

Abstract
Alström syndrome is a rare autosomal recessive ciliopathy caused by mutations in the ALMS1 gene. Hallmark characteristics include childhood onset of severe retinal degeneration, sensorineural hearing loss, obesity, insulin-resistant diabetes, and cardiomyopathy. Here we comprehensively characterize the auditory and otologic manifestations in a prospective case series of 38 individuals, aged 1.7-37.9 years, with genetically confirmed Alström syndrome. Hearing loss was preceded by retinal dystrophy in all cases, and had an average age of detection of 7.45 years (range 1.5-15). Audiometric assessments showed mean pure tone averages (0.5, 1, 2, 4 kHz) of 48.6 and 47.5 dB HL in the right and left ears, respectively. Hearing was within normal limits for only 8/74 ears (11%). For the 66 ears with hearing loss, the degree was mild (12%), moderate (54%), or severe (8%). Type of hearing loss was predominantly sensorineural (77%), while three ears had mixed loss, no ears had conductive loss, and type of hearing loss was indeterminate for the remaining 12 ears. Serial audiograms available for 33 patients showed hearing loss progression of approximately 10-15 dB/decade. Our data show that hearing loss associated with Alström syndrome begins in childhood and is a predominantly symmetric, sensory hearing loss that may progress to a severe degree. Absent otoacoustic emissions, intact speech discrimination, and disproportionately normal auditory brainstem responses suggest an outer hair cell site of lesion. These findings indicate that individuals with Alström syndrome would benefit from sound amplification and if necessary, cochlear implantation.
Keywords: Alström, ALMS1, ciliopathy, sensory hearing loss
Introduction
Alström syndrome (AS) (OMIM #203800) is a rare autosomal recessive ciliopathy first described in 1959 [Alström et al., 1959]. It is caused by mutations in the ALMS1 gene (2p13) that encodes the ciliary protein ALMS1 [Collin et al., 1999; Orphanet 1997; Marshall et al., 2005]. Prevalence of AS is estimated at 1 to 9 per 1,000,000 persons [Marshall et al. 2005], with approximately 700 cases described to date. AS is characterized by progressive sensorineural hearing loss (SNHL), severe retinal degeneration, childhood-onset obesity, insulin-resistant diabetes, hypertriglyceridemia, cardiomyopathy, hepatorenal and pulmonary disease, and normal intelligence [Marshall et al. 2005], though with developmental delay given auditory and visual deficits. Diagnosis of AS is made based on clinical diagnostic criteria and confirmed by mutations in ALMS1 [Marshall et al. 2005; Ozantürk et al., 2015]. Typically, patients present within the first years of life with retinal degeneration that progresses to blindness in the second decade. Nystagmus and photophobia are common. Impaired vision is most often followed or accompanied by a post-lingual SNHL, though cases with hearing loss at birth have been reported5. Life expectance rarely exceeds 50 years of age [Marshall et al., 2011], due to complications of cardiomyopathy, diabetes, hypertriglyceridemia and progressive multi-organ dysfunction associated with systemic fibrosis [Álvarez-Satta, et al., 2015]. From an otologic standpoint, chronic recurrent otitis media (ROM) as late as the third decade of life is commonly reported, with a higher than estimated prevalence of approximately 42-54% [Marshall et al., 2005; Marshall et al., 2012].
While progressive decline in pure tone sensitivity has been documented in patients with AS [Bahmad Jr. et al., 2014; Welsh et al., 2007], the auditory profile has not been fully characterized. Distortion product otoacoustic emissions (DPOAEs) and auditory brainstem responses (ABRs), which are critical assessments for discerning sensory versus neural hearing loss, have been reported for two siblings [Bahmad Jr. et al., 2014], but have not been reported for a cohort study.
Here we present comprehensive audiologic data and otologic findings on 38 individuals with AS, all prospectively evaluated at a tertiary clinical research center. In addition, we report the progression rate of hearing loss in AS calculated based on our analysis of historical audiograms of patients.
Methods
Patients
Between the dates of February 2013 and June 2014, 38 patients with AS were evaluated at the National Institutes of Health (NIH) Clinical Center, Bethesda, Maryland, under the NIH protocol “Clinical and Molecular Investigations into Ciliopathies” (NCT00068224), approved by the Institutional Review Board of the National Human Genome Research Institute (NHGRI). All 38 patients fulfilled the clinical diagnostic criteria for AS and underwent sequencing of the ALMS1 gene. Patients or their parents gave written informed consent.
Audiologic Evaluations
Age- and ability-appropriate audiologic evaluations including air- and bone-conduction pure-tone thresholds, speech thresholds, and evaluation of word recognition ability in quiet were conducted at the NIH Clinical Center using a Grason Stadler (Eden Prairie, MN, USA) diagnostic audiometer (GSI-61) in sound suites meeting American National Standards Institute (ANSI 2010) criteria. Hearing asymmetry was defined as >10 dB difference in pure tone thresholds between ears at two or more frequencies [Mazzoli et al., 2003]. Middle ear function was assessed using 226 Hz tympanometry. DPOAEs were evaluated in quarter octave bands over the frequency range 842-7996 Hz using an Otodynamics (Hatfield, Hertfordshire, UK) Echoport Otoacoustic Emission System supported by ILO V6 Clinical OAE software. DPOAEs were interpreted as present over a broad frequency range when the signal to noise ratio exceeded 6 dB for seven or more of the 14 tested frequency bands, and present at a limited number of frequencies when emissions were present at six or fewer bands. ABRs were recorded in response to separately presented condensation and rarefaction, air-conducted, broadband clicks delivered via insert earphones at 85/95 dB nHL at click repetition rates of 8.3 and 63.3/sec (GSI Audera, Eden Prairie, MN, USA) using an Fz to ipsilateral and contralateral earlobe surface electrode montage.
In addition, we obtained past audiometric records for review and included 125 historical audiograms from 33 patients in our analysis to examine longitudinal age-related trends and determine rate of progression of hearing loss in AS. Only behavioral data obtained with good reliability as judged by the assessing audiologist were included.
Otologic Evaluation
Detailed medical histories and comprehensive otologic examinations performed by a single otologist (HJK) included a head and neck examination as well as microscopic examination of both ears.
Data Interpretation and Analysis
Audiometric data gathered at the NIH visit were designated as the reference audiogram and used for all cross sectional analyses. Degree of hearing loss was based on air conduction pure tone thresholds. Classifications were as follows: within normal limits (WNL) when ≤20 dB HL, mildly reduced when >20-40 dB HL, moderately reduced when >40-70 dB HL, severely reduced when >70-95 dB HL and profoundly reduced when >95 dB HL [Mazzoli et al., 2003]. Overall degree of hearing loss was based on a four frequency pure-tone average (4F-PTA; 0.5/1/2/4 kHz). Type of hearing loss was based on the difference between the air- and bone-conduction thresholds for a three frequency pure-tone average (3F-PTA; 0.5/1/2 kHz). Hearing loss was classified as sensorineural when the air-conduction 3F-PTA >20 dB HL and 3F-PTA air-bone difference <10 dB; conductive when the air-conduction 3F-PTA >20 dB HL, the bone conduction 3F-PTA ≤ 20 dB HL and the 3F-PTA air-bone difference ≥10 dB; and mixed when both the air- and bone-conduction 3F-PTA >20 dB HL and the 3F-PTA air-bone difference were ≥10 dB [Mazzoli et al., 2003]. For cases in which sufficient or ear specific bone conduction thresholds could not be obtained, the hearing loss type was classified as indeterminate. Audiogram configurations were defined as flat (difference between low [0.25/0.5 kHz] and high [4/8 kHz] frequency PTA <15 dB), gently sloping downward (difference between low and high frequency PTA = 15-29 dB, or low frequency ascending (low frequency PTA more than 15 dB greater than the high frequency PTA]). Word recognition scores were evaluated as proportionate or disproportionate to the degree of hearing loss, based on the 3F-PTA using ranges established by Dubno et al., 1995. Peak static compliance and middle ear pressure were interpreted as WNL/low/high and WNL/negative/positive, respectively, using age appropriate norms [Margolis et al., 1987]. ABR latencies were interpreted as WNL/late based on normative data from Schwartz et al., 1989.
Data Analysis
Data were analyzed using Microsoft Excel (version 14.5.1) and GraphPad Prism 6.0h (La Jolla, CA, USA). Descriptive statistics were computed for hearing loss type and degree, tympanometry and patterns of hearing loss progression. Simple linear regressions were used to examine hearing thresholds by air conduction as a function of age. Fisher's exact test was used to determine if there was an association between history of ROM and AS. Paired Student's t-tests were conducted to compare hearing sensitivity between the right and left ears for the 4F-PTA. One-way analysis of variance (ANOVA) was used to investigate differences between the 4F-PTA for decade-based age groups. Post-hoc analyses investigating group differences were conducted using Tukey's multiple comparisons test. Statistical significance was set at p ≤0.05.
Results
Patient Characteristics and Clinical Findings
The patient cohort included 20 females and 18 males; ages at the time of NIH examination ranged from 1.7 to 37.9 years (M 16.0, SD 10.5). One family contributed three siblings, and four families contributed two siblings each, including two sets of dizygotic twins, leaving 32 independent families. There were 12 patients aged 1-10, 14 patients aged 11-20, five patients aged 21-30, and six patients aged 31-40 years old. Races included 35 Caucasians and three African-Americans. Bi-allelic pathological variants of the ALMS1 gene were identified in 31 of the 32 families; in one family the second pathological variant could not be identified (Supplemental Table 1). All pathological variants were null, resulting in frame shifting or stop codons. Eight families were homozygous while 23 were compound heterozygous (Supplemental Table 1).
The average age of HL detection occurred at 7.45 years (range 1.5-15). For 15 patients who had neonatal hearing screening information available, all but one patient passed. A history of sinusitis and nasal congestion were reported in 19/38 (50%) and 18/38 (47.4%) patients, respectively (Table I). Of note, a history of frequent otitis media was reported by 35 of 38 patients (92%); 19/38 (50%) had a history of pressure equalization tube placement.
Table I.
Clinical history of otologic and upper respiratory symptoms.
| Clinical History | Group Size (n) | Percentage |
|---|---|---|
| Subjective hearing loss | 25/38 | 65.7% |
| Tinnitus | 6/28 | 21.4% |
| Aural fullness | 7/28 | 25% |
| Vertigo | 0/28 | 0% |
| Dysequilibrium | 9/28 | 32% |
| Frequent otitis media | 35/39 | 92% |
| History of PE tubes | 19/38 | 50% |
| Sinusitis | 19/38 | 50% |
| Nasal Congestion | 18/38 | 47.4% |
| Rhinorrhea | 18/38 | 47.4% |
Data are presented as number of persons with finding/number assessed
On microscopic ear examination (HJK), four patients had serous middle ear effusions, five patients had patent pressure equalization tubes (9 ears), and seven patients showed evidence of myringosclerosis. All patients had nystagmus and evidence of retinal dystrophy. Cranial nerve evaluation was otherwise within normal limits.
In all patients, vision loss preceded hearing loss. While none of the patients had a history of vertigo, nine reported intermittent dysequilibrium. Other relevant clinical findings identified during clinical examination are presented in Table I.
Cross Sectional Analysis of Auditory Function
Hearing thresholds were obtained on 37/38 patients (74/76 ears) at the NIH visit. One patient, from one of the twin sibships, did not cooperate with the audiologic evaluations due to significant developmental delay caused by perinatal asphyxia. Hearing was symmetrical in 28/29 patients with sufficient pure tone thresholds to make this determination. One patient, aged 10 years, had a 15 dB difference between ears at two frequencies (3 and 8 kHz), although her hearing was classified as normal in both ears based on the 4F-PTA. Pure tone configurations were flat in 36 ears, gently sloping in 23 ears, and low frequency ascending in one ear. There was not sufficient pure tone data to calculate configuration for the remaining ears (n = 14).
Hearing thresholds were normal in 10.8% (8/74) of ears (Table II). Degree of hearing loss was mild in 12.2% (9/74) of ears, moderate in 54.1% (40/74) of ears, and severe in 8.1% (6/74) of ears. Degree of hearing loss could not be determined in 14.9% of ears, all of which were in the youngest age group (1-10 years), due to insufficient hearing threshold data. Normal hearing and mild hearing loss were observed in the youngest two age groups (1-10 and 11-20 years), but not in the older groups (21-30 and 31-40 years). Of the 66 ears with hearing loss, the type was sensorineural for 77.3% (51/66), mixed for 4.5% (3/66) (Table III), and no ears had pure conductive hearing loss. Sensorineural hearing loss was observed in all age groups, and was the only type measured in the older two age groups. Type of hearing loss was unknown for 18.2% (12/66) of ears, all of which were in the youngest group (1-10 years) of patients.
Table II. Degree of hearing loss for 74 ears, 37 participants.
| Classification of Hearing Loss Degree | Total Group 37 pts 74 ears | Age Groups | |||
|---|---|---|---|---|---|
|
| |||||
| 1-10 Years 12 pts 24 ears | 11-20 Years 14 pts 28 ears | 21-30 Years 5 pts 10 ears | 31-40 Years 6 pts 12 ears | ||
| Normal | 8 (10.8%) | 6 (25%) | 2 (7.1%) | 0 | 0 |
| Mild | 9 (12.2%) | 1 (4.2%) | 8 (28.6%) | 0 | 0 |
| Moderate | 40 (54.1%) | 6 (25%) | 17 (60.1%) | 10 (100%) | 7 (58%) |
| Severe | 6 (8.1%) | 0 | 1 (3.6%) | 0 | 5(42%) |
| Unknown | 11 (14.9%) | 11 (45.8%) | 0 | 0 | 0 |
Data are presented as n=ears (%); pts, participants
Table III.
Type of hearing loss, classified by age group for the 33 participants (66 ears) with hearing loss.
| Classification of Hearing Loss Type | Total 33 pts 66 ears | Age Groups | |||
|---|---|---|---|---|---|
|
| |||||
| 1-10 Years 9 pts 18 ears | 11-20Years 13 pts 26 ears | 21-30 Years 5 pts 10 ears | 31-40 Years 6 pts 12 ears | ||
| Known type | |||||
| Conductive | 0 | 0 | 0 | 0 | 0 |
| Mixed | 3 (4.5%) | 2 (11%) | 1 (4%) | 0 | 0 |
| Sensorineural | 51 (77.3%) | 4 (22%) | 25 (6%) | 10 (100%) | 12 (100%) |
| Unknown type | 12 (18.2%) | 12 (67%) | 0 | 0 | 0 |
Data are presented as n=ears (%); pts, participants
Analysis of this cross sectional data shows, on average, a 10 dB decline in pure tone thresholds between decade-based age groups (Figure 1). This is captured in mean audiograms for the decade-based age groups (Figure 2, right ear data only). Mean 4F-PTAs for the entire group were 48.6 (SD=20.0) dB HL and 47.5 (SD=19.7) dB HL in the right and left ears, respectively, and were not significantly different between ears (paired t-test, t(30) = 1.793, p= 0.0831, r2=0.0968). In light of this, both right and left ear 4F-PTA data were treated as a single dependent variable within each age group. An ordinary one-way ANOVA revealed a significant difference in 4F-PTA between all decade-based age groups (F(3, 4) = 2660, p<0.0001, r2 = 0.9995).
Figure 1.

Pure tone thresholds plotted by age for each ear (right=red, left=blue, n=37) based on evaluation at NIH clinical center. R2 and slope of the regression line are shown for the right ear; p<0.0001 for individual frequencies and the four-frequency pure tone average (4F-PTA). Dotted line at 20 dB HL represents the upper limit of normal hearing. R2 and slope of the regression line for the left ear are not significantly different (data not shown). Dotted line at 20 dB HL represents the upper limit of normal hearing. [Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/journal/10.1002(ISSN)1552-4833]
Figure 2.

Mean and standard deviation of air conduction pure tone thresholds for decade-based age groups. Data are for right ear only.
Word recognition scores could be obtained on 58 ears, and ranged from 40-100% (M 85.9%, SD 14.7) (Figure 3). All word recognition scores were proportionate to the degree of hearing loss with the exception of a single instance in which the right ear 3F-PTA was 5 dB HL and word recognition was 92%, both of which are in the clinically normal range.
Figure 3.
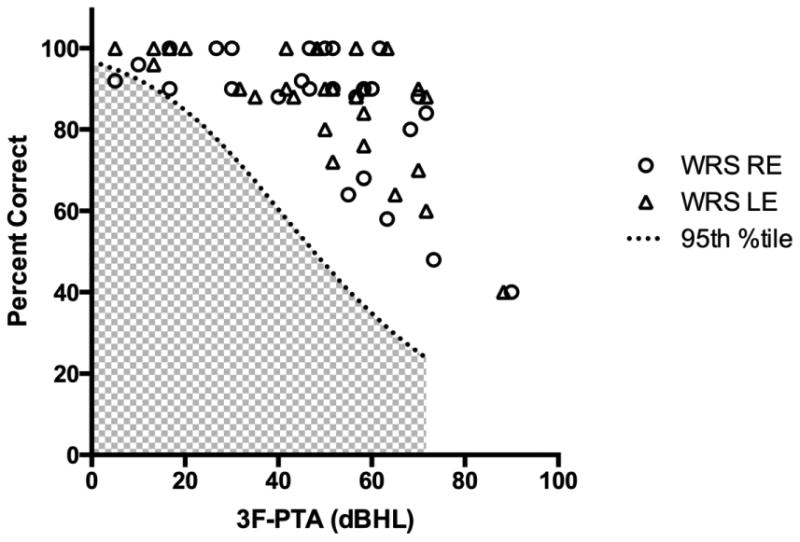
Maximum word recognition scores (WRS) shown as percent correct plotted as a function of the three-frequency (0.5, 1 and 2 kHz) pure tone average (3F-PTA) for the right and left ears. The dotted line indicates the 95th percentile confidence limit for the maximum WRS as a function of the 3F-PTA; scores in the shaded area are considered disproportionately poor in relation to the degree of hearing loss [Dubno et al., 1995].
Tympanometry demonstrated normal peak static compliance for 74.3% (55/74) of ears (Table IV). There was negative middle ear pressure with normal mobility in 12.2% (9/74). Flat tympanograms were observed in 13.5% (10/74); in 9 cases this finding was associated with patent pressure equalization tubes.
Table IV. Auditory test findings and interpretation.
| Test | Finding | Number of ears (percentage) | Interpretation |
|---|---|---|---|
| Tympanometry – Middle ear function | |||
| Normal | 55/74 (74.3%) | Normal middle ear function | |
| Negative Pressure | 9/74 (12.2%) | Eustachian tube dysfunction | |
| Flat – Normal volume | 1/74 (1.4%) | Stiff middle ear system | |
| Flat – Large volume | 9/74 (12.2%) | Patent ventilation tube | |
| DPOAE – Cochlear OHC function | |||
| Present: broad range | 12/64 (18.8%) | Intact OHC function | |
| Present: limited range | 3/64 (4.7%) | Partial OHC function | |
| Absent* | 46/64 (71.8%) | OHC dysfunction | |
| Noisy | 3/64 (4.7%) | Uninterpretable | |
| ABR – Auditory nerve and auditory brainstem function | |||
| Normal latencies | 48/60 (80%) | Normal auditory nerve/auditory brainstem function | |
| Borderline abnormal** | 5/60 (8.3%) | Attributable to degree of HL | |
| Delayed interpeak latencies | 6/60 (10%) | Retrocochlear | |
| Absent | 1/60 (1.6%) | Attributable to degree of HL | |
Data are presented as number of ears (%);
an additional eight ears with absent DPOAEs and abnormal tympanometry were not included in this count as indicative of OHC dysfunction;
borderline abnormalities included absent waves I or III (n=4 ears) and delayed wave III with all other findings normal (n=1); DPOAE, distortion product otoacoustic emission; OHC, outer hair cells; ABR, auditory brainstem response; HL, hearing loss
DPOAEs were absent in 71.2% (46/64) of ears (Table IV), all of which had hearing loss of mild or greater degree. DPOAEs were present across a broad frequency range in 12 ears, which included normal hearing (n=8), a mild hearing loss (n=2) or no threshold data (n=2). In the three ears with DPOAEs present over a limited frequency range, two had a mild hearing loss and one had mildly negative middle ear pressure with no behavioral threshold data.
ABRs were evaluated in 30 patients (60 ears) (Table IV). The majority (80%, 48/60 ears) had normal absolute and interpeak latencies for ABR peaks I, III and V. An additional 8% (5/60 ears) had minor ABR changes attributable to the degree of hearing loss such as missing wave I, or, in one case, a delay in wave III latency with normal interpeak latencies. One ear had no response on the ABR in the presence of a severe to profound sensorineural hearing loss. In general, the ABRs were more robust than expected for the degree of hearing loss. Four of six ears with severe hearing loss and 36/40 ears with moderate hearing loss had normal ABRs (data not shown). Abnormalities in both absolute and interpeak latencies suggestive of retrocochlear auditory dysfunction were present for 10% (6 ears, 4 patients), four ears with moderate and two with severe hearing loss. Brain MRI performed on one of these patients did not show evidence of structural changes.
Longitudinal Analysis of Hearing Sensitivity
Simple linear regressions approximating change in air conduction pure tone thresholds (based on 125 historical and 37 prospective audiograms; time between first and last audiograms M=7.77 years, SD=7.94, range=0.64-29.51 years) revealed significant hearing loss progression with age at a rate of 10-15 dB/decade up to the fourth decade of life for individual frequencies 0.5, 1, 2, 4 and 8 kHz and the 4F-PTA (Figure 4). Specifically, for the 4F-PTA, the best fit slope was 1.179 +/- 0.129 (95% CI 0.93 to 1.43) and 1.208 +/- 0.125 (95% CI 0.96 to 1.45) for right and left ears, respectively. Best fit slopes for individual frequencies are shown in Figure 4.
Figure 4.
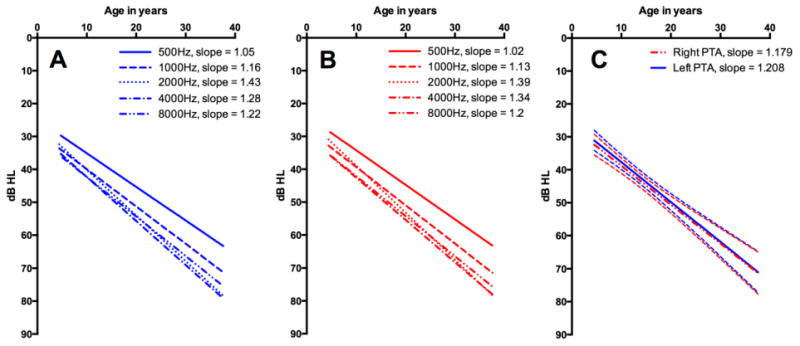
Linear regression analysis of 162 audiograms (125 historical, 37 NIH) obtained from patient population at frequencies 0.5, 1, 2, 4, and 8 kHz. (A) Left ear linear regression analysis. (B) Right ear linear regression analysis. (C) Linear regression analysis of 4F-pure tone averages for both right and left ears with 95% CI represented by dashed lines. Approximately 10-15 dB hearing loss per decade was observed across all frequencies. [Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/journal/10.1002(ISSN)1552-4833]
Evaluation of patients' historical records showed three with mixed loss, one with conductive loss, and one with hearing loss that varied between conductive and mixed. The remaining 27 patients had SNHL on all available audiograms. Of these, six patients with a history of conductive or mixed hearing loss, all had resolution of the conductive components and isolated SNHL at the time of NIH evaluation. For the three ears with mixed hearing loss on presentation to NIH, no historical audiograms with evidence of prior conductive or mixed loss were available or noted.
Discussion
In addition to their role in regulating embryonic development, primary cilia play a critical role in maintaining health of many tissues and cells in postnatal life including kidneys, retinal photoreceptor cells, olfactory receptor cells, and the sensory cells of the inner ear [Nauli et al., 2003]. Consistently, patients with multisystem ciliopathies such as Alström, Bardet-Biedl and Joubert syndromes often suffer from impairment of retinal, olfactory and auditory function. Progressive SNHL is a well-known feature of AS [Welsh et al., 2007]. However, age of onset and severity of hearing loss is variable. To date, the characteristic audiological findings in AS have not been systematically evaluated in a large patient series from a single center. In our cohort of 38 genetically confirmed AS patients, ages ranging from early childhood to adulthood, we were able to further define the characteristics and progression of auditory involvement in AS. Our longitudinal analysis of 125 historical audiograms and 37 audiograms obtained at the NIH demonstrated a predominantly symmetric, sensorineural hearing loss with a hearing loss progression of approximately 10-15 dB/decade. ANOVA showed a statistically significant difference in hearing levels between all age groups from the first through fifth decade of life. This supports the notion of worsening hearing level with increasing age. Hearing loss configuration was either flat or had a gently sloping pattern across the audiometric test frequency range for the majority of patients. For those 15 patients for whom newborn screening data were available, all but one passed the newborn hearing screening, consistent with a predominantly postnatal onset of the hearing loss. Since almost all patients with AS develop SNHL in childhood, it is important that they have close monitoring of their hearing status and receive prompt and proper aural amplification and accommodations. This is especially important for these patients with compromised vision for whom auditory cues are integral to safety, education, and communication.
Our patients demonstrated a history of frequent middle ear infections at a rate greater than that noted to occur in the general population (92% vs. 30%) [Hoffman et al., 2013]. Additionally, our patient group had a PET placement rate of 50% in contrast to one study which determined the average incidence of PET placement in developed countries to be 30% or less by age five [Djurhuus et al., 2014]. Despite these facts, the majority of our patients did not show a conductive component to their hearing loss, either in our cross sectional data (Table 3) or in the historical audiograms (data not shown). It should be noted, however, that the majority of historical audiograms did not include bone conduction threshold data. Only 3/74 ears showed a mixed loss and none showed a pure conductive loss at the time of the NIH audiogram, as most of the subjects had developed a sensorineural component of hearing loss. Our patients also reported 50% incidence of sinusitis. A recent clinical survey of this cohort with AS showed a history of bronchitis or pneumonia in 61 % of individuals. [Boerwinkle C, Marshall JD, Bryant J, Gahl WA, Olivier KN, Gunay-Aygun M. Respiratory Manifestations in 38 Patients with Alström Syndrome. Pediatric Pulmonology. 2016, In press.]
During differentiation in mouse lung development and repair after injury, primary cilia are transiently present and give way to motile cilia, suggesting that primary cilia may play a role in control programming of differentiation of motile ciliogenesis early in life [Jain et al., 2010]. It is plausible that the ciliopathy from ALMS1 mutation may delay or alter normal development of motile cilia of the respiratory epithelial cells found in the middle ears and paranasal sinuses. Perhaps this primary ciliary effect on the normal respiratory epithelium maturation may predispose patients with AS to recurrent upper and lower respiratory infection early in life, which they eventually outgrow as the respiratory epithelial lining develops fully.
The ALMS1 protein is ubiquitously expressed in fetal and postnatal tissues; at the subcellular level it localizes to the centrosome, basal bodies of the cilia, cytoplasm, cytoskeleton and microtubule organizing center [Collin et al., 2002; Hearn et al., 2002]. Although its function is not completely understood, current evidence suggests that ALMS1 plays roles in formation and maintenance of cilia, cell cycle regulation, endosomal trafficking, cell migration, and extracellular matrix production [Collin et al, 2012; Favaretto et al., 2014; Knorz et al., 2010; Leitch et al., 2007; Shenje et al., 2014; Zulato et al., 2011]. In the developing organ of Corti, ALMS1 is concentrated in basal bodies at the base of the cilia and supporting cells, in the basal bodies of the marginal cells in stria vascularis, and in the mesenchymal fibroblasts located on the spiral ligament and underside of the basilar membrane [Jagger et al., 2011]. Mice with defective ALMS1 show abnormalities of the shape and orientation of stereocilliary bundles and adult animals lose outer hair cells and develop progressive large vacuoles in the stria vascularis [Jagger et al., 2011; Nadol Jr. et al., 2015]. Additionally, Jagger et al., observed an accelerated loss of cochlear outer hair cells in mature Alms-/- mice as compared to mature control mice, while Nadol et al. observed loss of both outer and inner hair cells in humans with AS. The combination of our findings on pure tone audiometry, absent DPOAEs, proportionate word recognition scores, and robust ABRs suggest an outer hair cell and/or stria vascularis site of lesion, consistent with findings in the Alms1-disrupted animal models [Jagger et al., 2011]. It should be noted however, that the endocochlear potentials within four test mice were within the normal range, despite the presence of vacuolar degeneration, which further confounds the role of the stria vascularis in the pathophysiology.
We identified robust ABRs in spite of significant sensorineural hearing loss indicating that hearing loss in AS is predominantly cochlear-based. Abnormal ABR findings that could not be explained by the degree of hearing loss were observed in only 4 patients (6 ears). MRI of the brain and temporal bone available in one of these patients with an abnormal ABR demonstrated normal anatomy. Abnormal ABRs in these 4 patients raises the possibility of retrocochlear auditory dysfunction in a subset of patients with AS. These patients in our cohort may have central auditory nervous system involvement similar to the subset of patients with AS with structural brain abnormalities reported by Citton et al., 2013. Most neurons and glial cells possess non-motile solitary primary cilia, which are involved in cell signaling receptors and processes [Narita et al., 2015].
The presence of a functioning cochlear nerve and retrocochlear pathway are important parameters with implications for the approach to management of hearing loss. The robust ABRs and proportionate word recognition scores indicate that AS patients should benefit from traditional aural amplification and if necessary, cochlear implantation. Twenty-seven of the 38 patients in our cohort use hearing aids and there is at least one report of successful cochlear implantation in a patient with AS [Florentzson et al., 2010].
Based on clinical examination and the relative lack of vertigo and imbalance, there is little evidence in our population to suggest significant vestibular involvement. Dedicated vestibular assessments were not conducted and further research on the role of ALSM1 in the vestibular system is needed. However, given the presence of spontaneous nystagmus, a comprehensive vestibular assessment would be limited.
Limitations of this study
It is important to note that there was variability among the number of historical audiograms available for each patient, with some patients lacking data altogether and others with multiple hearing tests. It is possible this may have an effect on our data analysis. Additionally, the number of patients older than 30 years is limited (6 of 38), preventing further analysis of hearing levels with older age. In our cohort conductive hearing loss was rare and patients did not have evidence of active ear infections at the time of NIH examination, perhaps due to the fact that ill patients were not allowed to travel to the NIH.
Conclusion
Hearing loss onset in AS is typically post-lingual beginning in childhood. It is largely a symmetric sensory loss that progresses typically to a moderate severity at approximately 10-15 dB per decade. Despite the high incidence of otitis media in AS, the presence of a conductive component to the hearing loss was uncommon in our cohort. Absent DPOAEs, predominantly normal ABRs, and previously reported data from animal models indicate that dysfunction of outer hair cells, stria vascularis, or both is the primary origin of hearing loss in AS. This suggests that AS patients are typically good candidates for traditional aural amplification or, in cases of severe to profound SNHL, cochlear implantation.
Supplementary Material
Acknowledgments
We thank Alström Syndrome International (ASI) for their extensive support and the patients and their families who generously participated in this investigation. Support was provided by the Intramural Research Programs of the National Human Genome Research Institute (NIH intramural grant Z99 HG999999, the National Institute on Deafness and Other Communication Disorders (NIH intramural grant DC000064), and the NIH Clinical Center (NIH grant HD36878), Bethesda, MD, USA.
Footnotes
This paper is dedicated to the memory of our colleague, Jan Davis Marshall, who worked passionately on Alstrom syndrome for 23 years making a huge difference in the lives of patients with Alstrom and their families around the world.
Conflicts of Interest: None
References
- Alström CH, Hallgren B, Nilsson LB, Åsander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness. A specific syndrome (not hitherto described) distinct from Laurence-Moon-Biedl syndrome. A clinical endocrinologic and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand. 1959;34(Suppl. 129):1–35. [PubMed] [Google Scholar]
- Álvarez-Satta M, Castro-Sánchez S, Valverde D. Alström syndrome: current perspectives. Appl Clin Genet. 2015;8:171–179. doi: 10.2147/TACG.S56612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahmad F, Jr, Costa CS, Alves T, Santos M, Filho B, Jairo de V, Lucas M, Marshall J. Familial Alström syndrome: a rare cause of bilateral progressive hearing loss. Braz J Otorhinolaryngol. 2014;80(2):99–104. doi: 10.5935/1808-8694.20140023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citton V, Favaro A, Bettini V, Gabrieli J, Milan G, Greggio NA, Marshall JD, Naggert JK, Manara R, Maffei P. Brain involvement in Alström syndrome. Orphanet J Rare Dis. 2013;8:24. doi: 10.1186/1750-1172-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin GB, Marshall JD, Boerkoel CF, Levin AV, Weksberg R, Greenberg J, Michaud JL, Naggert JK, Nishina PM. Alström syndrome: further evidence for linkage to human chromosome 2p13. Hum Genet. 1999;105:474–479. doi: 10.1007/s004390051133. [DOI] [PubMed] [Google Scholar]
- Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel C, Sicolo N, Martin M, Nishina PM, Naggert JK. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74–78. doi: 10.1038/ng867. [DOI] [PubMed] [Google Scholar]
- Collin GB, Marshall JD, King BL, Milan G, Maffei P, Jagger DJ, Naggert JK. The Alström syndrome protein, ALMS1, interacts with [alpha]-actinin and components of the endosome recycling pathway. PLoS One. 2012;7:e37925. doi: 10.1371/journal.pone.0037925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djurhuus BD, Skytthe A, Christensen K, Faber CE. Increasing rate of middle ear ventilation tube insertion in children in Denmark. Int J Pediatr Otorhinolaryngol. 2014;78(9):1541–1544. doi: 10.1016/j.ijporl.2014.06.034. [DOI] [PubMed] [Google Scholar]
- Dubno JR, Lee FS, Klein AJ, Matthews LJ, La CF. Confidence limits for maximum word-recognition scores. J Speech Hear Res. 1995;38:490–502. doi: 10.1044/jshr.3802.490. [DOI] [PubMed] [Google Scholar]
- Favaretto F, Milan G, Collin GB, Marshall JD, Stasi F, Maffei P, Vettor R, Naggert JK. GLUT4 defects in adipose tissue are early signs of metabolic alterations in Alms1GT/GT, a mouse model for obesity and insulin resistance. PLoS One. 2014;9:e109540. doi: 10.1371/journal.pone.0109540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florentzson R, Hallén K, Möller C. The first clinical experience. Stockholm, Sweden: 10th International CI Conference; 2010. Alström syndrome and cochlear implantation. [Google Scholar]
- Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, White C, Connolly V, Taylor JFN, Russell-Eggitt I, Bonneau D, Walker M, Wilson DI. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alström syndrome. Nat Genet. 2002;31:79–83. doi: 10.1038/ng874. [DOI] [PubMed] [Google Scholar]
- Hoffman HJ, Daly KA, Bainbridge KE, Casselbrant ML, Homøe P, Kvestad E, Kvaerner KJ, Vernacchio L. Panel 1: Epidemiology, natural history, and risk factors. Otolaryngol Head Neck Surg. 2013;148(4 Suppl):E1–E25. doi: 10.1177/0194599812460984. [DOI] [PubMed] [Google Scholar]
- Jain R, Pan J, Driscoll JA, Wisner JW, Huang T, Gunsten SP, You Y, Brody SL. Temporal relationship between primary and motile ciliogenesis in airway epithelial cells. Am J Respir Cell Mol Biol. 2010;43:731–739. doi: 10.1165/rcmb.2009-0328OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagger D, Collin G, Kelly J, Towers E, Nevill G, Longo-Guess C, Benson J, Halsey K, Dolan D, Marshall J, Naggert J, Forge A. Alström Syndrome protein ALMS1 localizes to basal bodies of cochlear hair cells and regulates cilium-dependent planar cell polarity. Hum Mol Genet. 2011;20:466–481. doi: 10.1093/hmg/ddq493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorz VJ, Spalluto C, Lessard M, Purvis TL, Adigun FF, Collin GB, Hanley NA, Wilson DI, Hearn T. Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol Biol Cell. 2010;21:3617–3629. doi: 10.1091/mbc.E10-03-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch CC, Lodh S, Prieto-Echagüe V, Badano JL, Zaghloul NA. Basal body proteins regulate Notch signaling via endosomal trafficking. J Cell Sci. 2014;127:2407–2419. doi: 10.1242/jcs.130344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, Wu H, Hong N, Glynne R. A role for Alström syndrome protein, alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 2007;3:e8. doi: 10.1371/journal.pgen.0030008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis RH, Heller JW. Screening tympanometry: criteria for medical referral. Audiology. 1987;26:197–208. doi: 10.3109/00206098709081549. [DOI] [PubMed] [Google Scholar]
- Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, Carey C, Macdermott S, Russell-Eggitt I, Shea SE, Davis J, Beck S, Shatirishvili G, Mihai CM, Hoeltzenbein M, Pozzan GB, Hopkinson I, Sicolo N, Naggert JK, Nishina PM. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675–683. doi: 10.1001/archinte.165.6.675. [DOI] [PubMed] [Google Scholar]
- Marshall JD, Maffei P, Collin GB, Naggert JK. Alström syndrome: genetics and clinical overview. Curr Genomics. 2011;12:225–235. doi: 10.2174/138920211795677912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JD, Paisey RB, Carey C, Macdermott S. Alström syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2003. Feb 7, pp. 1993–2016. [Updated 2012 May 31] [Google Scholar]
- Mazzoli M, Van Camp G, Newton V, Giarbini N, Declau F, Parving A. Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment. Audiological Medicine. 2003;1:148–150. [Google Scholar]
- Nadol JB, Jr, Marshall JD, Bronson RT. Histopathology of the human inner ear in Alström's syndrome. Audiol Neurotol. 2015;20:267–272. doi: 10.1159/000381935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K, Takeda S. Cilia in the choroid plexus: their roles in hydrocephalus and beyond. Front in Cell Neurosci. 2015;9:39. doi: 10.3389/fncel.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AEH, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Ozantürk A, Marshall JD, Collin GB, Düzenli S, Marshall RP, Candan Ş, Tos T, Esen İ, Taşkesen M, Çayır A, Öztürk Ş, Üstün İ, Ataman E, Karaca E, Özdemir TR, Erol İ, Eroğlu FK, Torun D, Parıltay E, Yılmaz-Güleç E, Karaca E, Atabek ME, Elçioğlu N, Satman İ, Möller C, Muller J, Naggert JK, Özgül RK. The phenotypic and molecular genetic spectrum of Alström syndrome in 44 Turkish kindreds and a literature review of Alström syndrome in Turkey. J Hum Genet. 2015;60:51. doi: 10.1038/jhg.2014.101. [DOI] [PubMed] [Google Scholar]
- Schwartz DM, Pratt RE, Jr, Schwartz JA. Auditory brainstem response in preterm infants: evidence of peripheral maturity. Ear Hear. 1989;10:14–22. doi: 10.1097/00003446-198902000-00003. [DOI] [PubMed] [Google Scholar]
- Shenje LT, Andersen P, Halushka MK, Lui C, Fernandez L, Collin GB, Amat-Alarcon N, Meschino W, Cutz E, Chang K, Yonescu R, Batista DA. Mutations in Alström protein impair terminal differentiation of cardiomyocytes. Nat Commun. 2014;5:3416. doi: 10.1038/ncomms4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh LW. Alström syndrome: progressive deafness and blindness. Ann Otol Rhinol Laryngol. 2007;116:281–285. doi: 10.1177/000348940711600411. [DOI] [PubMed] [Google Scholar]
- Zulato E, Favaretto F, Veronese C, Campanaro S, Marshall JD, Romano S, Cabrelle A, Collin GB, Zavan B, Belloni AS, Rampazzo E, Naggert JK. ALMS1-deficient fibroblasts over-express extra-cellular matrix components, display cell cycle delay and are resistant to apoptosis. PLoS One. 2011;6:e19081. doi: 10.1371/journal.pone.0019081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.