

Abstract
Background
Cigarette smoking plays a major role in cardiovascular diseases. The acute effects of cigarette smoking produce central nervous system–mediated activation of the sympathetic nervous system. The overactive sympathetic nervous system stimulates the secretion of serotonin (5‐HT) and catecholamine into blood at supraphysiological levels. The correlation between these pathological conditions induced by smoking and the increased risk of thrombosis has not been thoroughly investigated. The goal of our study was to explore cigarette smoking–associated changes in platelet biology mediated by elevated 5‐HT and catecholamine levels in blood plasma.
Methods and Results
Using blood samples collected from healthy nonsmokers and smokers (15 minutes after smoking), we determined that cigarette smoking increased the plasma 5‐HT/catecholamine concentration by several fold and the percent aggregation of platelets 2‐fold. Liquid chromatography–tandem mass spectrometry analysis of proteins eluted from platelet plasma membranes of smokers and nonsmokers demonstrated that GTPase‐activating proteins and proteins participating in the actin cytoskeletal network were differentially and significantly elevated in smokers' platelet membranes compared with those of nonsmokers. Interestingly, Matrix‐assisted laser desorption/ionization–mass spectrometry analyses of the glycans eluted from platelet plasma membranes of the smokers demonstrated that the level and structures of glycans are different from the nonsmokers' platelet surface glycans. Pharmacological blockade of 5‐HT or catecholamine receptors counteracted the 5‐HT/catecholamine‐mediated aggregation and altered the level and composition of glycan on platelet surfaces.
Conclusions
Based on our findings, we propose that smoking‐associated 5‐HT/catecholamine signaling accelerates the trafficking dynamics of platelets, and this remodels the surface proteins and glycans and predisposes platelets to hyperactive levels. Smokers' platelets also had correspondingly higher resting concentrations of intracellular calcium and transglutaminase activity. These findings suggest a link among smoking, platelet 5‐HT, catecholamine signaling, and their downstream effectors—including phospholipase C and inositol‐1,4,5‐triphosphate pathways—resulting in an increased tonic level of platelet activation in smokers.
Keywords: catecholamine, circulation, human, receptor blocker, serotonin, smoking
Subject Categories: Cell Signalling/Signal Transduction, Mechanisms, Platelets, Proteomics, Translational Studies
Introduction
Cigarette smoking is the most alterable risk factor contributing to premature morbidity and mortality in the United States, accounting for ≈430 000 deaths annually.1 Smoking is a major cause of cardiovascular disease (CVD) and acts synergistically with other risk factors to substantially increase the risk of CVD.2 In 2000, >1 in 10 deaths worldwide from CVDs were attributed to smoking,3 and in the United States, smoking accounted for 33% of all deaths from CVD and 20% of deaths from ischemic heart disease in persons >35 years.4 As many as 30% of all deaths from coronary heart disease each year in the United States are attributable to cigarette smoking, and the risk is strongly dose dependent.5 Smoking also nearly doubles the risk of ischemic stroke and increases the risk of peripheral vascular disease, cancer, chronic lung disease, and many other chronic diseases. These facts emphasize the significant effects of cigarette smoking on overall health.
Clinical and preclinical studies report that the acute effects of cigarette smoking include central nervous system–mediated (CNS‐mediated) activation of the sympathetic nervous system,6, 7, 8 which stimulates secretion of prothrombotic molecules, such as serotonin (5‐HT), from the enterochromaffin cells of intestines and catecholamines, epinephrine, norepinephrine, and dopamine from adrenal glands and various tissues into the blood at supraphysiological levels.9, 10 In addition to smoking, episodic incidents, such as stress or anxiety, or antihypertensive drugs produce an overactive sympathetic nervous system.11, 12, 13 The individual effects of catecholamines and 5‐HT on cardiovascular conditions have been well established in in vitro study models.14, 15, 16, 17, 18, 19 However, their additive effects on platelet biology have not been evaluated thoroughly.
An overactive sympathetic nervous system stimulates the secretion of 5‐HT and catecholamine from peripheral tissues to the circulation. On stimulation, enterochromaffin cells of the intestine secrete 5‐HT and adrenal glands, and other tissues secrete catecholamine into the bloodstream. From the circulation, 5‐HT and catecholamine are taken up by specific transporters expressed on platelets and endothelial cells. In addition, platelet cell‐signaling responses are initiated by binding of 5‐HT and catecholamines to G protein–coupled receptors on platelet surfaces, specifically, the Gq‐coupled 5HT2A, Gs‐coupled β2, and Gi‐coupled α2 adrenergic receptors (Figure 1). Activation of each of these receptors has been shown to regulate platelet aggregation.14, 15, 16, 17, 18, 19 Platelet activation is also amplified by the exocytosis of alpha and dense granules, which store 5‐HT and catecholamine as well as procoagulant molecules.20 Whereas the G protein–dependent signals leading to platelet activation are well accepted, less appreciated is the coordinated interaction of these 5‐HT‐ and catecholamine‐initiated signals with 5‐HT uptake into the platelet cytoplasm, which ultimately allows transamidation of cytoplasmic 5‐HT to small GTPases to enhance secretion of platelet granules. The specific pathways are outlined as follows: Studies from others and our laboratory indicate that 5‐HT signaling through Gq activates phospholipase C (PLC) and results in the hydrolysis of PIP2 (phosphatidylinositol 4,5‐biphosphate) to IP3 (inositol‐1,4,5‐triphosphate).21, 22, 23 IP3 activates the serine/threonine protein kinase C family and facilitates Ca2+ mobilization (Figure 1). Catecholamine (norepinephrine/epinephrine) stimulates α2‐adrenoceptor Gi‐dependent signaling to inhibit adenylyl cyclase (AC) and activate phosphoinositide 3‐kinases, leading to biphasic aggregation of platelets.19 Norepinephrine signaling through this pathway also activates Src family kinases20, 22 to allow PLC‐dependent liberation of PIP2 and, ultimately, IP3, which facilitates Ca2+ mobilization.16, 22 Of particular relevance for our study, elevation of the cytoplasmic free Ca2+ concentration has been shown to activate transglutaminase (TGase), which transamidates 5‐HT to small GTP binding proteins (GTPases).16, 22, 23, 24, 25 Binding of epinephrine/norepinephrine to the platelet surface β2‐receptor signaling in platelets is more controversial but is conventionally known to signal through the Gs family of G proteins and activates AC to increase the intracellular concentration of the second messenger cAMP, a powerful inhibitor of platelet aggregation. High concentrations of cAMP resulting in protein kinase A activation have been shown to allow reorganization of the actin/myosin cytoskeletal network in a Rho‐GTP–dependent manner.14, 15, 18 Consequently, transamidation of Rho and Rab GTPases are important for platelet cytoskeletal reorganization and secretory behavior, respectively.
Figure 1.
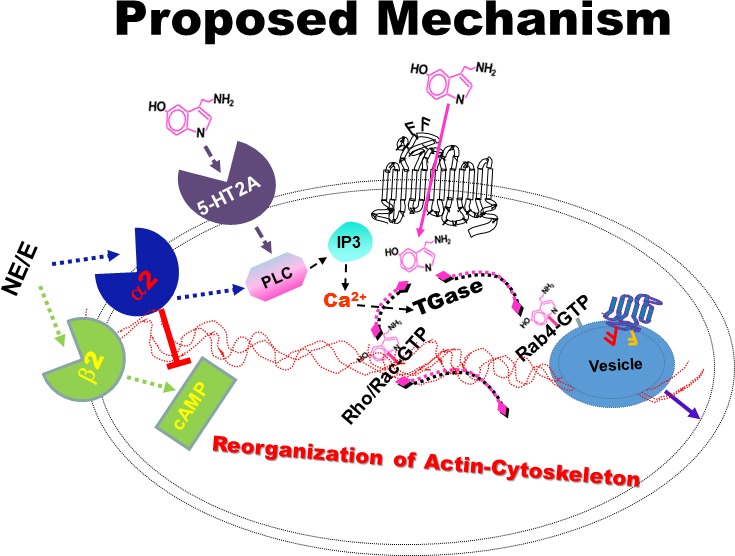
Proposed mechanism by which 5‐HT/catecholamine signaling facilitates the membrane trafficking of N‐glycan. 5‐HT and NE signaling activate 5‐HT2A and the α2 receptor, which elevates cytosolic free Ca2+/TGase via converting PIP2 (phosphatidylinositol 4,5‐biphosphate) to PIP3 pathway. Activated TGase serotonylates the small GTPases to place them in GTP, active form in which they facilitate the translocation of N‐glycan to the plasma membrane. E indicates epinephrine; IP3, inositol‐1,4,5‐triphosphate; NE, norepinephrine; PLC, phospholipase C; TGase, transglutaminase.
We hypothesize that following cigarette smoking, IP3 is activated by 5‐HT2A and α2‐receptor signals to alter the cytoskeletal network in platelets and to counteract the tonic inhibitory effect of cAMP on platelets. In the current study, we tested this hypothesis. Interestingly, liquid chromatography and mass spectrometry (MS) analysis of proteins eluted from platelet plasma membranes of smokers and nonsmokers demonstrated that GTPase‐activating proteins and proteins that participate in the actin cytoskeletal network were differentially and significantly elevated in smokers' platelet membranes compared with those of nonsmokers. Furthermore, MALDI (matrix‐assisted laser desorption/ionization) MS analysis determined that cigarette smoking elevated the surface level of double‐sialylated N‐glycan by 30%. These increases are due to the additive effect of 5‐HT and adrenergic receptor signaling. A mixture of 5‐HT‐ and catecholamine‐treated nonsmokers' platelets showed elevation of plasma glycan level and percent aggregation rates of platelets to the levels found in smokers' plasma recapitulates, whereas pharmacologic blockade of 5‐HT and adrenergic receptors reduced these levels to background. Taken together, these data suggest that smoking‐associated elevated levels of plasma 5‐HT and catecholamine make platelets prone to aggregation in part by changing the cytoskeletal network to accelerate the movement of N‐glycan to the platelet surface.
Methods
Participants
Blood samples were obtained from healthy male nonsmoking volunteers aged <30 years and from healthy young adult daily cigarette smokers. To avoid variations in platelet age during the menstrual cycle, only male smokers were included in our studies. The study was approved by the University of Arkansas for Medical Sciences (UAMS) institutional review board. The health status of participants was determined by their own physicians. Older participants (aged >30 years) were excluded to minimize variation in the platelet aggregation rate that occurs with age‐dependent changes in endothelium26, 27 and variation that may occur with advancing subclinical atherosclerosis.
Participants in these studies were recruited from among graduate school and medical school students. Their smoking habits were very similar: ∼10–12 cigarettes per day. All of our donors started smoking at college age (<20 years). The smokers' blood was withdrawn within 15 minutes of cigarette smoking. No distinctions were made with regard to race, ethnicity, or national origin, but the majority were white.
Participants signed the informed consent and then completed the health questionnaire and underwent blood pressure screening. Donors were excluded if they had hypertension or diabetes mellitus or were taking any of the following medications: α‐blockers, β‐blockers, aspirin, other platelet inhibitors, medications that alter serotonin metabolism or uptake, herbal supplements, or recreational drugs.
Blood Handling and Platelet Isolation
In total, 25 mL of peripheral venous blood was drawn into a heparin‐containing tube by venipuncture from an antecubital vein. Platelet‐rich plasma was prepared by adding one‐half volume of Tyrode‐HEPES buffer to blood and centrifugation of blood at 1.0×103 rotations per minute for 10 minutes. In each assay, a dilution of 300 000/μL of platelets in blood was used.10
Quantitative Measurement of 5‐HT and Catecholamine Levels by Competitive ELISA
Plasma 5‐HT and catecholamine levels were determined by a competitive ELISA technique, following the manufacturer's instructions (Rocky Mountain Diagnostics BA E‐5600; 5‐HT assays: Rocky Mountain Diagnostics BA E‐8900).
Aggregation Assay
For aggregation assays, platelets in plasma were prepared, and platelet counts were normalized (300 000/μL) using a Hemavet 950 system. The response to collagen (3 μg/mL) as a platelet agonist was monitored by light transmittance.28
Flow Cytometry
The level of platelet activation was assessed using fluorescein isothiocyanate–labeled P‐selectin antibodies. Platelets (300 000/μL) were incubated in antibodies, and at the end of the incubation, 300 μL of 2% formaldehyde in PBS was added to stop the reaction.
The level of serotonin transporter proteins on the plasma membrane of platelets was determined using a special antibody that was designed by our group and generated by Proteintech Group, Inc against a synthetic peptide corresponding to one of the extracellular loops of serotonin transporter.28
Fluorescence‐activated cell sorting was performed; the samples were gated for single platelets based on forward and side scatter profiles, and 20 000 events were recorded and read at the UAMS Flow Cytometry Core Facility.
Fura‐2‐AM Loading Platelets and Ca2+ Measurements
Isolated platelets (300 000/μL) were suspended in Tyrode‐HEPES buffer. Platelets were loaded with Fura‐2‐AM (Fura‐2‐acetoxymethyl ester; 2 μmol/L) for 45 minutes at room temperature while protected from light. Thereafter, the platelet suspension was centrifuged at 700g for 10 minutes, and the platelet pellet was washed and resuspended in Tyrode‐HEPES buffer and stored in the dark until the measurement was performed. A standard curve was prepared by using calcium calibration buffers containing 10‐fold concentrates of the K2EGTA and CaEGTA reagents, which were used for dissociation constant determination. Ca2+‐dependent fluorescence signals were obtained using excitation at 340 nm and 380 nm, and fluorescence intensity ratios were detected at 510 nm.16 The intracellular Ca2+ levels were calculated by the use of a general formula, as previously described by the manufacturer (Invitrogen). Briefly, the intracellular calcium ([Ca2+]i in nmol/L) was calculated according to the formula [Ca2+]i=k d×(F−F min)/(F max−F), where k d is the Fura dissociation constant. The F max value was obtained with Triton X‐100 (0.1%) in the presence of saturating calcium (CaCl2 1 mmol/L), whereas the F min value was obtained with the divalent chelator EGTA (10 mmol/L) plus Tris (20 mmol/L; pH 8).
TGase Assay
Tissue TGase enzymatic activity was assayed by measuring the incorporation of [1.4(n)‐3H]putrescine dihydrocloride (14.4 Ci/mmol per liter) into N,N'‐dimethylcasein, as described previously.16 Briefly, platelet (300 000/μL of platelets) homogenates were incubated for 90 minutes at 37°C with [1.4(n)‐3H]‐putrescine dihydrocloride (2.5 mmol/L, 3.97 mCi/mmol), dithiothreitol (3.8 mmol/L), CaCl2 (2.5 mmol/L), and dimethylcasein (5 mg/mL). Purified tissue TGase (1 μg) from guinea pig liver was used as a positive control. The reaction was stopped by the addition of 50% trichloroacetic acid and precipitated with 10% BSA overnight at 4°C. After several wash steps, the amount of putrescine incorporation into precipitated proteins was determined by scintillation counting.
Sample Preparation and Liquid Chromatography–MS/MS
A 100‐μg aliquot of each sample was reduced with 10 mmol/L dithiothreitol at 60°C and alkylated with 50 mmol/L iodoacetamide at room temperature. Proteins were digested in solution using 1 μg trypsin at 37°C for 18 hours, the digest was quenched with formic acid, and peptides were desalted using solid‐phase extraction with the Empore C18 plate (3M). A 2‐μg aliquot of each peptide sample was analyzed by nano–liquid chromatography–MS/MS using an Easy‐nLC 1000 HPLC system (ThermoFisher) interfaced to a Q Exactive HF tandem mass spectrometer. Peptides were loaded directly onto a 75 μm×50 cm analytical column and eluted using a 4‐hour gradient at 300 nL/min; the column was packed with PepMap C18 2 μm resin. The mass spectrometer was operated in data‐dependent mode, with MS and MS/MS performed in the orbitrap at 70 000 and 17 500 full width at half maximum resolution, respectively. The 15 most abundant ions were selected for MS/MS from each MS scan. Dynamic exclusion and repeat settings ensured each ion was selected only once and excluded for 15 seconds thereafter.
Data were processed using MaxQuant v1.5.3.1, which served several functions.29 Recalibrated MS and MS/MS data were searched against the combined forward and reverse Swissprot human protein database. The database was appended with common background proteins. Search parameters were precursor mass tolerance 10 ppm, product ion mass tolerance 20 ppm, fully tryptic peptides only, fixed modification of carbamidomethyl cysteine, variable modifications of oxidized methionine, and protein N‐terminal acetylation. Data were filtered at 1% protein and peptide false discovery rate and required at least 1 unique peptide per protein. The extracted ion peak area of detected peptides was used to calculate protein intensity.
Data Analysis
Nonlinear regression fits of experimental and calculated data were performed with Origin software, which uses the Marquardt‐Levenberg nonlinear least squares curve fitting algorithm. Each figure shows a representative experiment that was performed at least 3 times. The unit of analysis is the mean of these replications. The statistical analysis given in the Results section is from multiple experiments. A random‐effects model for Figure 1 data was used that takes into consideration repeated measures for participants in different experimental conditions, that is, 9 nonsmokers with and without a combination of 5‐HT and catecholamine and 7 smokers after smoking 4 hours and 5 minutes. It should be noted that 3 smokers were measured only 5 minutes after smoking. If statistical significance was achieved at the 5% level of significance, the Tukey multiple comparison procedure was used to determine significance when performing all pairwise contrasts among experimental group means. Means, standard deviations, and unit sample sizes were calculated for the experimental groups. For the Tukey multiple comparison procedure, the differences among group means, adjusted confidence limits for group differences, and adjusted P values are displayed.
Results
Cigarette Smoking Elevates Plasma 5‐HT and Catecholamine Concentrations to Supraphysiological Levels
The acute effects of cigarette smoking on plasma levels of 5‐HT, epinephrine, norepinephrine, and dopamine were measured in blood samples collected from young (aged <30 years) healthy male nonsmokers (n=15) and smokers (n=10) 15 minutes or 4 hours after smoking (Table 1). The variations in between the measurements are presented as the standard deviation in Table 1. ELISA analyses showed that the levels of epinephrine (1.63‐fold), norepinephrine (2.4‐fold), and dopamine (1.7‐fold) were elevated in plasma 15 minutes after smoking. Consistent with our preliminary findings, Gu et al,30 using metabolomics to assay 5‐HT in blood samples from cigarette smokers, showed that cigarette smoking increased 5‐HT plasma concentrations several fold. At 4 hours after smoking, plasma levels of 5‐HT were 1.6‐fold higher than those of nonsmokers; at 15 minutes after smoking, plasma levels of 5‐HT were 4.7‐fold higher than those of nonsmokers and were 3.2‐fold higher than those of smokers 4 hours after smoking.
Table 1.
Compresence of the Platelets Isolated From Smokers' and Nonsmokers' Blood Samples
| Presence in Blood, ng/mL | ||||
|---|---|---|---|---|
| 5‐HT | Norepinephrine | Epinephrine | Dopamine | |
| Nonsmoker (n=15) | 0.95±0.14 | 6.12±0.62 | 1.94±0.22 | 103.27±16 |
| Smoker, 4 h not smoked (n=10) | 1.49±0.4 | 10.54±1.31 | 2.27±0.17 | 126±14 |
| Smoker, 15 min after smoked (n=10) | 4.5±0.73 | 14.67±2.64 | 3.18±0.45 | 174.2±11.5 |
Cigarette Smoking Predisposes Platelets to a Prothrombotic State
We examined platelet aggregation rates in blood samples from smokers 15 minutes and 4 hours after smoking. We also determined the effects of treating platelets from nonsmokers with a combination of 5‐HT and catecholamine to simulate the signaling milieu present in plasma after cigarette smoking. The 5‐HT and catecholamine were freshly prepared from stock solutions (final concentrations: 4.5 ng/mL 5‐HT, 15 ng/mL norepinephrine, 175 ng/mL dopamine).
Platelet‐reactivity status was determined by measuring collagen‐induced aggregation with an aggregometer and with fluorescence‐activated cell sorting analysis for P‐selectin, a known marker for secretion of α‐granule, which indicates activated platelets (Figure 2). Percentage of aggregation for nonsmokers' platelets was 67±5%. For those who had not smoked for 4 hours, percentage of platelet aggregation was 71±4.0% (not significant), but 15 minutes after smoking a cigarette, percentage of aggregation was 80±4.0%. When nonsmokers' platelets were pretreated with 5‐HT and catecholamine, aggregation was 85±5.0% (n=10 each; Figure 2A and 2B). In sum, the percentage of platelet aggregation for smokers was significantly higher (22%) than that of nonsmokers, and it was insignificantly higher 15 minutes after smoking than after having abstained for 4 hours.
Figure 2.
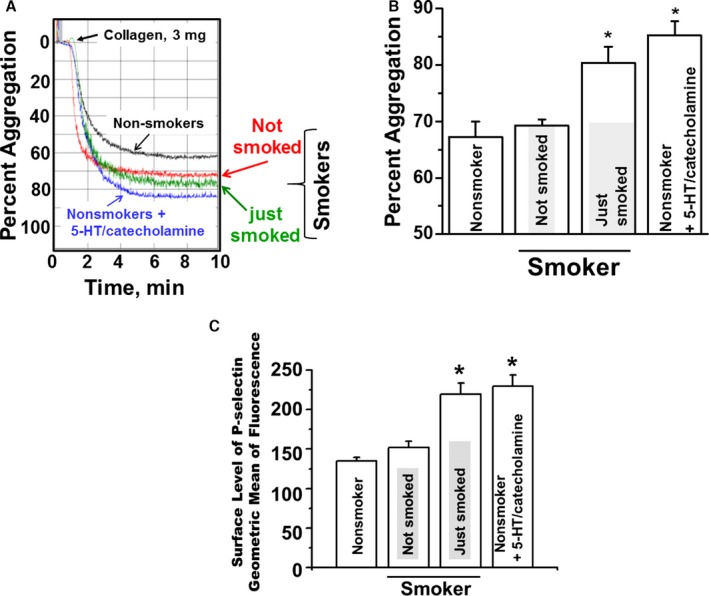
Aggregation rates of platelets from smoker and nonsmoker blood samples. A, Representative tracings of stirred platelet aggregation are shown. In response to stimulation with 3 μg/mL of collagen, smokers' platelets (300 000/μL) showed 20.5% higher aggregation than platelets isolated from nonsmokers' blood samples. B, When nonsmokers' platelets were treated with the mixture of 5‐HT/catecholamine, their aggregation rates were 28% higher than those of untreated platelets. Not smoked indicates platelets from smokers who abstained for 4 hours; just smoked indicates platelets from smokers 15 minutes after smoking; nonsmokers plus 5‐HT/catecholamine indicates platelets from nonsmokers treated in vitro with 5‐HT/catecholamine. C, P‐selectin flow cytometry. P‐selectin levels on platelets under conditions described in Figure 2B. Asterisks indicate statistically significant differences (P<0.01) between nonsmokers' platelets and smokers' or 5‐HT/catecholamine‐treated nonsmokers' platelets. All assays were performed in triplicate (n=10/group).
Platelets from all conditions also were examined using fluorescence‐activated cell sorting analysis to determine surface levels of the activation marker P‐selectin (Figure 2C). These findings agreed with the aggregometer data, indicating an increase in P‐selectin on platelet surfaces in 5‐HT/catecholamine‐treated platelets from nonsmokers or smokers just after smoking; P‐selectin levels modestly increased on platelets from abstinent smokers, but it was not statistically significant. Our preliminary results strongly indicate that smokers' platelets are prothrombotic before smoking, and this condition is elevated almost 2‐fold soon after smoking. The random‐effects model found significance among the experimental group means, and the subsequent Tukey multiple comparisons procedure found significance among all pairwise contrasts with the exception of nonsmokers versus smokers who had not smoked for 4 hours. The adjusted 95% familywise confidence intervals, standard errors, and adjusted P values for the differences in group means can be seen in Table 2. In addition, means, sample sizes, and standard deviations from platelet response to collagen stimulation is displayed in Table 3.
Table 2.
Tukey Procedure Contrasts
| Contrast | Estimate | SE | Lower CL | Upper CL | P Adjusted |
|---|---|---|---|---|---|
| Nonsmoker vs 5‐HT/catecholamine‐treated nonsmoker | −18.1 | 1.0 | −21.0 | −15.2 | 0.0 |
| Nonsmoker vs Smoker not smoked 4 h | −2.1 | 1.2 | −5.4 | 1.3 | 0.4 |
| Nonsmoker vs Smoker just smoked | −13.2 | 1.1 | −16.3 | −10.3 | 0.0 |
| 5‐HT/catecholamine‐treated nonsmoker vs smoker not smoked 4 h | 16.0 | 1.2 | 12.6 | 19.4 | 0.0 |
| 5‐HT/catecholamine‐treated nonsmoker vs smoker just smoked | 4.9 | 1.1 | 1.8 | 7.9 | 0.0 |
| Smoker just smoked vs not smoked 4 h | −11.1 | 1.1 | −14.2 | −8.1 | 0.0 |
CL indicates confidence limit.
Table 3.
Summary Statistic for Percent Aggregation of Platelets
| N | Mean | SD | |
|---|---|---|---|
| Group | |||
| Nonsmoker | 9 | 67.2 | 2.7 |
| 5‐HT/catecholamine‐treated nonsmoker | 9 | 85.3 | 2.5 |
| Smoker not smoked 4 h | 7 | 69.3 | 1.1 |
| Smoker just smoked | 10 | 80.4 | 2.8 |
| Overall | 35 | 76.1 | 8.0 |
5‐HT2A and Adrenergic Receptor Antagonists Counteract Enhanced Platelet Aggregation
We explored signaling pathways by which 5‐HT and catecholamine, at supraphysiological levels, facilitate membrane trafficking of N‐glycan to the platelet surface. Platelets isolated from nonsmokers' blood samples were pretreated with individual receptor antagonists and then treated with 5‐HT and catecholamines. The 5‐HT receptor (5‐HT2A) was blocked with sarpogrelate (10 μmol/L), and β2‐ or α2‐adrenergic receptors were blocked with propranolol (20 μmol/L) or atipamezole (1 μmol/L), respectively.8, 31, 32 Each receptor antagonist significantly decreased 5‐HT/catecholamine‐mediated platelet aggregation (reduced from 85.5±3.0% to 74±3.0%), but none decreased aggregation to the untreated level (Figure 3A and 3B). We then evaluated the combined effects of 3 blockers on aggregation of platelets treated with 5‐HT and catecholamines (as in the previous experiment). Pretreatment with the combination of 3 receptor antagonists resulted in 22% decreased aggregation (Figure 3C and 3D). Overall, aggregation without antagonists was 88±5.0% in 5‐HT/catecholamine‐treated platelets, but pretreatment with the 3 antagonists decreased aggregation to 67±3.0% (n=10). Although the individual receptor blockers decreased the aggregation rate and cell‐surface P‐selectin level, the mixture of blockers totally counteracted 5‐HT/catecholamine‐mediated platelet aggregation (Figure 3E). When platelets were first treated with 5‐HT and catecholamines and then with the blocker mixture, aggregation was not reduced (Figure 3F).
Figure 3.
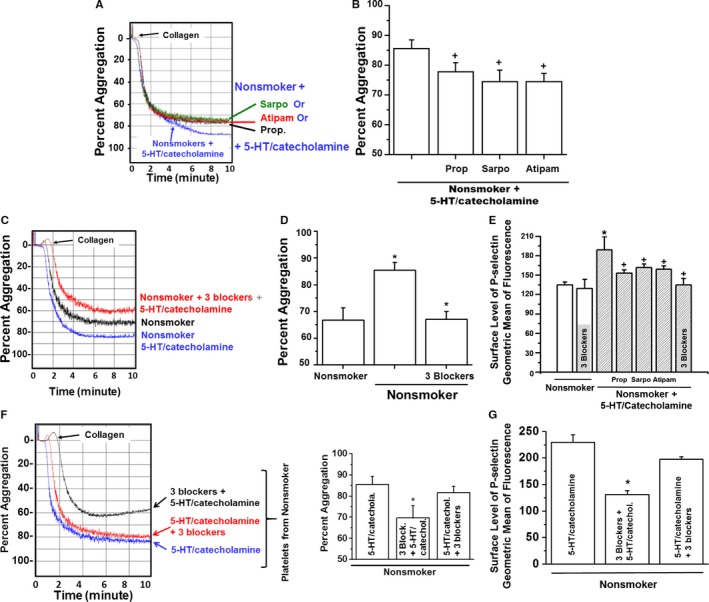
Effects of receptor antagonists on rates of platelet aggregation. Statistically significant differences are indicated between untreated and 5‐HT/catecholamine‐treated platelets (*) and receptor blocker–pretreated or only 5‐HT/catecholamine‐treated platelets (+). All assays were performed in triplicate (n=10 group). A, Platelets were first pretreated with the receptor blockers propranolol (Prop; β2‐blocker), sarpogrelate (Sarpo; 5‐HT2A blocker), or atipamezole (Atipam; α2‐blocker) for 15 minutes and then treated with 5‐HT/catecholamine for an additional 10 minutes. At the end of the treatments, platelets were stimulated with collagen, and their aggregation profiles were monitored in an aggregometer. B, Approximately 13% of the platelets from Sarpo‐ or Atipam‐treated platelets and 9% of Prop‐treated platelets were aggregated at the end of 10 minutes. The average of 4 measurements is presented in the bar graph. C, Additive effect of 3 blockers on 5‐HT/catecholamine‐treated platelet aggregation. Platelets were processed as describe in panel A then they were stimulated with the mixture of Sarpo, Prop, and Atipam for 15 minutes and then treated with 5‐HT/catecholamine for an additional 10 minutes. At the end of the treatments, platelets were stimulated with collagen, and their aggregation profile was monitored in an aggregometer. D, Approximately 21% of the platelets treated with the mixture of 3 drugs were aggregated at the end of 10 minutes. E, Each receptor blocker reduced the surface level of P‐selectin differently, but the mixture of 3 blockers totally counteracted the 5‐HT/catecholamine‐mediated elevation of P‐selectin level on platelet surfaces. The average of 4 measurements is presented in the bar graph. F, The combined effect of 3 blockers was evaluated by pretreating the platelets first with the mixture of the receptor blockers (Prop, Sarpo, and Atipam) for 15 minutes and then treated with 5‐HT/catecholamine for an additional 10 minutes or vice versa. At the end of the treatments, platelets were stimulated with collagen, and their aggregation profile was monitored in an aggregometer. Only ≈68% of the platelets from blocker mixture–treated platelets were aggregated at the end of 10 minutes; however, if the platelets were first treated with 5‐HT/catecholamine and then with the blocker mixture, this did not reduce the aggregation level. The average of 4 measurements is presented in the bar graph. G, The surface level of P‐selectin showed similar changes with the order of the treatments (blocker mixture and 5‐HT/catecholamine). The average of 4 measurements is presented in the bar graph.
The reversibility of 5‐HT/catecholamine‐mediated reactivity of platelets was tested by measuring nonsmokers' platelet aggregation after applying the antagonist combination either before or after treatment with 5‐HT/catecholamine treatment. The results of percentage of aggregation and platelet surface P‐selectin levels indicated that 5‐HT/catecholamine receptor blockade is effective only in preventing 5‐HT/catecholamine‐induced enhancement of aggregation if applied before 5‐HT/catecholamine pretreatment (Figure 3F and 3G).
Ca2+ Level and the TGase Activity of the 5‐HT/Catecholamine‐Treated Platelets
Stimulation of cells with high levels of 5‐HT is reported to induce the modification (transamidation) of small GTPases with 5‐HT (termed serotonylation).16, 22, 23, 25, 28 This modification may occur in a G protein–coupled receptor‐dependent manner via Ca2+ mobilization and activation of a Ca2+‐dependent enzyme, TGase in platelets. Consequently, we determined levels of free Ca2+ in our experimental models, as described previously.16 Briefly, platelets were loaded with 2 μmol/L Fura‐2‐AM, a calcium ion‐binding fluorochrome, characterized by distinct spectral properties in the presence of low and high concentrations of Ca2+. Fura‐2 does not interfere with platelet activation.16 We first compared levels of free intracellular Ca2+ in platelets from nonsmokers or smokers 15 minutes after smoking (Figure 4A). The level of free intracellular Ca2+ was 48±1.5% higher in platelets from smokers than in those from nonsmokers. Next, we analyzed the direct effects of 5‐HT and catecholamines and found that free intracellular Ca2+ was 36.5±2.63% higher in platelets treated with 5‐HT/catecholamine than in untreated platelets (Figure 4B).
Figure 4.
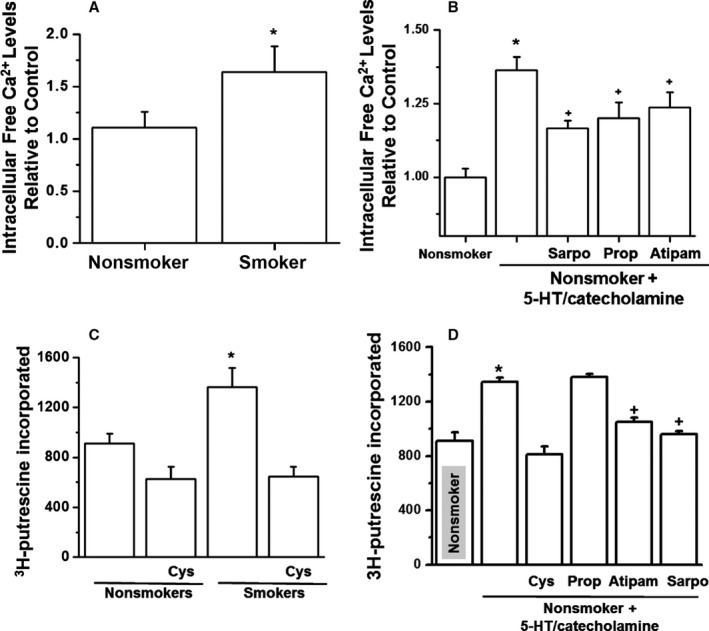
Ca2+ mobilization in platelets after 5‐HT/catecholamine treatment. Fura‐2‐AM (Fura‐2‐acetoxymethyl ester) was loaded in nonsmokers' platelets or those from smokers 15 minutes after smoking (A) or in nonsmokers' platelets treated with 5‐HT/catecholamine (B). The Ca2+ levels were 1.9‐fold higher in smokers' or 5‐HT/catecholamine‐treated nonsmokers' platelets than in untreated platelets. (C) The incorporation of 3H‐putrescine into N,N‐dimethylcasein was measured with the use of purified bovine transglutaminase (TGase), which served as a positive control. TGase activity in platelet lysates showed a 40% increase in smokers'. Similar elevation was noted in 5‐HT/catecholamine‐treated nonsmokers' platelets. (D) Atipam and Sarpo reduced the Ca level as well as TGase activity against 5‐HT/catecholamine treatment. Statistical differences are indicated between untreated and 5‐HT/catecholamine‐treated platelets (*) and receptor blocker and 5‐HT/catecholamine–treated or only 5‐HT/catecholamine‐treated platelets (+). All assays were performed in triplicate (n=10 group). Atipam indicates atipamezole; Cys, cystamine; Prop, propranolol; Sarpo, sarpogrelate.
TGase activity was determined by measuring the amount of 3H‐putrescine incorporation into precipitated platelet proteins with scintillation counting.16, 28 TGase purified from guinea pig liver was used as a positive control. TGase activity was 44% higher in 5‐HT/catecholamine‐treated nonsmokers' platelets and in platelets isolated from smokers' blood (Figure 4C and 4D, respectively). Pretreatment with the TGase inhibitor cystamine (10 μmol/L) reduced the TGase activity of 5‐HT/catecholamine‐treated platelets to that of untreated platelets.
Granulophysin Confirms Exocytosis of the Granules
Several studies indicate that platelet activation is associated with enhanced signaling through multiple pathways, including activation of small GTPase‐mediated exocytosis of granules. We examined the involvement of 3 receptors in 5‐HT/catecholamine‐mediated exocytosis of dense granules16 by monitoring the plasma membrane level of granulophysin, a marker of dense‐granule exocytosis, via flow cytometry (Figure 5). Surface granulophysin exposure was enhanced >2‐fold in platelets from smokers compared with those from nonsmokers (Figure 5A). Antagonists of 5‐HT and α2‐adrenergic receptors reduced the level of granulophysin on 5‐HT/catecholamine‐treated platelet surfaces (Figure 5B). In further support of our hypothesis, we found that blocking the 5‐HT/catecholamine‐mediated activity of TGase by cystamine reduced the surface level of granulophysin on platelets to the control level (Figure 5B).
Figure 5.
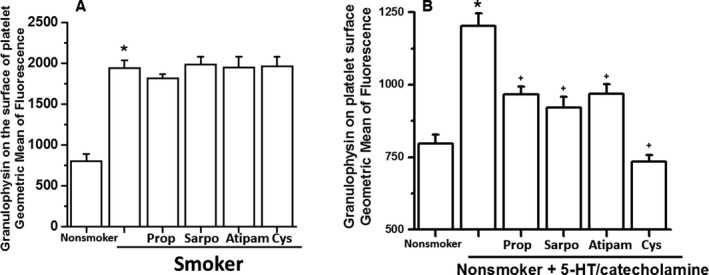
Level of granulophysin on the platelet surface. A, 5‐HT/catecholamine‐treated or ‐untreated platelets were evaluated by measuring the level of granulophysin, a surrogate marker for dense‐granule exocytosis. The flow cytometry data revealed a 50% elevation in the expression level of granulophysin on the surface of smokers' platelets. Treatment of the smokers' platelets with receptor blocker did not change the result. B, Each receptor blocker reduced the surface level of granulophysin differently, but the mixture of 3 receptor blockers totally counteracted the 5‐HT/catecholamine‐mediated elevation of granulophysin level on platelet surfaces. Statistical differences are indicated between untreated and 5‐HT/catecholamine‐treated platelets (*) and receptor blocker–pretreated or only 5‐HT/catecholamine‐treated platelets (+). All assays were performed in triplicate (n=10 group). Atipam indicates atipamezole; Cys, cystamine; Prop, propranolol; Sarpo, sarpogrelate.
Smoking‐Associated Changes on the Platelet Plasma Membrane Proteins
We performed proteomic analyses of plasma membranes isolated from platelets from smokers' and nonsmokers' blood samples. Platelet plasma membrane vesicles were isolated and reduced with dithiothreitol and alkylated with iodoacetamide then digested with trypsin (MS Biowork, MI). The digest was analyzed by nano LC/MS/MS interfaced to a tandem mass spectrometer. Data were processed and recalibrated MS and MS/MS data were searched in Swissprot human protein database. The database was appended with common background proteins.29
Interestingly, we identified some proteins on membranes of smokers' platelets whose levels were elevated several fold, compared with those of nonsmokers'. Some of these proteins are already reported to be involved in the reorganization of cytoskeletal proteins (Table 4).
Surface levels of glycoprotein P‐selectin are elevated on smokers' platelets and on nonsmokers' platelets that were treated with 5‐HT and catecholamines, indicating activation‐dependent membrane trafficking of the glycoprotein. We determined whether smoking‐mediated activation delivers only P‐selectin or also other glycoproteins to the plasma membranes of hyperactive platelets.
Plasma membranes were isolated from an equal number of platelets from blood samples of smokers and nonsmokers. Total glycoprotein was eluted from the plasma membranes isolated from an equal number of platelets from the smokers' and nonsmokers' pooled blood samples (n=3 per group) and analyzed by MALDI‐MS (Figure 6A and 6B). The content of glycoproteins differed substantially in the kind of N‐glycan present; nonsmokers' platelets had twice as many (≈41%) high‐mannose‐type structures on their surface as smokers' platelets (≈19%). In contrast, the surfaces of platelets isolated from smokers' blood samples had more total sialylated N‐glycan (≈74%) than the surfaces of platelets from nonsmokers (≈56%). Among the sialylated glycans, total monosialylated and disialylated species were higher for smokers than nonsmokers (monosialylated, 19% versus 8%; disialylated, 47% versus 36%) (Figure 6B). The relative abundance of N‐glycan released from the platelet surfaces was 30% higher for smokers' platelets than for nonsmokers' platelets (Figure 6A inset).31, 32, 33, 34 Consequently, our data indicate that smoking produces heightened platelet responsiveness (measured by stirred platelet aggregation) and increased levels of platelet‐activation markers and sialylated N‐glycan on platelet surfaces.
Figure 6.
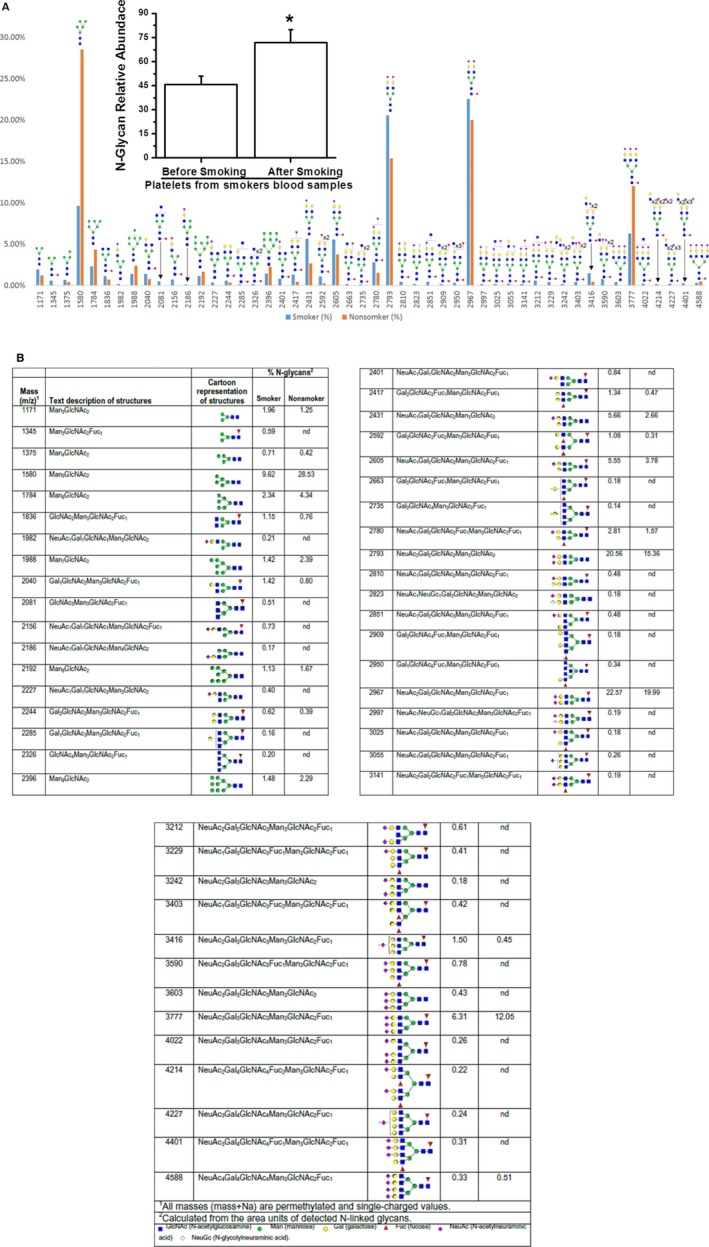
Full MALDI TOF/TOF MS of permethylated N‐glycan from plasma membrane vesicles of (A) nonsmokers' and smokers' platelets. Plasma membrane vesicles from nonsmokers' and smokers' platelets, in equal number, were isolated from blood pool (n=3 per group).8 The N‐linked glycans on membrane vesicles were released enzymatically by PNGase F. Released N‐glycan were permethylated and profiled by an LTQ Orbitrap Discovery mass spectrometer equipped with a nanospray ion source. The glycoproteins eluted from treated and untreated platelet plasma membrane showed differences in the type of N‐glycan. Smokers' platelet plasma membrane had more total sialylated N‐glycan (~74%) than untreated (~56%) platelet (inset). The relative abundance of sialylated N‐glycan on plasma membrane of smokers' platelets was 1.3‐fold higher than that on nonsmokers' platelets. The asterisk (*) indicates a statistically significant difference between nonsmokers' and smokers' platelets (P<0.007; results are representative of 3 individual experiments). (inset) Glycoproteins detected by MALDI TOF/TOF/MS. B, Glycans eluted from the plasma membrane of nonsmokers' and smokers' platelets.
Discussion
Cigarette smoking is a major cause of cardiovascular disease, stroke, aortic aneurysm, and peripheral artery diseases. The risk is seen both as an increased risk of acute thrombosis of narrowed vessels and as an increased degree of atherosclerosis in the blood vessels involved. The occurrence of myocardial infarction after smoking35 supports the role of smoking in thrombosis. In addition, abundant evidence demonstrates that smoking contributes to development of atherosclerotic plaque.35, 36, 37, 38, 39, 40
Cigarette smoking delivers several compounds into blood either directly40 or indirectly via central nervous system activation of the sympathetic nervous system6, 7 to stimulate the secretion of catecholamine and 5‐HT into the blood at supraphysiological levels.8, 9, 10 These compounds act on platelets through heterotrimeric G protein–coupled receptors such as 5‐HT2A, β2‐, and α2‐receptors. In this study, we evaluated the combined effect of 5‐HT and adrenergic signaling in cigarette smoking–mediated acute platelet activation, with the hypothesis that combined blockade of these platelet receptors in chronic smokers might reduce their risk of thrombosis.
Cigarette smoking acutely changes the glycan structure from high mannose to sialylated N‐glycan, indicating that remodeling the platelet surface with N‐glycan may establish a more adhesive environment and provide a mechanism by which smoking contributes to platelet activation and thrombosis. The major question in our studies is how the platelet surface glycan structure changes in 15 minutes.
Platelets contain a sufficient pool of sugar nucleotides and glycosyltransferases, which are normally sequestered in intracellular compartments and are released to the cell surface on activation.31, 41, 42, 43 We hypothesize that changes in glycan structures on the smokers' platelets are induced by the catalytic activities of glycosylation enzymes that, in response to smoking, are translocated to the plasma membrane. This hypothesis is supported by preliminary data demonstrating significantly elevated levels of specific proteins found exclusively on the surface of smokers' platelets including zyxin, profilin, G protein–signaling modulator 3, and RhoC (Table 4). All play critical roles in membrane trafficking of secretory vesicles by altering the actin–myosin network, binding G protein–coupled receptors, or activating small GTPases.44, 45, 46 Consistent with our findings, a recent study with the proteomic analysis of the hippocampus and cortex of mice treated for 6 months with nicotine also showed an elevation in the expression of spectrin, profilin, and zyxin.44, 46 Similar changes were determined on the surface of nonsmokers' platelets following a short treatment with 5‐HT/catecholamine at supraphysiological levels, suggesting the direct involvement of 5‐HT/catecholamine in translocation of N‐glycan to the plasma membrane. Furthermore, the receptor blockers against 5‐HT and adrenergic receptors provided evidence of the involvement of IP3 and AC pathways in N‐glycan movement to the platelet surface as well as the formation of hyperactive platelets.
When 5‐HT, epinephrine, and norepinephrine bind to the platelet surface, 5‐HT2A and β2‐ and α2‐adrenergic G protein–coupled receptors Gq, Gs, and Gi, respectively, are activated and transduce their signals on independent but interconnected pathways in promoting platelet aggregation (Figure 1). Activation of 5‐HT2A and catecholamine (norepinephrine/epinephrine) α2‐adrenergic receptor signaling activates PLC and results in hydrolysis of PIP2 to IP3 formation.18, 19, 20 IP3 activates serine/threonine protein kinase C and facilitates Ca2+ mobilization. Elevated free Ca2+ in the cytoplasm activates TGase, which modifies Rab4/Rho/Rac with 5‐HT (serotonylation).16, 21, 22, 23, 24, 25, 38 The α2‐adrenergic receptors, however, enhance aggregation by acting on 2 signaling pathways; these receptors stimulate PLC and reduce cAMP by inhibiting AC, an antiaggregation molecule (Figure 1). In contrast, the β2‐adrenergic receptor activates AC and increases the cAMP level; therefore, β2 signaling reduces platelet aggregation. Blocking β2 receptors with propranolol decreased the percentage of aggregation of platelets from the smokers' blood sample just after a smoked cigarette (Figure 1). These findings suggest that in the presence of 5‐HT/catecholamine at supraphysiological levels, the β2 receptor responds differently or α2 suppresses the protective role of cAMP by masking the β2 receptor.47, 48 As the proteomic data support by showing the changes in actin binding proteins in smokers' platelets, the actin–myosin (actomyosin) cytoskeletal network is remodeled in smokers' platelets, enhancing aggregation. Elevation in Ca2+ and TGase activity in smokers' platelets additionally implies the activation of small GTPases.
The Rab protein family of small GTP binding proteins regulates vesicular traffic.16, 21, 22, 23, 25, 28 In human platelets, Rab4, Rho, and Rac are reported to be modified with 5‐HT by TGase.16, 21, 22, 23, 25, 28 Rab4 is associated with early endosomes and regulates membrane recycling.16, 21, 22, 23, 25, 28 All Rab proteins contain highly conserved domains required for guanine nucleotide binding, GTP/GDP exchange, and GTP hydrolysis that are essential for their proper targeting and function.25 In heterologous and endogenous systems, we reported that TGase transamidates the GTP hydrolysis domain of Rab4 and Rac, which produced constitutively active Rab4 in heterologous cell culture using various mutant Rab proteins.16, 25, 28 These findings were further confirmed with radioimmunoprecipitation assays for the transamidation of Rab4 and Rac with 3H‐5HT and their GTP‐binding ability with35S‐γ‐GTP in in vitro study models.16, 25, 28 Rab4 facilitates the exocytosis of small vesicles in a GTP active form in which the small vesicles use the cytoskeletal network to produce vesicle translocation to the plasma membrane.21 Based on all of these current and previous findings, we hypothesize that plasma 5‐HT and catecholamine (at supraphysiological levels) in smokers' blood accelerates the translocation of vesicles to the platelet surface by keeping small GTPases in constitutively active (GTP‐bound) forms and by rearranging the cytoskeletal network.
Our data suggest a model in which the translocation of glycosylation enzymes to the platelet surface due to 5‐HT/catecholamine signaling is supplemented by microvesicle‐dependent delivery of molecules. It is reported that cigarette smoking induces the release of prothrombotic membrane microvesicles both in vitro49 and in vivo.50 Consequently, our future studies will investigate the impact of supplementation of microvesicles from smokers' versus nonsmokers' blood samples to provide additional mechanistic insights into these findings.
Findings from these studies will address the paradigm‐shifting question of how smoking alters the proteins and glycans on the surface of platelets and how these surface alterations predispose platelets to thrombus formation, a previously unrecognized but critical issue in the field. We suggest that reducing the activities of 5‐HTand adrenergic receptors restores the physiological profile on the platelet surface, which could provide a novel pharmacological strategy to prevent smoking‐associated thrombosis. In addition, we limited our study group to young male participants (aged <30 years) to eliminate additional smoking‐independent variables that may influence smoking‐associated membrane trafficking. Future studies should extend this study to other populations. Last, MALDI‐MS analysis found smoking‐associated changes on the platelet surface glycan structure from high‐mannose‐type glycan to sialylated N‐glycans; however, it is not known whether the high‐mannose‐type glycans protect resting platelets from aggregation. This will be an important area on which to focus in future studies.
Our studies tested the impact of individual receptor blockers before and after smoking and in nonsmokers' platelets following 5‐HT/catecholamine treatment. Individual blockers reduced the percentage of aggregation to a certain level, but the best result was achieved by the combination of these 3 blockers. Pretreatment with the 3 blockers before 5‐HT/catecholamine treatment reduced the percentage of aggregation significantly. Smoking‐mediated cardiovascular risks are characterized by the number and frequency of cigarettes smoked and the duration of smoking. Stopping cigarette smoking may substantially reduce risks of various CVDs, yet the threshold level of 5‐HT and catecholamine in smokers' plasma stays higher than that of nonsmokers.
Sources of Funding
The carbohydrate analyses in these studies were supported in part by the National Institutes of Health grants 1S10OD018530 and P41GM10349010 to the Complex Carbohydrate Research Center. These works were supported by NIH Heart Lung and Blood Institute HL108825 to Elliott and NIH Heart Lung and Blood Institute HL091196 to Kilic and American Heart Association (13GRNT17240014) to Kilic and Hornick Endowment for Research into Stroke and Related Disorders to Kilic.
Disclosures
None.
Acknowledgments
We gratefully acknowledge the TRI‐UAMS nurses, Shawna Owens and Susan Blair. We also extend our thanks to Andrea Harris at Flow Cytometry Core Facility and Richard Jones at MS Bioworks, LLC for working with us on the proteomic analysis of our samples.
(J Am Heart Assoc. 2017;6:e005465 DOI: 10.1161/JAHA.116.005465.)28522678
References
- 1. Nelson DE, Nelson DE, Kirkendall RS, Lawton RL, Chrismon JH, Merritt RK, Arday DA, Giovino GA. Surveillance for smoking–attributable mortality and years of potential life lost; by state—United States, 1990. MMWR CDC. 1994;43:1–8. [PubMed] [Google Scholar]
- 2. Anderson KM, Wilson PW, Odell PM, Kannel WD. An updated coronary risk profile: a statement for health professionals. Circulation. 1991;83:356–362. [DOI] [PubMed] [Google Scholar]
- 3. Ezzati M, Henley SJ, Thun MJ, Lopez AD. Role of smoking in global and regional cardiovascular mortality. Circulation. 2005;112:489–497. [DOI] [PubMed] [Google Scholar]
- 4. Centers for Disease Control and Prevention . Smoking‐attributable mortality, years of potential life lost, and productivity losses—United States, 2000–2004. Morb Mortal Wkly Rep. 2008;57:1226–1228. [PubMed] [Google Scholar]
- 5. Ockene IS, Miller NH. Cigarette smoking, cardiovascular disease, and stroke: a statement for healthcare professionals from the American Heart Association. American Heart Association Task Force on Risk Reduction. Circulation. 1997;96:3243–3247. [DOI] [PubMed] [Google Scholar]
- 6. Peterson DF, Coote JH, Gilbey MP, Futuro‐Neto HA. Differential pattern of sympathetic outflow during upper airway stimulation with smoke. Am J Physiol. 1983;245:433–437. [DOI] [PubMed] [Google Scholar]
- 7. Porchet HC, Benowitz NL, Sheiner LB, Copeland JR. Apparent tolerance to the acute effect of nicotine results in part from distribution kinetics. J Clin Invest. 1987;80:1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parati G, Esler M. The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur Heart J. 2012;33:1058–1066. [DOI] [PubMed] [Google Scholar]
- 9. Siess W, Lorenz R, Roth P, Weber PC. Plasma catecholamines, platelet aggregation and associated thromboxane formation after physical exercise, smoking or norepinephrine infusion. Circulation. 1982;66:44–48. [DOI] [PubMed] [Google Scholar]
- 10. Brenner B, Harney JT, Ahmed BA, Jeffus BC, Unal R, Mehta JL, Kilic F. Plasma serotonin levels and the platelet serotonin transporter. J Neurochem. 2007;102:206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grassi G, Grassi G, Seravalle G, Trevano FQ, Dell'oro R, Bolla G, Cuspidi C, Arenare F, Mancia G. Neurogenic abnormalities in masked hypertension. Hypertension. 2007;50:537–542. [DOI] [PubMed] [Google Scholar]
- 12. Martin CA, McGrath BP. White‐coat hypertension. Clin Exp Pharmacol Physiol. 2014;41:22–29. [DOI] [PubMed] [Google Scholar]
- 13. Middlekauff HR, Park J, Agrawal H, Gornbein JA. Abnormal sympathetic nerve activity in women exposed to cigarette smoke: a potential mechanism to explain increased cardiac risk. Am J Physiol Heart Circ Physiol. 2013;305:1560–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Woulfe DS. Platelet G protein‐coupled receptors in hemostasis and thrombosis. J Thromb Haemost. 2005;310:2193–2200. [DOI] [PubMed] [Google Scholar]
- 15. Smyth SS, Smyth SS, Woulfe DS, Weitz JI, Gachet C, Conley PB, Goodman SG, Roe MT, Kuliopulos A, Moliterno DJ, French PA, Steinhubl SR, Becker RC. G‐protein‐coupled receptors as signaling targets for antiplatelet therapy. Arterioscler Thromb Vasc Biol. 2009;29:449–457. [DOI] [PubMed] [Google Scholar]
- 16. Ziu E, Freyaldenhoven S, Mercado CP, Ahmed BA, Preeti P, Lensing S, Ware J, Kilic F. Down‐regulation of the serotonin transporter in hyperreactive platelets counteracts the pro‐thrombotic effect of serotonin. J Mol Cell Cardiol. 2012;52:1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fraer M, Kilic F. Serotonin: a different player in hypertension‐induced thrombosis. AHA‐Hypertension. 2015;65:942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Millan MJ, Marin P, Bockaert J, Mannoury la Cour C. Signaling at G‐protein‐coupled serotonin receptors: recent advances and future research directions. Trends Pharmacol Sci. 2008;29:454–464. [DOI] [PubMed] [Google Scholar]
- 19. Dowal L, Flaumenhaft R. Targeting platelet G‐protein coupled receptors (GPCRs): looking beyond conventional GPCR antagonism. Curr Vasc Pharmacol. 2010;8:140–154. [DOI] [PubMed] [Google Scholar]
- 20. McNicol A, Israel SJ. Platelets and anti‐platelet therapy. J Pharmacol Sci. 2003;93:381–396. [DOI] [PubMed] [Google Scholar]
- 21. Mercado C, Ziu E, Kilic F. Communication between 5‐HT and small GTPases. Curr Opin Pharmacol. 2011;11:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walther DJ, Peter JU, Winter S, Holtje M, Paulmann N, Grohmann M, Vowinckel J, Alamo‐Bethencourt V, Wilhelm CS, Ahnert‐Hilger G, Bader M. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha‐granule release. Cell. 2003;115:851–862. [DOI] [PubMed] [Google Scholar]
- 23. Shirakawa R, Yoshioka A, Horiuchi H, Nishioka H, Tabuchi A, Kita T. Small GTPase Rab4 regulates Ca2+‐induced alpha‐granule secretion in platelets. J Biol Chem. 2000;275:33844–33849. [DOI] [PubMed] [Google Scholar]
- 24. Senis YA, Mazharian A, Mori J. Src family kinases: at the forefront of platelet activation. Blood. 2014;124:2013–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahmed BA, Jeffus B, Harney JT, Bukhari IS, Unal R, Lupashin VV, van der Sluijs P, Kilic F. Serotonin transamidates Rab4 and facilitates binding to the C‐terminus of hSERT. J Biol Chem. 2008;283:9388–9398. [DOI] [PubMed] [Google Scholar]
- 26. Pittilo M. Cigarette smoking, endothelial injury and cardiovascular disease. Int J Exp Pathol. 2000;81:219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zeiher AM, Schächinger V, Minners J. Long‐term cigarette smoking impairs endothelium‐dependent coronary arterialvasodilator function. J Circulation. 1995;92:1094–1100. [DOI] [PubMed] [Google Scholar]
- 28. Li Y, Hadden C, Cooper A, Ahmed A, Wu H, Lupashin VV, Mayeux PR, Kilic F. Sepsis‐induced elevation in plasma serotonin facilitates endothelial hyperpermeability. Sci Rep. 2016;9:22747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 30. Gu F, Derkach A, Freedman ND, Landi MT, Albanes D, Weinstein SJ, Mondul AM, Matthews CE, Guertin KA, Xiao Q, Zheng W, Shu XO, Sampson JN, Moore SC, Caporaso NE. Cigarette smoking behaviour and blood metabolomics. Int J Epidemiol 2016;45:1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mercado CP, Quintero MV, Li Y, Singh P, Byrd AK, Talabnin K, Ishihara M, Azadi P, Rusch NJ, Kuberan B, Maroteaux L, Kilic F. A serotonin‐induced N‐glycan switch regulates platelet aggregation. Sci Rep. 2013;30:2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kjeldsen SE, Weder AB, Egan B, Neubig R, Zweifler AJ, Julius S. Effect of circulating epinephrine on platelet function and hematocrit. Hypertension. 1995;25:1096–1105. [DOI] [PubMed] [Google Scholar]
- 33. Anumula KR, Taylor PB. A comprehensive procedure for preparation of partially methylated alditol acetates from glycoprotein carbohydrates. Anal Biochem. 1992;203:101–108. [DOI] [PubMed] [Google Scholar]
- 34. Domon B, Costello CE. Structure elucidation of glycosphingolipids and gangliosides using high‐performance tandem mass spectrometry. Biochemistry. 1998;27:1534–1543. [DOI] [PubMed] [Google Scholar]
- 35. US Department of Health and Human Services . The health benefits of smoking cessation a report of the surgeon general. Atlanta: US Department of Health and Human Services, Public Health Service, Centers for Disease Control, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 1990. DHHS Publication No. (CDC) 90‐8416. [Google Scholar]
- 36. Strong JP, Richards ML. Cigarette smoking and atherosclerosis in autopsied men. Atherosclerosis. 1976;23:451–476. [DOI] [PubMed] [Google Scholar]
- 37. Auerbach O, Garfinkel L. Atherosclerosis and aneurysm of aorta in relation to smoking habits and age. Chest. 1980;78:805–809. [DOI] [PubMed] [Google Scholar]
- 38. Solberg LA, Strong JP. Risk factors and atherosclerotic lesions: a review of autopsy studies. Arteriosclerosis. 1983;3:187–198. [DOI] [PubMed] [Google Scholar]
- 39. US Department of Health and Human Services .The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2004. [Google Scholar]
- 40. Hoffmann D, Hoffmann I. The changing cigarette, 1950–1995. J Toxicol Environ Health. 1997;50:307–364. [DOI] [PubMed] [Google Scholar]
- 41. Mercado C, Kilic F. Molecular mechanisms of SERT in platelets: regulation of plasma serotonin levels. Mol Interventions. 2010;10:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee‐Sundlov MM, Ashline DJ, Hanneman AJ, Grozovsky R, Reinhold VN, Hoffmeister KM, Lau JT. Circulating blood and platelets supply glycosyltransferases that enable extrinsic extracellular glycosylation. Glycobiology. 2017;27:188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wandall HH, Rumjantseva V, Sørensen AL, Patel‐Hett S, Josefsson EC, Bennett EP, Italiano JE Jr, Clausen H, Hartwig JH, Hoffmeister KM. The origin and function of platelet glycosyltransferases. Blood. 2012;120:626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reinhard M, Jouvenal K, Tripier D, Walter U. Identification, purification, and characterization of a zyxinrelated protein that binds the focal adhesion and microfilament protein VASP (vasodilator‐stimulated phosphoprotein). Proc Natl Acad Sci USA. 1995;92:7956–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giguère PM, Gall BJ, Ezekwe EA Jr, Laroche G, Buckley BK, Kebaier C, Wilson JE, Ting JP, Siderovski DP, Duncan JA. G Protein signaling modulator‐3 inhibits the inflammasome activity of NLRP3. J Biol Chem. 2014;289:33245–33257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wallace SW, Magalhaes A, Hall A. The Rho target PRK2 regulates apical junction formation in human bronchial epithelial cells. Mol Cell Biol. 2011;31:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matsuura K, Otani M, Takano M, Kadoyama K, Matsuyama S. The influence of chronic nicotine treatment on proteins expressed in the mouse hippocampus and cortex. Eur J Pharmacol. 2016;780:16–25. [DOI] [PubMed] [Google Scholar]
- 48. Keularts IM, van Gorp RM, Feijge MA, Vuist WM, Heemskerk JW. α(2A)‐adrenergic receptor stimulation potentiates calcium release in platelets by modulating cAMP levels. J Biol Chem. 2000;275:1763–1772. [DOI] [PubMed] [Google Scholar]
- 49. Li M, Yu D, Williams KJ, Liu ML. Tobacco smoke induces the generation of procoagulant microvesicles from human monocyte/macrophages. Arterioscler Thromb Vasc Biol. 2010;30:1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Badrnya S, Baumgartner R, Assinger A. Smoking alters circulating plasma microvesicle pattern and microRNA signatures. Thromb Haemost. 2014;112:128–136. [DOI] [PubMed] [Google Scholar]