

Abstract
Metastatic castration-resistant prostate cancer (mCRPC) is the most aggressive and deadly form of prostate cancer. It is characterized by the overexpression of epidermal growth factor receptors whose signals are mediated by small monomeric G proteins of the Ras superfamily. These require polyisoprenylation for functional activity. Polyisoprenylated cysteinyl amide inhibitors (PCAIs) of polyisoprenylated methylated protein methyl esterase (PMPMEase) were developed as potential targeted therapies to mitigate excessive growth signaling in mCRPC either by inhibiting PMPMEase and/or perturbing the polyisoprenylation-dependent functional interactions. We investigated the effects of PCAIs on the viability of prostate cancer PC 3, DU 145, MDA PCa 2b, LNCaP and 22Rv1 cells, determined the effect of the PCAIs on PC 3 cell proliferation, survival and caspase-mediated apoptotic cell death. Metastatic PC 3 and DU 145 cell migration and invasion in the presence of NSL-BA-040 were determined using the scratch and matrigel invasion assays. We further investigated the effect of NSL-BA-040 on F-actin organization in TagRFP F-actin marker-transfected metastatic PC 3 cells. The PCAIs suppress mCRPC cell viability with EC50 values ranging from 1.3 to 4.0 µM for the most potent of the PCAIs against PC 3, DU 145, MDA PCa 2b, LNCaP and 22Rv cells. PCAIs induced apoptotic cell death in PC 3 and DU 145 cells as determined by annexin V/propidium iodide flow cytometry analysis through the activation of caspases 3 and 8 while also inhibiting migration and invasion through the disruption of F-actin organization. Taken together, our studies show the anti-cancer effects on mCRPC cells through induction of caspase-mediated apoptosis and F-actin-mediated inhibition of cell motility and invasion thereby indicating the anti-tumor and anti-metastatic potential of the PCAIs.
Keywords: F-actin, cell motility, polyisoprenylated protein, prostate cancer, G proteins
Introduction
Metastatic castration-resistant prostate cancer (mCRPC) remains an incurable disease that poses a major challenge to therapy. Consequently, the vast majority of the 26,120 expected deaths in 2016 [1-3] are likely to be from mCRPC given that this form of the disease is resistant to the androgen-deprivation therapy (ADT) [4]. Lack of understanding of the molecular phenomena driving PCa metastasis implies that therapies targeting the ‘drivers’ are limited. In the absence of new effective therapies, the survival rates are expected to remain low. Current chemotherapies such as Docetaxel, mitoxantrone and cabazitaxel only extend the lives of men with mCRPC by 2 to 4 months [5,6] while also causing severe adverse effects [7]. Abiraterone, enzalutamide or sipuleucel-T are also used without much success [8]. Therefore, identifying the molecular mechanisms involved in the promotion of mCRPC is imperative for the development of targeted and more effective therapies.
The epidermal growth factor receptor (EGFR) and its ligands are overexpressed and assume the autocrine control in mCRPC tumor growth [9,10], suggesting that EGFR signaling plays an important role in PCa progression to the metastatic disease. It has also been shown that cancer cells overexpress EGFR as a resistance mechanism to monoclonal antibodies and tyrosine kinase inhibitors therapies that block EGFR [11]. Downstream mediators of EGFR signaling such as Ras have been shown to be hyperactive and appear to drive PCa metastasis [12]. Furthermore, cancers with activated Ras do not respond to anti-EGFR drugs. This, in addition to the EGFR overexpression, makes Ras a very important target in the search for more effective mCRPC therapies. The Ras superfamily of GTPases consists of Ras and Rho subfamilies that regulate cell proliferation, differentiation, apoptosis and cytoskeletal organization involved in cell migration and invasion [13]. Oncogenic Ras, together with c-myc, has been reported to confer metastatic capability on cells in culture while Rho subfamily members are implicated in cancer metastasis [14]. The Rho subfamily includes Rho, Rac and Cdc42 that regulate assembly and disassembly of actin cytoskeleton required for cell migration and invasion [14]. Cell movement is regulated through cell-substrate adhesion, cell-cell adhesion, vesicular trafficking and transcription [14]. Rho A regulates stress fibers and the formation of focal adhesions, Rac 1 stimulates actin polymerization and formation of lamellipodia which then initiate cell movement [15] while Cdc42 regulates filopodia formation and directs cell movement [14].
A major post-translational pathway that is vital for the functions of Ras proteins is polyisoprenylation [16]. The modifications facilitate Ras proteins’ association with the inner surfaces of the plasma membranes [17]. The polyisoprenylation pathway terminates in the only reversible step regulated by polyisoprenylated protein methyl transferase (PPMTase) [18] and polyisoprenylated methylated protein methyl esterase (PMPMEase) [19]. Work in our laboratory [20,21] showed that inhibition of PMPMEase with synthetic and natural small molecules results in cancer cell death, inhibition of cancer cell migration as well as significant changes in the expression of cancer-related genes [22,23]. Cushman et al (2013) showed that knockdown of PMPMEase (CES1) diminished Rho A demethylation, inhibited migration and altered the morphology of breast cancer MDA-MB-231 cells [24]. Our previous studies indicate that castration-resistant PCa cell lines have high levels of PMPMEase expression and activity [21] that when inhibited results in diminished cell viability and migration [21]. In this study, we investigate the effects of PCAIs against the molecular processes that drive mCRPC.
Materials and methods
Cell lines and cell culture
Human androgen-sensitive PCa, LNCaP and 22Rv1 cells, and castration-resistant PC 3, DU 145 and the African American androgen-independent MDA PCa 2b cell lines were obtained from American Type Culture Collection (Manassas, VA). LNCaP and 22Rv1 were maintained in RPMI 1640, PC 3 were cultured in Ham’s F12K while DU145 were maintained in MEM. All media for these cell lines, except MDA PCa 2b, were supplemented with 10% fetal bovine serum (FBS) and PSN (100 U/mL penicillin, 50 μg/mL streptomycin and 50 μg/mL neomycin) from Life Technologies (Grand Island, NY). MDA PCa 2b cells were cultured in Ham’s F12K supplemented with 20% FBS, 25 ng/mL cholera toxin, 10 ng/mL mouse epidermal growth factor, 0.005 mM phosphoethanolamine, 100 pg/mL hydrocortisone and 45 nM selenious acid and 0.005 mg/mL bovine insulin. All cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. pCMVLifeAct-TagRFP plasmid expressing F-actin marker was obtained from ibidi (Madison, WI), Lipofectamine 3000 was obtained from Life Technologies Corporation (Grand Island, NY) and used to transfect plasmid into PCa cells. G 418 disulfate salt from Sigma (St Louis, MO) was used to select for transfected cells since pCMVLifeAct-TagRFP plasmid has neor gene. BD matrigel-coated 24 well inserts were obtained from BD Biosciences. The polyisoprenylated cysteinyl amide inhibitors (PCAIs), NSL-RD-036 (NSL-BA-036), NSL-BA-040, NSL-BA-055 NSL-BA-056 as well as the non-farnesylated analog, NSL-100 were synthesized in our lab as previously described [20,25].
Effect of the PCAIs on the viability and proliferation of PCa cells
The human PCa cell lines were seeded at a density of 2×104/well in 96 well plates and allowed to attach over 24 h in media containing 5% FBS except for the MDA PCa 2b cells where 10% FBS was used. These were then treated for 48 h with 1 to 20 μM of the PCAIs dissolved in 1 μL of acetone. Control cells were treated with an equal volume of acetone. Cell Titre-Blue solution (20 μL) was then added to each well and incubated for 1-2 h after which the cell viability was determined as previously described [23]. To determine the effect of PCAIs on PC 3 cell proliferation, 2×104 cells were plated into a 24-well plate and allowed to attach overnight. These were then treated with 0.1 to 2.0 μM of the PCAIs for 48 h. Viable cells were counted using a Countess II Automated Cell Counter from Life Technologies (Grand Island, NY).
Effect of PCAIs on PC 3 colony formation
Metastatic PC 3 cells were plated at a density of 1.5×105/mL in T-25 flasks and allowed to attach overnight. The cells were treated with NSL-BA-040 (1.0-4.0 μM) for 48 h after which they were trypsinized, counted and re-plated in 6-well plates at a density of 500-1000 cells per well and maintained in complete growth media for 14 days. They were then fixed with a solution of acetic acid in methanol (1:7) and stained with 1% crystal violet solution in methanol. Colonies consisting of 50 or more cells were counted with a stereomicroscope. The results were expressed as a percentage of the controls.
Effect of PCAIs on PC 3 spheroid formation
Metastatic PC 3 cells were seeded in round bottom 96 well Nunclon Sphera microplates at 5000 cells per well. They were then treated with 1-5 μM of NSL-BA-040 or NSL-BA-055 for 48 h after which 10 μL of acridine orange/ethidium bromide (AO/EB) (100 μg/mL) was added followed by incubation at 37°C for 15 min. Images were taken with Nikon Eclipse Ti 100 inverted fluorescent microscope (Nikon Instruments Inc., Melville, NY).
Apoptotic-effect of PCAIs on metastatic PCa cell lines
The mechanism by which the PCAIs induce cell death was determined using the AO/EB staining, annexin V/Propidium iodide flow cytometry and caspase 3/7 activation assays. PC 3 cells were plated in 24 wells at 5×104 per well and allowed to attach overnight. They were treated with 0.5 to 5.0 μM NSL-BA-040 for 48 h after which 10 μL AO/EB solution (100 μg/mL) was added to each treatment and allowed to incubate for 15 min. Changes in the nuclei after treatment were visualized and pictures taken with Nikon Eclipse Ti 100 inverted fluorescent microscope (Nikon Instruments Inc., Melville, NY). For the flow cytometry analysis, 1 mL of the metastatic PC 3 and DU 145 cells were seeded at 1×105 cell per mL in six well plates and left overnight to attach. These were then treated with 1 to 8 μM NSL-BA-040 in media supplemented with 5% FBS for 48 h. Apoptosis was determined using ApopNexin FITC Apoptosis Detection Kit (EMD Millipore, Temecula CA) according to the manufacturer’s protocol. Briefly, cells were trypsinized and centrifuged at 500×g for 5 min. Cell pellets were washed twice with 1×PBS and re-suspended in binding buffer. Annexin FITC and propidium iodide were added to the cell suspensions and incubated for 10 min at room temperature in the dark. Analysis was done using the FAS Calibur with Cell Quest Pro software (BD Biosciences NJ).
Caspase activation was determined using the CaspaTagCaspase-3/7 in situ Assay Kit, Fluorescein (EMD Millipore, Temecula CA) according to the manufacturer’s instructions. PC 3 cells were treated with PCAIs (1-10 μM) or 200 nM staurosporin (positive control) for 48 h. The cells were incubated with 30×FLICA reagent in treatment media for 1 h at 37°C under 5% CO2. FLICA-containing media was removed and Hoechst solution (0.5% v/v in 1×PBS) added to the CaspaTag-labeled cells to stain the nuclei.Cells were washed with 1×wash buffer and fixative added. Fluorescent images were obtainedwith the Nikon Eclipse Ti 100 inverted microscope (Nikon Instruments Inc., Melville, NY) equipped with a Nikon DS Qi2 digital camera and S Plan Fluor ELWD 20×Ph1 ADM objective with a numerical aperture of 0.45. The effect of PCAIs treatment on caspase activation was further investigated by Western blotting. Cells were plated in 60 mm dishes at 3.5×105 per mL and treated with NSL-BA-040 (5 μM) for 16 h after which they were lysed on ice with RIPA lysis buffer supplemented with 1×protease inhibitors cocktail (Sigma, St. Louis, MO) and the protein concentrations determined using the Bradford assay. Aliquots of each sample containing 50 μg of protein were resolved on 10-20% gradient SDS-PAGE (Bio-Rad) and transferred onto PVDF membranes. After blocking in 3% non-fat dried milk (Sigma, St Louis MO), membranes were incubated at 4°C overnight with caspase 3, 7 (Cell Signaling Technology, Danvers, MA) and cleaved caspase 8 (Sigma, St Louis MO) primary antibodies. After incubating at room temperature for 1 h with horseradish peroxidase-conjugated anti-rabbit IgG from Santa Cruz Biotech (Santa Cruz, CA), Clarity Western ECL Substrate was added and the chemiluminescence images obtained using the ChemiDoc Imaging Systems (Bio-Rad, CA).
Effect of PCAIs on PCa cell migration and invasion
The ability of the PCAIs to inhibit PCa cell migration and invasion were determined using the wound-healing and matrigel invasion methods. Cells were seeded in ibidi cell culture inserts, allowed to grow to confluence and serum-starved for 24 h. After treatment with 0.5 to 2 μM NSL-BA-040, wound closure was monitored by capturing images with the Nikon Eclipse Ti 100 inverted microscope (Nikon Instruments Inc., Melville, NY) at 0, and 24 h post-treatment and the images analyzed as previously described [21]. In order to determine the effects on cell invasion, matrigel-coated 24-well inserts (Corning) with 8 μm pores were used. PC 3 or DU 145 cells (1×105) were seeded into the inserts containing 500 μL of media supplemented with 1% FBS while 750 μL of media supplemented with 10% FBS was added to the lower chamber for chemo-attraction. After 24 h, cells that had migrated through the membrane lining of the insert were fixed with 3.7% formaldehyde in 1×PBS and stained with 1% crystal violet in methanol. Microscope images of the stained cells were obtained with an Olympus DP70 microscope using 4×objective. The captured images invaded cells counted and expressed as percentages of the control.
Effect of PCAIs on F-actin organization in metastatic PC 3 cells
PC 3 cells were seeded in 6-well plates in media containing 10% FBS for 24 h. These were transfected with 1 μg of pCMVLifeAct-TagRFP F-actin marker plasmid using 4 μL of lipofectamine 3000 (Life Technologies Corporation Grand Island, NY) per well Transfected cells were selected with G 418 (300 ug/mL) for a month. PC 3 cells expressing the LifeAct -TagRFP F-actin marker were plated in 24-well plates at 5×104/well and allowed to attach. After cell had attached, they were treated with 1.0-10 μM of NSL-BA-040 and imaged for its effects on F-actin organization using Nikon Eclipse Ti 100 inverted microscope (Nikon Instruments Inc., Melville, NY) for 48 h.
Statistical analysis
All results were expressed as the means ± SEM. Statistical analysis was performed with Graph Pad Prism software (San Diego, CA). One-way ANOVA analyses with Bonferroni’s post hoc test were used for comparisons between multiple groups. The Student’s t-test was used for statistical comparison between two groups. p values ≤0.05 was considered significant.
Results
PCAIs inhibit the viability and proliferation of PCa cells
All four PCAIs (NSL-RD-036, NSL-BA-040, NSL-BA-055 and NSL-BA-056) used in this study were effective at inducing cell death in virtually the PCa cell lines at concentrations of about 10 to 20 µM. The most potent of the PCAIs were NSL-BA-040 and NSL-BA-055 with EC50 values ranging from 2.0-to 5.0 μM and 1.5 to 4.3 µM, respectively for the different cell lines. NSL-RD-036 (EC50 values of 4.7 to 13 µM) and NSL-BA-056 (EC50 values 5.1 to 14.8 µM) were also very effective against the PCa cell lines (Figure 1 and Table 1). The non-farnesylated analog of the PCAIs, NSL-100 was not effective at inhibiting the viabilities of the cells (Figure 1). As shown in Figure 1, docetaxel, currently used clinically to manage metastatic prostate, was much less effective against the PCa cell lines when compared to the PCAIs with EC50 in excess of 50 μM (Table 1). This implies that docetaxel is at least 11- to 33-fold and 3- to 10-fold less effective than the most and least potent of the tested PCAIs. PC 3 cell growth and proliferation was significantly inhibited (P<0.01) in a concentration-dependent manner by all the PCAIs tested. As shown in Figure 1F, 2 µM of NSL-RD-036 and NSL-BA-040 completely blocked PC 3 cell proliferation. When we conducted the colony-forming assay to determine the ability of NSL-BA-040 to attenuate PC 3 cell growth relapse, the compounds inhibited colony formation in a concentration dependent manner (P<0.001) (Figure 2A, 2B).
Figure 1.
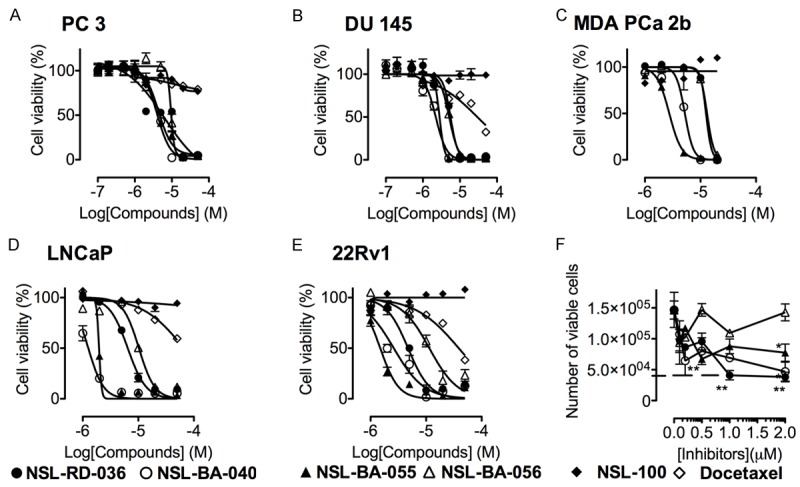
PCAIs-induced-PCa cell death is polyisoprenylation-dependent. (A) PC 3, (B), DU145 (C) African American MDA PCa 2b (D) LNCaP and (E) 22Rv1 were seeded in 96-wellsand treated with 0 to 50 μM of the respective PCAIs, docetaxel or with 0-100 μM of the non-farnesylated analogs of the PCAIs as described in the methods. After 48 h, the cell viabilities were determined using Cell Titre Blue and the number of viable cells counted for the effect on cell proliferation (F). Each point represents the mean ± SEM of 4 determinations. *P<0.05, **P<0.01, ***P<0.001 compared to the untreated cells using Student’s t-test.
Table 1.
EC50 values for the PCAIs inhibition of PCa cell viabilities as determined from the data in Figure 1
| COMPOUND | EC50 (μM) | ||||
|---|---|---|---|---|---|
|
| |||||
| PC 3 | DU 145 | MDA PCa 2b | LNCaP | 22Rv1 | |
| NSL-RD-036 | 6.3 | 5.6 | 12.5 | 5.4 | 4.7 |
| NSL-BA-040 | 4.0 | 2.2 | 5.2 | 1.3 | 2.4 |
| NSL-BA-055 | 4.3 | 2.8 | 2.7 | 2.0 | 1.5 |
| NSL-BA-056 | 9.4 | 5.1 | 13.1 | 10.3 | 14.8 |
| NSL-100 | >50.0 | >50.0 | >50.0 | >50.0 | >50.0 |
| Docetaxel | >50.0 | >50.0 | >50.0 | >50.0 | >50.0 |
Figure 2.
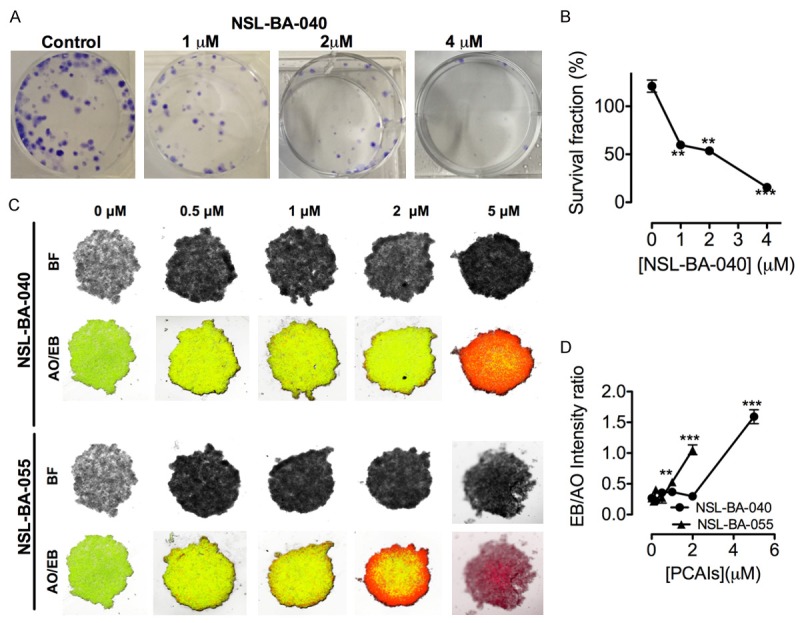
PCAIs inhibit colony formation and induce apoptosis in spheroids. Treatment with NSL-BA-040 for 48 h inhibited PC 3 colony formation (A) as shown in the concentration-dependent reduction in survival fraction (B). Images of PC 3 spheroids treated with the indicated concentrations of the PCAIs for 48 h were captured following staining with acridine orange and ethidium bromide (AO/EB), the former turning live cells green and the latter turning apoptotic cells red (C). The graph shows the ratio of TRITC to FITC intensity (D). The ratio could not be obtained for spheroids treated with 5 mM NSL-BA-055 due to the absence of live cells. Each point represents the mean ± SEM of 4 determinations. *P<0.05. **P<0.01***P<0.001 compared to the controls using Student’s t-test.
PCAIs induce cell death in PC 3 spheroids
The ability of PCAIs to potentially induce apoptosis in tumor cells was assessed in vitro by treating PC 3 spheroids with NSL-BA-040 and NSL-BA-055. The PCAIs induced concentration-dependent cell death in the PC 3 spheroids as revealed by the progressive increase in ethidium bromide staining. By comparison, the untreated control spheroids were predominantly green due to the exclusion of ethidium bromide by the intact live cells (Figure 2C). The increasing intensity ratios of EB over AO with increasing PCAIs concentrations (Figure 2D) is an indication of the increasing proportion of dead over live cells in the spheroids since AO stains live cell nuclei green and EB stain the nuclei of dead cells red.
NSL-BA-040 induces apoptotic cell death in metastatic PC 3 and DU 145 cells
Apoptotic cell death is associated with changes in cell morphology and significant changes in DNA. Therefore, we determined the mechanism of the PCAIs-induced cell death by treating PC 3 cells with NSL-BA-040 and stained with acridine orange and ethidium bromide (AO/EB) dye. AO stains live cell nuclei green while EB stains the nuclei of dead cells red. Changes in nuclei morphology and color have been used to demonstrate apoptotic cell death [26]. Treatment with increasing concentrations of NSL-BA-040 (1.0-2.0 μM) revealed progressive decline in the number of live cells as shown by decrease in AO-labeled nuclei and an increase in the number of EB-labeled nuclei.
The apoptotic effect of NSL-BA-040 was further studied using flow cytometry analysis. Propidium iodide and FITC annexin V staining of DU 145 and PC 3 cells treated with NSL-BA-040 were analyzed with a flow cytometer. A concentration-dependent induction of apoptosis was observed as shown in Figure 3A and 3B. The populations of early (20%) and late apoptotic (8%) cells were significantly increased (20%) with the treatment compared to the control. Activation of executioner caspases 3/7 in apoptotic cell death has been shown in several studies [27]. We thus investigated the involvement of caspase 3/7 in the apoptotic effect of NSL-BA-040 in PC 3 cells using the CaspaTagTMCaspase-3/7 In Situ Assay (EMD Millipore). PC 3 cells treated with NSL-BA-040 (5 μM), showed prominent activation of Caspase-3/7, as indicated by intense green fluorescence in the cells compared to the control cells. The other PCAIs at similar concentrations were less effective at activating the caspases 3/7 as shown in Figure 4A. This indicates that NSL-BA-040 induces apoptotic cell death in PC 3 cells through activation of Caspase-3/7.Western blot analysis revealed that treatment with NSL-BA-040 (5 μM) resulted in a one-fold decrease in un-cleaved caspase 3 and 7 in both cell lines (Figure 4B and 4C). Cleaved caspase 8 showed a two-fold increase with treatment in DU 145 cells and a slight increase in PC 3 cells (Figure 4D and 4E).
Figure 3.
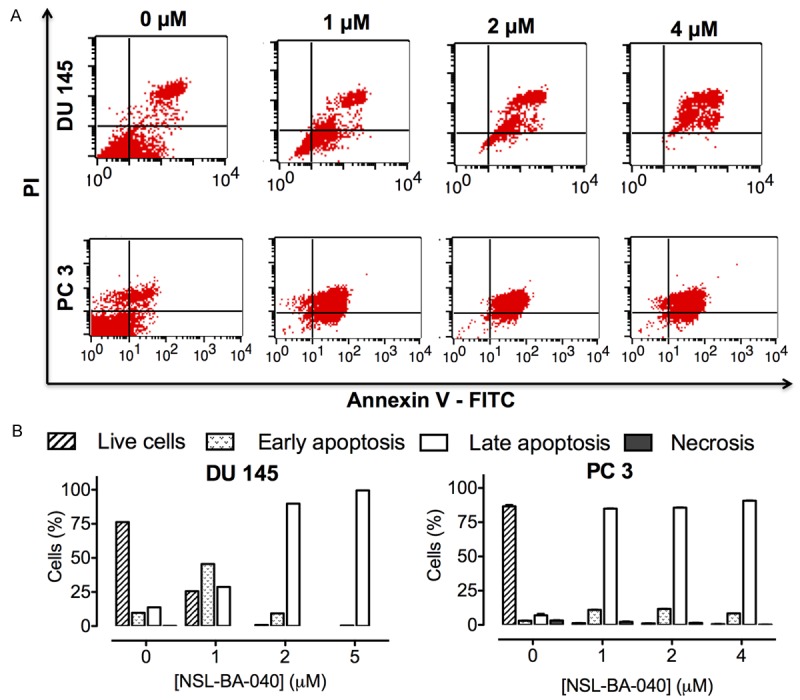
NSL-BA-040induces apoptosis in metastatic PCa cells. (A) DU 145 and PC 3 cells were treated with the indicated concentrations of NSL-BA-040 for 48 h, stained with annexin V and propidium iodide and analyzed using flow cytometry as described in the methods (B) Bar graphs showing the distribution of cells as determined by flow cytometry analysis.
Figure 4.
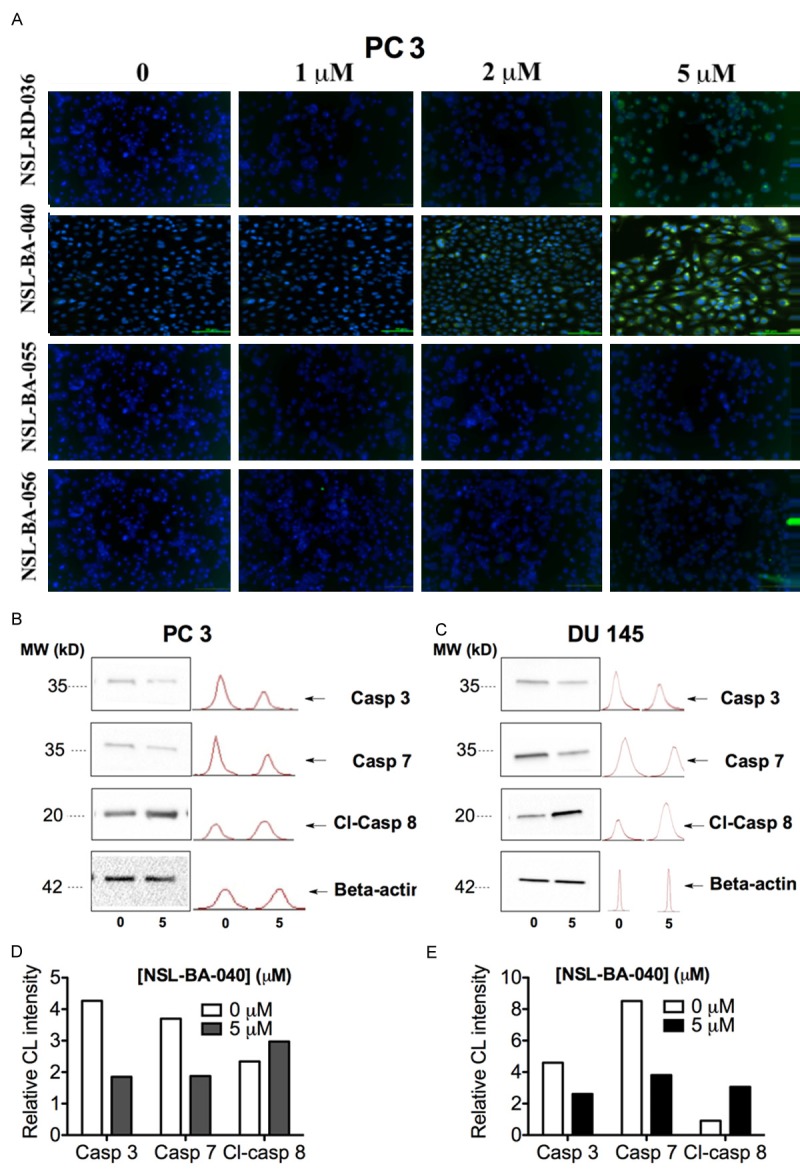
PCAIs-induced apoptosis in PCa cells lines is associated with caspase 3, 7 and 8 activation. A: Caspase 3/7 activation was observed after treating PC 3 cells with 1 to 5 μM PCAIs for 48 h and then reacting with the fluorescent caspase 3/7 irreversible inhibitor; green FLICA. Images were taken with Nikon DS Qi2 digital camera linked to a Nikon Eclipse Ti 100 inverted microscope. B, D: Western blot analysis showing the expression of caspase 3, 7 and cleaved caspase 8 in DU 145 and PC 3 cells after treatment with 5 μM NSL-BA-040. C, E: Bar graph showing the chemiluminescence intensity (CL) of the protein bands relative to control.
NSL-BA-040 inhibits migration and invasion of metastatic PC 3 and DU 145
Malignancy in PCa is dependent on the migration and invasion of nearby and distant tissues. The wound-healing assay was used to investigate the ability the PCAIs to limit the motility of PC 3 cells. As shown in Figure 5A and 5B, the number of cells that migrated into the wound area following treatment with NSL-BA-040 decreased significantly in a concentration dependent manner when compared to the control. NSL-BA-040 at 0.5, 1.0 and 2.0 μM, inhibited PC 3 migration by 23%, 35% and 40%, respectively compared to the control (Figure 5A and 5B). Using the matrigel invasion assay, it was determined that NSL-BA-040 significantly inhibited PC 3 and DU 145 cell invasion at sub-micro-molar concentrations. NSL-BA-040 at 0.1 μM reduced number of PC 3 and DU 145 cells that invaded through the matrigel by 27% and 23%, respectively. At the higher concentration of 2.0 μM, PC 3 and DU 145 cell invasion was reduced by 91% and 94%, respectively when compared to the controls (Figure 5C and 5D). These results indicate that at non-cytotoxic concentrations, NSL-BA-040 inhibits migration and invasion of highly metastatic PCa cells in a concentration-dependent manner.
Figure 5.
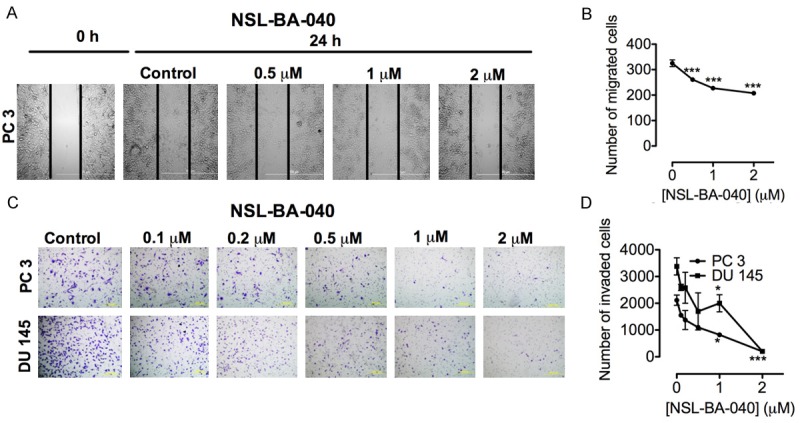
NSL-BA-040 inhibits migration and invasion of PC 3 and DU 145 cells. A: Wounds were created in monolayers of cells and treated with the indicated concentrations of NSL-BA-040 for 24 h as described in the methods. Images were then taken showing the inhibition PC 3 cell migration. B: NSL-BA-040 significantly decreased the number of cells that migrated into the wound compared to the control in a concentration dependent manner. C: PC 3 and Du145 cells were seeded in matrigel-coated inserts and treated with or without NSL-BA-040. After 24 h invaded cells were fixed, stained and counted. D: The number of cells that invaded through matrigel decreased significantly with the treatment compared to the respective untreated controls. Each point represents the mean ± SEM of 3 determinations. *P<0.05, **P<0.01 and ***P<0.001 versus untreated control cells compared by Student’s t-test.
NSL-BA-040 disrupts F-actin organization and morphology of PC 3 cells
PC 3 cells expressing the TagRFP-actin marker protein showed significant alterations in F-actin organization upon exposure to NSL-BA-040. As shown in Figure 6, the control cells showed intact well-organized F-actin and normal cell shape. However, after treatment with 1.0-5.0 µM NSL-BA-040, the cells rounded up and had no visible or collapsed F- actin filaments.
Figure 6.
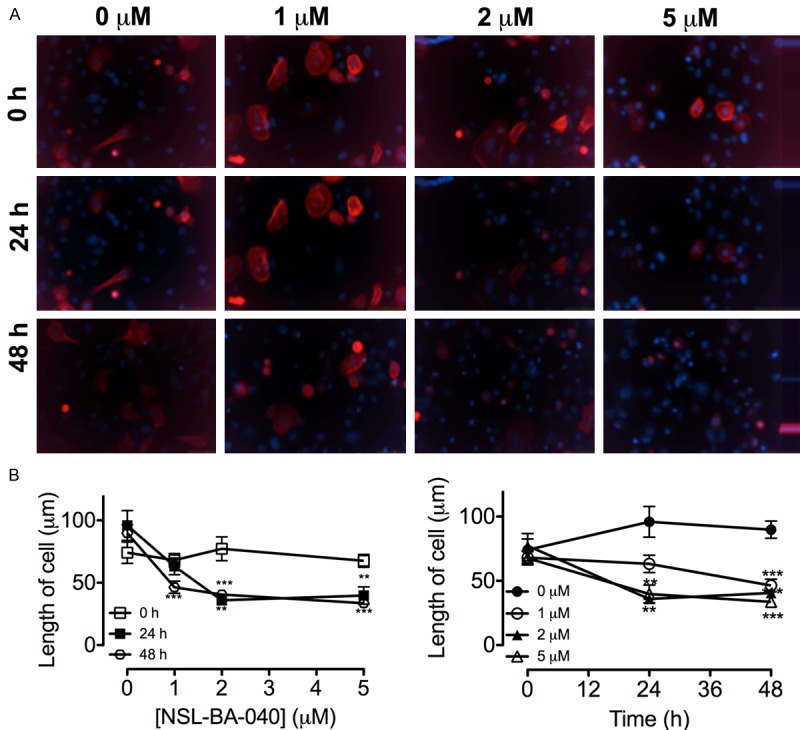
NSL-BA-040 disrupts F-actin organization in TagRFP F-actin marker transfected PC 3 cells. A: PC 3 cells expressing the LifeAct-TagRFP F-actin marker was treated with 1 to 5 μM of NSL-BA-040 as described in the methods. The fluorescent images were captured using Nikon DS Qi2 digital camera connected to Nikon Eclipse Ti 100 inverted microscope. B: The length of cells decreased significantly with the treatment compared to the control. Each point represents the mean ± SEM (n = 25). **P<0.01 and ***P<0.001.
Discussion
Several studies to elucidate the molecular mechanisms that underlie PCa relapse and metastasis have revealed a strong correlation with amplification-driven activation of the EGFR signaling pathway [28]. However, mCRPC is resistant to the tyrosine kinase inhibitors that target EGFR [29]. Polyisoprenylated proteins such as Ras, Rho and related monomeric G proteins mediate EFGR signaling pathways. Activation of Ras GTPases has been reported to be essential for PCa metastasis as it drives the over expression of EGFR ligands that promote the tumor growth [10]. Consequently, polyisoprenylated small molecules such as the PCAIs have the potential to mitigate the hyperactivity of monomeric G-proteins by disrupting the metabolism and/or the polyisoprenylation-dependent protein-protein interactions of polyisoprenylated proteins [20]. The potential effectiveness of the PCAIs is clearly demonstrated by the fact that the least potent of the PCAIs, NSL-BA-056, was 3-10 fold more effective against the viability of the PCa cell lines than docetaxel, the treatment of choice for mCRPC [30]. Previous studies from our laboratory revealed that the PCAIs are 10-fold more potent than farnesyl thiosalicylic (FTS), a small polyisoprenylated molecules that has been demonstrated to dislodge K-Ras from cell membranes leading to apoptosis, [20,31]. In light of these, the PCAIs could be acting in similar fashion by disrupting the interactions between Ras and Rho family of monomeric G-proteins and their effector. This is because Ras and Rho are known to promote cell growth, proliferation, survival, apoptosis and metastasis [32,33]. These effects on the PCa cells are consistent with an earlier study by Aguilar et al (2014) showing the effects of the PCAIs on pancreatic cancer cell viability and cell death [20]. The polyisoprenyl moiety is essential for the observed effects since the non-farnesylated cysteinyl amide analogs of the PCAIs were not effective against the PCa cells. This further demonstrates the possible interference with polyisoprenylated protein-mediated processes.
Further investigations to determine the mechanisms of cell death indicate that the PCAIs induce apoptotic cell death, triggered either through the intrinsic or extrinsic pathway. The extrinsic pathway involves the activation of the death receptors and subsequent activation of caspase 8 while the intrinsic pathway involves the mitochondria and activation of caspase 9 [34]. Activated caspases 8 and 9 from their respective pathways activate caspases 3 and 7, resulting in apoptosis. The observed activation of caspase 8 following treatment with the PCAIs is therefore consistent with the observed activation of caspase 3/7. This indicates that the apoptotic effect of the PCAIs on metastatic PC 3 and DU 145 prostate cells involve the extrinsic pathway. The apoptotic cell death induced by the PCAIs in PCa cells is potentially of immense therapeutic significance given that the transitions from androgen-dependence, through androgen-independence to mCRPC transition is associated with profound resistance to current therapies [35,36]. Androgen-independent PCa cell lines have also been reported to evade apoptosis after ADT which drives their continuous survival and proliferation [37]. The ability of PCAIs to induce significant cell death in androgen-dependent and metastatic androgen-independent as well as African American prostate cancer cell lines makes them a potential single therapeutic agent that can target and eliminate incurable metastatic forms of prostate cancer. This is significant because African American men have higher risks of being diagnosed and dying from metastatic advanced PCa than other ethnic groups [38].
The aberrant activities of the polyisoprenylated Rho A, Rho C, Rac 1 and Cdc42 proteins have also been implicated in invasion and metastasis in prostate and other cancers [39]. Reorganization of actin cytoskeleton, focal adhesion formation and cell migration, which constitute the hallmarks of metastasis, depend mostly on Rho signaling [39]. However, polyisoprenylation is necessary for Rho and related proteins to be functional [40]. In the presence of the PCAIs, the invasive and migratory ability of metastatic PCa cell lines, PC 3 and DU 145, were significantly inhibited. This suggests that PCAIs probably interfere with Rho interactions, resulting in the disruption of Rho-mediated migratory and invasive properties in these metastatic cell lines. The inhibitory effect of the PCAIs on invasion and migration is further supported in the TagRFP F-actin marker transfected metastatic PC 3 cells, which revealed complete disruption of F-actin organization with corresponding morphological changes on treatment with the PCAIs. Similar morphological changes were reported by Cushman et al in the triple negative breast cancer cell line (MDA-MB 231), following knockdown of CES1/PMPMEase, indicating the importance of PMPMEase in Rho methylation and F-actin cytoskeletal function [24].
Conclusions
Taken together, our data show that the PCAIs have the potential to limit PCa tumor growth by inducing apoptotic cell death through the activation of caspases 8 and then 3/7 as well as inhibit metastasis by disrupting F-actin-mediated migration. The effects of the PCAIs are most likely through the disruption of polyisoprenylated protein signaling. This is significant since EGFR is frequently overexpressed in mCRPC and the Ras proteins require polyisoprenylation to mediate the EGFR signaling.
Acknowledgements
The research reported in this publication was supported by the National Cancer Institute (NCI) and National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under Grant SC1CA190505 and by the National Institute On Minority Health and Health Disparities (NIMHD) of the NIH under Award Number G12 MD007582 (previous award NIH/NCRR/RCMI G12 RR03020) and the National Institute On Minority Health and Health Disparities (NIMHD) of the National Institutes of Health (NIH) under Award Number G12 MD007582 (previous award NIH/NCRR/RCMI G12 RR03020 The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure of conflict of interest
None.
References
- 1.Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, Bronson RT, Ryeom S, Hahn WC, Loda M, Cichowski K. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16:286–294. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonarakis ES, Carducci MA, Eisenberger MA. Novel targeted therapeutics for metastatic castration-resistant prostate cancer. Cancer Lett. 2010;291:1–13. doi: 10.1016/j.canlet.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 4.Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA Cancer J Clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- 5.Kawalec P, Paszulewicz A, Holko P, Pilc A. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. A systematic review and meta-analysis. Arch Med Sci. 2012;8:767–775. doi: 10.5114/aoms.2012.31610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonpavde G, Di Lorenzo G, Higano CS, Kantoff PW, Madan R, Shore ND. The role of sipuleucel-T in therapy for castration-resistant prostate cancer: a critical analysis of the literature. Eur Urol. 2012;61:639–647. doi: 10.1016/j.eururo.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 7.Patel PH, Kockler DR. Sipuleucel-T: a vaccine for metastatic, asymptomatic, androgenindependent prostate cancer. Ann Pharmacother. 2008;42:91–98. doi: 10.1345/aph.1K429. [DOI] [PubMed] [Google Scholar]
- 8.Sartor O, Gillessen S. Treatment sequencing in metastatic castrate-resistant prostate cancer. Asian J Androl. 2014;16:426–431. doi: 10.4103/1008-682X.126378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gan Y, Shi C, Inge L, Hibner M, Balducci J, Huang Y. Differential roles of ERK and Akt pathways in regulation of EGFR-mediated signaling and motility in prostate cancer cells. Oncogene. 2010;29:4947–4958. doi: 10.1038/onc.2010.240. [DOI] [PubMed] [Google Scholar]
- 10.Schulze A, Nicke B, Warne PH, Tomlinson S, Downward J. The transcriptional response to Raf activation is almost completely dependent on Mitogen-activated Protein Kinase Kinase activity and shows a major autocrine component. Mol Biol Cell. 2004;15:3450–3463. doi: 10.1091/mbc.E03-11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 12.Schulze A, Nicke B, Warne PH, Tomlinson S, Downward J. The transcriptional response to Raf activation is almost completely dependent on Mitogen-activated Protein Kinase Kinase activity and shows a major autocrine component. Mol Biol Cell. 2004;15:3450–3463. doi: 10.1091/mbc.E03-11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernandez-Alcoceba R, del Peso L, Lacal JC. The Ras family of GTPases in cancer cell invasion. Cellular and Molecular Life Sciences: CMLS. 2000;57:65–76. doi: 10.1007/s000180050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Mol Life Sci. 2010;8:23. doi: 10.1186/1478-811X-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, Mareel M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004;18:1016–1018. doi: 10.1096/fj.03-1110fje. [DOI] [PubMed] [Google Scholar]
- 16.Gelb MH, Brunsveld L, Hrycyna CA, Michaelis S, Tamanoi F, Van Voorhis WC, Waldmann H. Therapeutic intervention based on protein prenylation and associated modifications. Nat Chem Biol. 2006;2:518–528. doi: 10.1038/nchembio818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M, Casey PJ. Protein prenylation: unique fats make their mark on biology. Nature reviews. Nat Rev Mol Cell Biol. 2016;17:110–122. doi: 10.1038/nrm.2015.11. [DOI] [PubMed] [Google Scholar]
- 18.Oboh OT, Lamango NS. Liver prenylated methylated protein methyl esterase is the same enzyme as Sus scrofa carboxylesterase. J Biochem Mol Toxicol. 2008;22:51–62. doi: 10.1002/jbt.20214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamango NS. Liver prenylated methylated protein methyl esterase is an organophosphatesensitive enzyme. J Biochem Mol Toxicol. 2005;19:347–357. doi: 10.1002/jbt.20100. [DOI] [PubMed] [Google Scholar]
- 20.Aguilar BJ, Nkembo AT, Duverna R, Poku RA, Amissah F, Ablordeppey SY, Lamango NS. Polyisoprenylated methylated protein methyl esterase: a putative biomarker and therapeutic target for pancreatic cancer. Eur J Med Chem. 2014;81:323–333. doi: 10.1016/j.ejmech.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poku RA, Amissah F, Duverna R, Aguilar BJ, Kiros GE, Lamango NS. Polyisoprenylated methylated protein methyl esterase as a putative drug target for androgen-insensitive prostate cancer. Ecancermedicalscience. 2014;8:459. doi: 10.3332/ecancer.2014.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amissah F, Duverna R, Aguilar BJ, Poku RA, Kiros GE, Lamango NS. Polyisoprenylated methylated protein methyl esterase overexpression and hyperactivity promotes lung cancer progression. Am J Cancer Res. 2014;4:116–134. [PMC free article] [PubMed] [Google Scholar]
- 23.Amissah F, Taylor S, Duverna R, Ayuk-Takem LT, Lamango NS. Regulation of polyisoprenylated methylated protein methyl esterase by polyunsaturated fatty acids and prostaglandins. Eur J Lipid Sci Technol. 2011;113:1321–1331. doi: 10.1002/ejlt.201100030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cushman I, Cushman SM, Potter PM, Casey PJ. Control of RhoA methylation by carboxylesterase I. J Biol Chem. 2013;288:19177–19183. doi: 10.1074/jbc.M113.467407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nkembo AT, Ntantie E, Salako OO, Amissah F, Poku RA, Latinwo LM, Lamango NS. The antiangiogenic effects of polyisoprenylated cysteinyl amide inhibitors in HUVEC, chick embryo and zebrafish is dependent on the polyisoprenyl moiety. Oncotarget. 2016;7:68194–68205. doi: 10.18632/oncotarget.11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S, Zhao Q, Xiang H, Liu M, Zhang Q, Xue W, Song B. Antiproliferative activity and apoptosis-inducing mechanism of constituents from Toona sinensis on human cancer cells. Cancer Cell Int. 2013;13:12. doi: 10.1186/1475-2867-13-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winter RN, Kramer A, Borkowski A, Kyprianou N. Loss of caspase-1 and caspase-3 protein expression in human prostate cancer. Cancer Res. 2001;61:1227–1232. [PubMed] [Google Scholar]
- 28.Schlomm T, Kirstein P, Iwers L, Daniel B, Steuber T, Walz J, Chun FH, Haese A, Kollermann J, Graefen M, Huland H, Sauter G, Simon R, Erbersdobler A. Clinical significance of epidermal growth factor receptor protein overexpression and gene copy number gains in prostate cancer. Clin Cancer Res. 2007;13:6579–6584. doi: 10.1158/1078-0432.CCR-07-1257. [DOI] [PubMed] [Google Scholar]
- 29.Siu MK, Abou-Kheir W, Yin JJ, Chang YS, Barrett B, Suau F, Casey O, Chen WY, Fang L, Hynes P, Hsieh YY, Liu YN, Huang J, Kelly K. Loss of EGFR signaling regulated miR-203 promotes prostate cancer bone metastasis and tyrosine kinase inhibitors resistance. Oncotarget. 2014;5:3770–3784. doi: 10.18632/oncotarget.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang P, Henning SM, Magyar CE, Elshimali Y, Heber D, Vadgama JV. Green tea and quercetin sensitize PC-3 xenograft prostate tumors to docetaxel chemotherapy. J Exp Clin Cancer Res. 2016;35:73. doi: 10.1186/s13046-016-0351-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reif S, Weis B, Aeed H, Gana-Weis M, Zaidel L, Avni Y, Romanelli RG, Pinzani M, Kloog Y, Bruck R. The Ras antagonist, farnesylthiosalicylic acid (FTS), inhibits experimentally-induced liver cirrhosis in rats. J Hepatol. 1999;31:1053–1061. doi: 10.1016/s0168-8278(99)80318-3. [DOI] [PubMed] [Google Scholar]
- 32.Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta. 2009;1796:91–98. doi: 10.1016/j.bbcan.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 33.Aznar S, Lacal JC. Rho signals to cell growth and apoptosis. Cancer Lett. 2001;165:1–10. doi: 10.1016/s0304-3835(01)00412-8. [DOI] [PubMed] [Google Scholar]
- 34.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thirugnanam S, Xu L, Ramaswamy K, Gnanasekar M. Glycyrrhizin induces apoptosis in prostate cancer cell lines DU-145 and LNCaP. Oncol Rep. 2008;20:1387–1392. [PubMed] [Google Scholar]
- 36.Rothermund CA, Kondrikov D, Lin MF, Vishwanatha JK. Regulation of Bcl-2 during androgen-unresponsive progression of prostate cancer. Prostate Cancer Prostatic Dis. 2002;5:236–245. doi: 10.1038/sj.pcan.4500582. [DOI] [PubMed] [Google Scholar]
- 37.Laufer M, Denmeade SR, Sinibaldi VJ, Carducci MA, Eisenberger MA. Complete androgen blockade for prostate cancer: what went wrong? J Urol. 2000;164:3–9. [PubMed] [Google Scholar]
- 38.Thompson IM, Ankerst DP, Chi C, Goodman PJ, Tangen CM, Lucia MS, Feng Z, Parnes HL, Coltman CA Jr. Assessing prostate cancer risk: results from the Prostate Cancer Prevention Trial. J Natl Cancer Inst. 2006;98:529–534. doi: 10.1093/jnci/djj131. [DOI] [PubMed] [Google Scholar]
- 39.Chen M, Knifley T, Subramanian T, Spielmann HP, O’Connor KL. Use of synthetic isoprenoids to target protein prenylation and Rho GTPases in breast cancer invasion. PloS One. 2014;9:e89892. doi: 10.1371/journal.pone.0089892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–255. [PMC free article] [PubMed] [Google Scholar]