965) and has affinity for α1‐, α2A‐adrenoceptors and muscarinic receptors [α2A‐ Ki 5.0 μM determined from radioligand binding experiments, Cottingham et al., 2011; IC50 values from radioligand binding experiments (α1‐adrenceptors 0.57 μM, α2A‐adrenoceptors 9.2 μM, muscarinic receptors 0.6 μM, Wong et al., 1982)]. Phenoxybenzamine belongs to a class of drugs called β‐haloalkylamines, which include dibenamine that cause irreversible blockade of receptors through alkylation through a highly reactive ethyleniminium intermediate (Khan and Furchgott, 1982). They are able to block α‐adrenoceptors, muscarinic, histamine and serotonin receptors for several hours after washout (Khan and Furchgott, 1982) but not β1‐ or β2‐adrenoceptors (Gille et al., 1985). Incubation and washout of human right atrium with phenoxybenzamine causes a ~1.2 log increase in potency of (−)‐noradrenaline (Gille et al., 1985).
In view of the mixed pharmacology of desipramine and phenoxybenzamine, the prevention of cardiostimulant effects of mirabegron in their presence could be due to a number of mechanisms. It is possible that the cardiostimulant effect of mirabegron involves its uptake into sympathetic nerve terminals, which is no longer possible in the presence of desipramine or phenoxybenzamine‐treated tissues (Figure 6). However, both desipramine and phenoxybenzamine also block α‐adrenoceptors, and therefore, we cannot rule out the possibility that mirabegron blocks prejunctional α‐adrenoceptors so that they have reduced autoinhibitory capacity on the release of noradrenaline from sympathetic nerve terminals. Recent studies have also demonstrated that mirabegron is an antagonist at α1A‐ and α1D‐adrenoceptors with an affinity of approximately 1 μM (Alexandre et al., 2016), that is, concentrations close to its affinity at β3‐adrenoceptors but at least 10‐fold higher than its peak plasma concentrations after therapeutic dosing (Krauwinkel et al., 2012).
Figure 6.
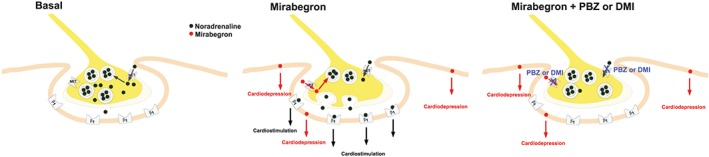
Proposed effects of mirabegron in human heart. In sympathetic nerve terminals, noradrenaline is stored in storage vesicles (Basal). Synaptic noradrenaline is a substrate for neuronal uptake in the neuron by the noradrenaline transporter (NET, neuronal uptake, Chen et al., 2004). It is proposed that mirabegron is taken up into sympathetic nerve terminals by the NET transporter, causes release of noradrenaline from storage vesicles by exocytosis which in turn activates β1‐adrenoceptors (β1) on post‐junctional membranes of the myocardial cell. Mirabegron causes cardiodepression by an unknown mechanism (see text for further discussion). Thus mirabegron simultaneously causes cardiostimulation by an indirect mechanism and cardiodepression. The cardiostimulant, but not the cardiodepressant, effects of mirabegron can be prevented by neuronal uptake blockers phenoxybenzamine (PBZ) or desipramine (DMI) or by blockade of β1‐adrenoceptors.
A limitation of the current study is that direct measurements of noradrenaline outflow from sympathetic nerves in right atrium (Abadie et al., 1996) were not determined. These would provide additional and definitive evidence whether the cardiostimulant effect of mirabegron is mediated through release of noradrenaline from sympathetic nerve terminals.
In summary, we have, in this study, observed separate cardiostimulant and cardiodepressant effects of mirabegron in human isolated right atrium, unrelated to the activation of β3‐adrenoceptors. Instead, mirabegron activated β1‐adrenoceptors, possibly through release of noradrenaline. Our evidence supports previous research showing that β3‐adrenoceptors do not modulate contractility of human hearts. The development of other selective β3‐adrenoceptor agonists, vibegron (Di Salvo et al., 2017), ritobegron and solabegron (Michel and Korstanje, 2016), motivated by the identity of the β3‐adrenoceptor as a therapeutic target to manage overactive bladder syndrome, provides further opportunities to investigate human heart β3‐adrenoceptor function. Our studies may be helpful for the design and testing of future β3‐adrenoceptor agonists intended for clinical use.
Author contributions
W.M. did the study design, carried out experiments, analysed data, maintained data base, interpreted data, draft manuscript, approved submission version and is accountable. M.M. and A.K. performed the conception of study, study design, draft manuscript, approved submission version and is accountable. X.W.L. carried out the review and assessment of arrhythmias in patients prior to heart surgery and approved submission version. P.M. did the conception of study, study design, carried out experiments, interpreted data, draft manuscript, approved submission version and is accountable.
Conflict of interest
M.C.M. has been a consultant to and has received research support from Astellas; he is a consultant for, shareholder of and has received research support from Velicept Therapeutics. W.M., X.W.L., A.J.K. and P.M. declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
M.W. is a New Investigator of The Prince Charles Hospital Foundation and was additionally supported in part by an Experienced Researcher Grant awarded to P.M. by The Prince Charles Hospital Foundation. We thank the heart surgeons of the Prince Charles Hospital (Andrew Clarke, Cheng He, Homayoun Jalali, Dong Kang, Rishen Naidoo, Anil Prabhu, Peter Tesar, Bruce Thomson, Douglas Wall and Livia Williams) who carefully excised heart preparations, and we thank all surgical theatre staff who assisted with coordination of collection of human heart samples.
Mo, W. , Michel, M. C. , Lee, X. W. , Kaumann, A. J. , and Molenaar, P. (2017) The β3‐adrenoceptor agonist mirabegron increases human atrial force through β1‐adrenoceptors: an indirect mechanism?. British Journal of Pharmacology, 174: 2706–2715. doi: 10.1111/bph.13897.
References
- Abadie C, Foucart S, Pagé P, Nadeau R (1996). Modulation of noradrenaline release from isolated human atrial appendages. J Auton Nerv Syst 61: 269–276. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre EC, Kiguti LR, Calmasini FB, Silva FH, da Silva KP, Ferreira R et al. (2016). Mirabegron relaxes urethral smooth muscle by a dual mechanism involving β3‐adrenoceptor activation and α1‐adrenoceptor blockade. Br J Pharmacol 173: 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG (2010). A full pharmacological analysis of the three turkey β‐adrenoceptors and comparison with the human β‐adrenoceptors. PLoS One 5: e15487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bönisch H, Trendelenburg U (1988). The mechanism of action of indirectly acting sympathomimetic amines In Catecholamines I. In: Trendelenburg U, Weiner (eds). Handbook of Experimental Pharmacology 90. Springer‐Verlag Berlin Heidelberg: New York, London, Paris, Tokyo, pp. 247–277. [Google Scholar]
- Buxton BF, Jones CR, Molenaar P, Summers RJ (1987). Characterization and autoradiographic localization of β‐adrenoceptor subtypes in human cardiac tissues. Br J Pharmacol 92: 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelore MR, Deng L, Tota L, Guan XM, Amend A, Liu Y et al. (1999). Potent and selective human β3‐adrenergic receptor antagonists. J Pharmacol Exp Ther 290: 649–655. [PubMed] [Google Scholar]
- Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P et al. (2009). Molecular architecture of the human sinus node. Insights into the function of the cardiac pacemaker Circulation 119: 1562–1572. [DOI] [PubMed] [Google Scholar]
- Chapple CR, Cardozo L, Nitti VW, Siddiqui E, Michel MC (2014). Mirabegron in overactive bladder: a review of efficacy, safety, and tolerability. NeurourolUrodyn 33: 17–30. [DOI] [PubMed] [Google Scholar]
- Chen N‐H, Reith ME, Quick MW (2004). Synaptic uptake and beyond: the sodium‐ and chloride‐dependent neurotransmitter transporter family SLC6. Pflügers Arch 446: 519–531. [DOI] [PubMed] [Google Scholar]
- Christ T, Molenaar P, Klenowski PM, Ravens U, Kaumann AJ (2011). Human atrial β1L‐adrenoceptor but not β3‐adrenoceptor activation increases force and Ca2+ current at physiological temperature. Br J Pharmacol 162: 823–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottingham C, Chen Y, Jiao K, Wang Q (2011). The antidepressant desipramine is an arrestin‐biased ligand at the α2A‐adrenergic receptor driving receptor down‐regulation in vitro and in vivo . J Biol Chem 286: 36063–36075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis WG (1970). A comparison of the local anaesthetic‐, “quinidine‐like”‐ and adrenergic β‐blocking‐activities of five β‐receptor antagonists. J Pharm Pharmacol 22: 284–290. [DOI] [PubMed] [Google Scholar]
- Di Salvo J, Nagabukuro H, Wickham LA, Abbadie C, DeMartino JA, Fitzmaurice A et al. (2017). Pharmacological characterization of a novel beta 3 adrenergic agonist, vibegron: evaluation of antimuscarinic receptor selectivity for combination therapy for overactive bladder. J Pharmacol Exp Ther 360: 346–355. [DOI] [PubMed] [Google Scholar]
- Fitzgerald JD, O'Donnell SR (1971). Pharmacology of 4‐hydroxypropranolol, a metabolite of propranolol. Br J Pharmacol 43: 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JD, Wale JL, Austin M (1972). The haemodynamic effects of (±)‐propranolol, dexpropranolol, oxprenolol, practolol and sotalol in anaesthetised dogs. Eur J Pharmacol 17: 123–134. [DOI] [PubMed] [Google Scholar]
- Gauthier C, Tavernier G, Charpentier F, Langin D, Le Marec H (1996). Functional β3‐adrenoceptor in the human heart. J Clin Invest 98: 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille E, Lemoine H, Ehle B, Kaumann AJ (1985). The affinity of (−)‐propranolol for β1‐ and β2‐adrenoceptors of human heart. Differential antagonism of the positive inotropic effects and adenylate cyclase stimulation by (−)‐noradrenaline and (−)‐adrenaline. Naunyn Schmiedebergs Arch Pharmacol 331: 60–70. [DOI] [PubMed] [Google Scholar]
- Häggendal J, Lindqvist M (1963). Behaviour and monoamine levels during long‐term administration of reserpine to rabbits. Acta Physiol Scand 57: 431–436. [DOI] [PubMed] [Google Scholar]
- Hall JA, Kaumann AJ, Brown MJ (1990). Selective beta 1‐adrenoceptor blockade enhances positive inotropic response to endogenous catecholamines mediated through beta 2‐adrenoceptors in human atrial myocardium. Circ Res 66: 1610–1623. [DOI] [PubMed] [Google Scholar]
- Iversen LL (1965). Inhibition of noradrenaline uptake by drugs. J Pharm Pharmacol 17: 62–64. [DOI] [PubMed] [Google Scholar]
- Kaumann AJ (1986). The β1‐adrenoceptor antagonist CGP 20712A unmasks β2‐adrenoceptors activated by (−)‐adrenaline in rat sinoatrial node. Naunyn Schmiedebergs Arch Pharmacol 332: 406–409. [DOI] [PubMed] [Google Scholar]
- Kaumann AJ, Lemoine H (1987). β2‐Adrenoceptor‐mediated positive inotropic effect of adrenaline in human myocardium. Quantitative discrepancies with binding and adenylate cyclase stimulation. Naunyn Schmiedebergs Arch Pharmacol 335: 403–411. [DOI] [PubMed] [Google Scholar]
- Khan MT, Furchgott RF (1982). Interactions of phenoxybenzamine and nicotinic agonists at the nicotinic receptor in the cat adrenal medulla. J Pharmacol Exp Ther 221: 117–122. [PubMed] [Google Scholar]
- Krauwinkel W, van Dijk J, Schaddelee M, Eltink C, Meijer J, Strabach G et al. (2012). Pharmacokinetic properties of mirabegron, a β3‐adrenoceptor agonist: results from two phase I, randomized, multiple‐dose studies in healthy young and elderly men and women. Clin Ther 34: 2144–2160. [DOI] [PubMed] [Google Scholar]
- Langslet A (1970). Membrane stabilization and cardiac effects of d,l‐propranolol, d‐propranolol and chlorpromazine. Eur J Pharmacol 13: 6–14. [DOI] [PubMed] [Google Scholar]
- Lemoine H, Ehle B, Kaumann AJ (1985). Direct labelling of β2‐adrenoceptors. Comparison of binding potency of 3H‐ICI 118,551 and blocking potency of ICI 118,551. Naunyn Schmiedebergs Arch Pharmacol 331: 40–51. [DOI] [PubMed] [Google Scholar]
- Michel MC, Korstanje C (2016). β3‐Adrenoceptor agonists for overactive bladder syndrome: role of translational pharmacology in a repositioning clinical drug development project. Pharmacol Ther 159: 66–82. [DOI] [PubMed] [Google Scholar]
- Molenaar P, Christ T, Ravens U, Kaumann A (2006). Carvedilol blocks β2‐ more than β1‐adrenoceptors in human heart. Cardiovasc Res 69: 128–139. [DOI] [PubMed] [Google Scholar]
- Molenaar P, Savarimuthu SM, Sarsero D, Chen L, Semmler AB, Carle A et al. (2007). (−)‐Adrenaline elicits positive inotropic, lusitropic and biochemical effects through β2‐adrenoceptors in human atrial myocardium from nonfailing and failing hearts, consistent with Gs coupling but not with Gi coupling. Naunyn Schmiedebergs Arch Pharmacol 375: 11–28. [DOI] [PubMed] [Google Scholar]
- Nayler WG, Chipperfield D, Lowe TE (1969). The negative inotropic effect of adrenergic beta‐receptor blocking drugs on human heart muscle. Cardiovasc Res 3: 30–36. [DOI] [PubMed] [Google Scholar]
- Pesce G, Adler‐Graschinsky E (1983). Comparison of the effects of β‐phenylethylamine and d‐amphetamine on rat isolated atria. J Pharmacol Exp Ther 227: 205–214. [PubMed] [Google Scholar]
- Skeberdis VA, Gendviliene V, Zablockaite D, Treinys R, Macianskiene R, Bogdelis A et al. (2008). β3‐adrenergic receptor activation increases human atrial tissue contractility and stimulates the L‐type Ca2+ current. J Clin Invest 118: 3219–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svalǿ J, Hordling J, Bouchelouche K, Andersson K‐E, Korstanje C, Bouchelouche P (2013). The novel β3‐adrenoceptor agonist mirabegron reduces carbachol‐induced contractile activity in detrusor tissue from patients with bladder outflow obstruction with or without detrusor overactivity. Eur J Pharmacol 699: 101–105. [DOI] [PubMed] [Google Scholar]
- Takasu T, Ukai M, Sato S, Matsui T, Nagase I, Maruyama T et al. (2007). Effect of (R)‐2‐(2‐aminothiazol‐4‐yl)‐4′‐{2‐[(2‐hydroxy‐2‐ phenylethyl)amino]ethyl} Acetanilide (YM178), a novel selective β3‐adrenoceptor agonist, on bladder function. J Pharmacol Exp Ther 321: 642–647. [DOI] [PubMed] [Google Scholar]
- Van Zwieten PA (1969). Decrease in ionic permeability of the cell membrane in guinea‐pig atrial tissue by treatment with antifibrillatory agents and hexobarbitone, determined by means of 86Rb. Br J Pharmacol 35: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeck B, Widmark E (1985). The interaction of ephedrine with β‐adrenoceptors in tracheal, cardiac and skeletal muscles. Clin Exp Pharmacol Physiol 12: 439–442. [DOI] [PubMed] [Google Scholar]
- Wong DT, Threlkeld PG, Best KL, Bymaster FP (1982). A new inhibitor of norepinephrine uptake devoid of affinity for receptors in rat brain. J Pharmacol Exp Ther 222: 61–65. [PubMed] [Google Scholar]
- Wuest M, Eichhorn B, Grimm MO, Wirth MP, Ravens U, Kaumann AJ (2009). Catecholamines relax detrusor through β2‐adrenoceptors in mouse and β3‐adrenoceptors in man. J Pharmacol Exp Ther 328: 213–222. [DOI] [PubMed] [Google Scholar]