

Abstract
COX metabolites of 8,9-EET, previously observed as potent mitogenic lipid mediators, were synthesized for the first time by using two synthetic approaches. These synthetic materials allow for structural confirmation of COX metabolites of 8,9-EET and further study of their biological roles.
Introduction
The omega-6 arachidonic acid (AA) is metabolized by three major classes of enzymes, namely cyclooxygenases (COX), lipoxygenases (LOX), and cytochrome P450s (CYP), to primarily produce biologically active prostaglandins (PGs), hydroperoxyeicosatetraenoates (HPETEs), and epoxyeicosatrienoates (EETs), respectively1–3. These metabolites may further be metabolized by COX, LOX, and CYP as well as other enzymes, giving rise to secondary metabolites with alternative and/or more potent biological activities.
The CYP450 monooxygenase pathway produces epoxyeicosatrienoic acids (EETs), which have a broad spectrum of biological activities including anti-inflammation, anti-hypertension, analgesia, and cardioprotection.4 Yet EETs could also be mildly proangiogenic, protumorigenic, and prometastatic.5–9 Previous studies have suggested that the protumorigenic and prometastastic activity attributed to EETs may actually be mediated by their corresponding downstream metabolites and not by EETs. EETs are metabolically unstable in vivo and are primarily but not exclusively metabolized by soluble epoxide hydrolase (sEH) to their corresponding 1,2-diols, which are generally biologically less active.10 Besides, EETs has been shown to be metabolized by CYP450, COX, LOX and other enzymes, producing secondary metabolites that have less well understood biological activity. For example, Teder et al.11 recently showed that 14,15-EET is a substrate for human 15-LOX-1 and murine 12-LOX, giving a mixture of isomers containing a hydroperoxy group at positions 5, 8 and 12. However, these metabolites have not been found in vivo yet and their biological significance remains unknown.
Metabolism of EETs has also been demonstrated to occur through CYP450 enzymes, leading to the formation of diepoxyeicosadienoic acids12 and hydroxyepoxyeicosatrienoic acids.13 Diepoxyeicosadienoic acids are further hydrolyzed by sEH into corresponding tetraols or dihydroxytetrahydrofuranes,12,14 which were shown to be vasoactive15 and influence intracellular calcium concentration. Hydroxyepoxyeicosatrienoic acids, formed by ω or ω-1 CYP450 hydroxylation of EETs, were shown to be high affinity (low nM) ligands of peroxisome proliferator-activated receptor-α (PPAR-α) and might play a role in transcription regulation.16
Results and discussion
It has been reported that 5,6- and 8,9-EET can be metabolized by COX,17–20 however little is known about biological roles of these metabolites. It has been shown that COX metabolism of 5,6-EET results in formation of vasoactive 5,6-epoxy-PGE1 and inactive 5-hydroxy-PGI1.17 Two studies have also demonstrated metabolism of 8,9-EET by COX, leading to the formation of the 11-hydroxy-8,9-EET (8,9,11-EHET) which has been shown to be a renal vasoconstrictor and a potent glomerular mitogen, at least an order of magnitude more potent than the 8,9-EET.18,20
Despite the limited evidence describing the biological activity arising from COX products of EET, we have recently shown through use of COX and sEH inhibitors that the tumor growth and metastasis attributed to EETs is highly dependent on COX. In this study, dual inhibition of COX and sEH suppressed tumor growth and metastasis whereas inhibition of sEH alone has an opposite effect.21 This may suggest that the protumorigenic and metastastic activity is due to the downstream metabolites formed from COX. Results from this study motivated us to further investigate the link between the COX and sEH metabolic pathways by characterizing some of the products that form from EETs by the action of the COX enzymes.
Zhang et al. were the first to show that 8,9-EET metabolized by COX from ram seminal vesicles to form hydroxylated products.20 The authors showed that while COX-1 catalyzed hydroxylation of 8R,9S-EET to form two products (11R- and supposedly 15R-(OH)-8R,9S-EETs), only one product was observed for the 8S,9R-EET enantiomer (11R-(OH)-8S,9R-EET). Thus three products were expected from enzymatic hydroxylation of (±)-8,9-EET by COX. To assure accurate structural elucidation of the COX products of 8,9-EET by comparison of the UHPLC retention times and coelution experiments with synthetic standards, a UHPLC method able to separate diastereomers of 11- and 15-EHET needed to be developed. The requirement for a method which can separate all isomers of a biological product is not always met in the scientific literature. However, separation of all isomers is essential for structural confirmation and, in the absence of such an LC–MS method, comparing retention times and/or coelution with synthetic standards does not prove the identity of the biological product.
Since a total of 8 isomeric products are possible for the hydroxylation reaction of (±)-8,9-EET with COX enzymes (2 regioisomers × 2 diastereomers × 2 enantiomers), we decided to perform the determination of absolute stereochemistry in two steps. First, we developed a LC-MS method able to separate the regioisomers and diastereoisomers of the hydroxylated products of 8,9-EET then determined the relative stereochemistry of products corresponding to each chromatographic peak. The question of absolute stereochemistry of the enzymatic products was then addressed by analyzing separately product mixtures obtained from reactions of enantiomerically pure 8R,9S- and 8S,9R-EETs with COX enzymes.
Since COX catalyzed enzymatic hydroxylation of (±)-8,9-EET was expected to produce only three hydroxylated products, an alternative source of both diastereomers of 11- and 15-EHETs was needed. Free radical oxidation of (±)-8,9-EET in biomimetic fashion might be such a source, however the hydrogen atom abstraction at the bisallylic position at C-13 followed by the addition of molecule of oxygen at C-11 or C-15 may give mixture of kinetic (Z,E) and thermodynamic (E,E) conjugated diene products depending on the hydrogen atom donor present in the reaction mixture.22,23 Thus, an appropriate hydrogen donor/radical carrier that allows for selective hydrogen abstraction at bisallylic position at C-13 and trapping of the kinetic Z,E-conjugated diene peroxyls have to be used. Punta et al. showed that free radical autoxidation of methyl linoleate in presence of N-methyl-benzohydroxamic acid (NMBHA) as hydrogen donor/radical carrier lead to exclusive formation of the 9- and 13-trans,cis hydroperoxides.24 Subjection of (±)-8,9-EET to this reaction conditions followed by in situ hydroperoxide reduction with triphenylphosphine resulted in racemic mixture of isomeric hydroxy EETs (Scheme 1) in roughly equimolar ratio.
Scheme 1.
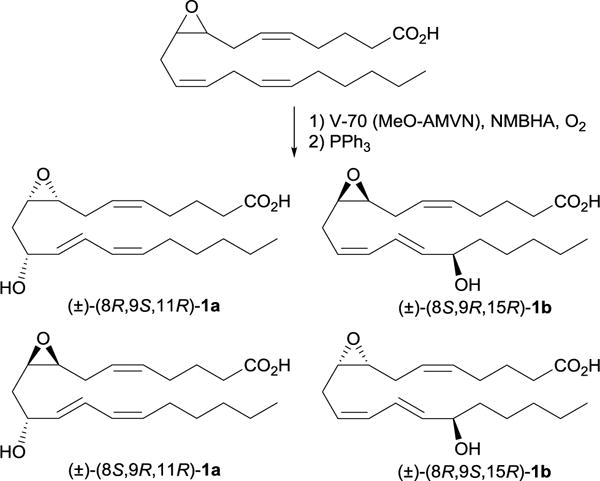
Radical biomimetic oxidation of (±)-8,9-EET produce all 8 isomeric EHETs
The mixture of eight isomeric EHETs (2 regioisomers × 2 diastereomers × 2 enantiomers) produced from autoxidation was then used as a standard to optimize the UHPLC conditions to resolve the diastereomers of both 11- and 15-EHET (Figure S1). Unlike the results from Zhang et al., which demonstrated a total of three COX products, the enzymatic hydroxylation of (±)-8,9-EET with both COX-1 and COX-2 isoforms produced four products. The enzymatic products all had similar retention times to the autoxidation products, as observed in Figure S2. Furthermore, all enzymatic products had the same MS/MS spectra as the autoxidation products. Fragmentation patterns of the enzymatic products at 10.49 and 10.84 min were almost identical to the first two chemically produced isomeric EHETs, having the characteristic fragment ion m/z 235.1351 resulting from an α-fragmentation at C15 (see Supporting Information). Observation of this fragment suggests that they are diastereomeric 15-EHETs. Similarly, the presence of the characteristic fragment ion m/z 183.1040 is indicative of α-fragmentation at C11, and suggests that the peaks eluting at 14.88 and 15.33 min are 11-EHETs.
The autoxidation of 8,9-EET produced a mixture having same products as the biological oxidation of 8,9-EET, based on retention time and MS/MS fragmentation. Here, we present an alternative individual total synthesis of 11- and 15-EHET regioisomers, for use as analytical standards for structural elucidation and in in vivo biological assays to understand their biological activity.
Our retrosynthetic analysis of 8R,9S,11R-EHET began with the disconnection of the conjugated diene giving vinyl iodide 2 and propargylic alcohol 3 as potential precursors (Scheme 2). Further simplification of the propargylic alcohol 3 via standard functional group manipulations led to triyne 4 as a possible intermediate. Finally disconnection of the Csp-Csp3 bond as indicated in Scheme 2 revealed known building blocks: methyl 7-bromohept-2-ynoate 5 and diynol 6.
Scheme 2.
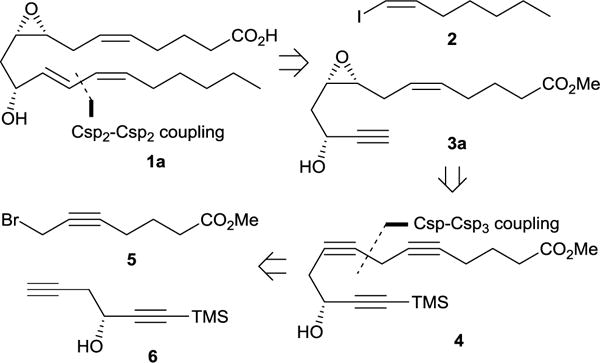
Retrosynthetic analysis for 8R,9S,11R-EHET
The synthesis of 8R,9S,11R-EHET commenced by the construction of the coupling partners 5 and 6. Although methyl 7-bromohept-5-ynoate 5 is commercially available it could be easily prepared from much cheaper hexynoic acid with 53% overall yield in three steps as shown in Scheme 3. Racemic diynol 6 was obtained by treatment of the trimethylsilylpropynal with freshly prepared propargylzinc bromide. Lipase-catalyzed kinetic resolution of the racemic alcohol 6 provided the R enantiomer of the alcohol 6 in 45% yield and >96% ee. With 5 and 6 in hand the next step was to perform their coupling under standard copper-catalysis conditions which yielded the triynol 4 in 63% yield. Selective partial hydrogenation of two sterically unhindered triple bonds in the presence of Brown catalyst Ni2B25 resulted in the formation of dienol 8. Stereoselective hydroxyl directed vanadium-catalyzed epoxidation26 of 8 followed by cleavage of trimethylsilyl group gave the diastereomeric mixture of epoxyalcohols 3 in 71% overall yield. The diastereoselectivity of epoxidation of 8 is considerably lower compared to epoxidation of similar substrates bearing an alkyl substituent instead of alkynyl. This could be explained in terms of van der Waals repulsive forces between axial substituents for chair-like transition state proposed by Mihelich et al.26 The van der Waals radius of acetylene carbons (1.7 Å) is considerably smaller compared to the methyl group (2 Å) which translates to weaker van der Waals repulsive forces and consequently a smaller free activation energy difference between transition states A and B and poorer diastereoselectivity (Figure 1).
Scheme 3.
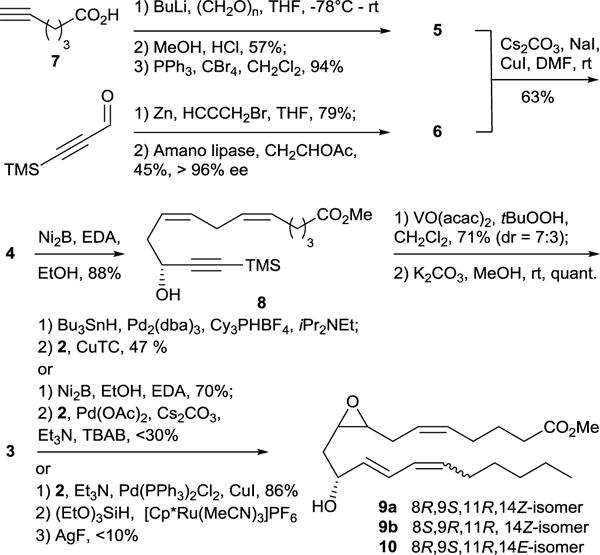
Synthesis of 8R,9S,11R- and 8S,9R,11R-EHET methyl esters
Figure 1.
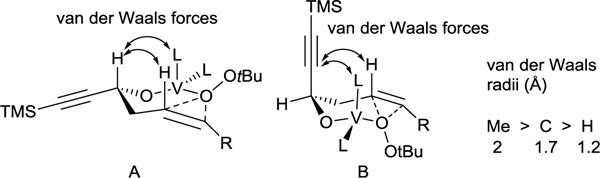
Transition states for epoxidation of 8.
For the transformation of 3 into desired EHET methyl ester we exclusively considered reactions that do not require protection of the hydroxy group present in 3. We first tested the approach using Heck reaction as a key step. Partial hydrogenation of the terminal alkyne in 3 was performed in presence of the Brown catalyst. Although the over reduction of the terminal alkyne could not be completely prevented in this case, it was significantly diminished by shortening reaction times to few minutes. Thus an inseparable mixture of the allylic alcohol with the over reduced product was then subjected to the Heck reaction with vinyl iodide 7 under Jeffery conditions27 to give the desired 8R,9S,11R-EHET methyl ester together with small amounts of trans-trans diastereoisomer 10. The best yield of pure 9 obtained from this reaction after HPLC purification was 30%. However, high variability in the reaction yield was noticed when repeating the reaction, which pushed us to look for alternative approaches.
trans-Hydrosilylation/protodesilylation approach for the synthesis of (E)-olefins28,29 from corresponding alkynes has gained much attention over the past decade because of extremely mild reaction conditions and functional group tolerability that allowed for its use in numerous total syntheses of natural products.30 After establishment of this method in our laboratory on a model compound (Z)-pentadec-9-en-7-yn-6-ol, its applicability to the synthesis of 8R,9S,11R-EHET was tested. Unfortunately, hydrosilylation of the Sonogashira product of 3a and 2 followed by protodesilylation of the intermediate with AgF resulted in formation of the complex mixture containing less than 10% of the desired product 9.
We next turned our attention to the one-pot tandem Pd-catalyzed hydrostannylation/Stille coupling procedure.31 Surprisingly, the reaction between 3 and 2 under these conditions yielded a complex mixture of isomeric products in 20% overall yield (conditions are not shown on the scheme). The NMR analysis of the major product after purification by HPLC revealed it to be the trans-trans isomer 10 (Scheme 3) rather than the desired trans-cis product 9. Complex mixture of products obtained in this reaction is an indicator of poor regio- and stereoselectivity of the first hydrostannylation step, which can be improved by using alternative reaction conditions such as described by Darwish et al.32 Poor reaction yield on the other hand can be improved by using copper-based catalytic systems,33 which revealed to be superior to palladium based catalysts in multiple cases. Thus hydrostannylation of 3 under Darwish conditions followed by CuTC mediated Stille coupling provided diastereomeric mixture of EHET methyl esters 9 in 47 % yield for two steps. 8R,9S,11R-EHET methyl ester 9a and 8S,9R,11R-EHET methyl ester 9b were separated by preparative reverse phase HPLC.
The retrosynthetic analysis for 8R,9S,15R-EHET is presented by Scheme 4 and begins with disconnection of the conjugated diene to give known vinyl iodide 1134 and epoxydiyne 12 as possible precursors. 12 can be further retrosynthetically disconnected to hexynoic acid and epoxy alcohol 13 that in turn can be traced retrosynthetically to propargyl bromide 14 and propargyl alcohol as starting materials.
Scheme 4.
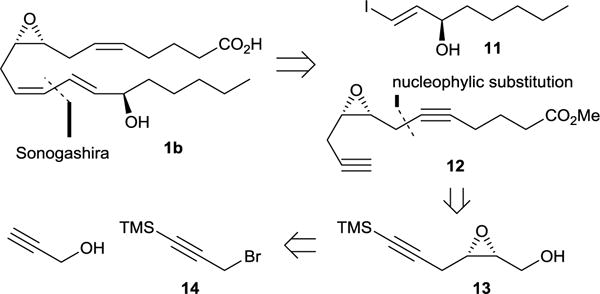
Retrosynthetic analysis for 8R,9S,15R-EHET
The synthesis of 8R,9S,15R-EHET began by copper catalyzed coupling between propargyl bromide 14 and propargyl alcohol to give a diynol intermediate which was partially hydrogenated to the allylic alcohol 15 in the presence of nickel boride (Scheme 5). Sharpless asymmetric epoxidation of 15 proceeded smoothly and provided the corresponding epoxyalcohol in 66% yield and 64% ee. The transformation of 13 into intermediate 12 turned out to be troublesome because the alkylation yield of hexynoyc acid dilithium salt with the triflate derived from 13 was very sensitive to the THF/HMPA ratio, the sequence of reagent addition, and the quenching procedure. Moreover, the triflate derived from 13 is relatively unstable, especially as a solution in THF, and it should be used as soon as produced. It is also worth mentioning that the formation of hexynoic acid dilithium salt in the THF/HMPA mixture results in larger quantities of side products and lower yield of the desired product as compared to the alternative procedure where the same salt is preformed in pure THF followed by the addition of HMPA. Thus the alkylation of hexynoic acid dilithium salt with freshly prepared triflate of 13 followed by esterification with trimethylsilyl diazomethane and cleavage of trimethylsilyl group under basic conditions gave the desired diyne 12 in 42% overall yield for three steps. Sonogashira coupling between 12 and vinyl iodide 11 proceeded smoothly at room temperature forming the intermediate 16 which was further transformed into the final product 8R,9S,15R-EHET methyl ester by partial hydrogenation in presence of Brown catalyst.25
Scheme 5.
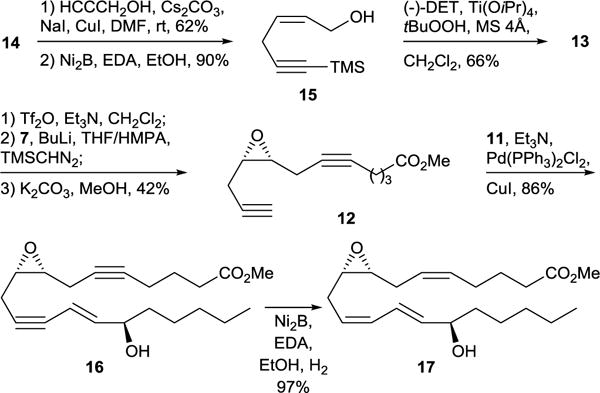
Synthesis of 8R,9S,15R-EHET methyl ester
Synthesized EHET methyl esters were converted into corresponding free acids by hydrolysis with either sodium hydroxide or carboxylesterase 2 (CES 2). Synthetic 8R,9S,11R-EHET, 8S,9R,11R-EHET and 8R,9S,15R-EHET have indistinguishable retention times as compared to the COX metabolites eluting at 14.88, 15.33 and 10.84 min, respectively. Moreover, the synthetic EHETs co-elute with COX products when injected together, as presented in Figures S4 and S5. The MS/MS fragmentations between the synthesized products and those produced from COX enzymes are identical (See supporting information). Overall, the identical retention times and identical fragmentation patterns between the synthetic 11- and 15-EHET and enzymatically produced EHETs provide strong evidence for regio- and relative stereochemistry assignment of EHETs corresponding to each chromatographic peak.
To elucidate absolute stereochemistry of the enzymatic products, COX mediated hydroxylations of enantiomerically pure 8R,9S- and 8S,9R-EETs were performed separately (see SI). Since the stereochemistry at epoxide is fixed, only regio- and diastereomeric products are expected in these reactions. Known absolute stereochemistry at the epoxide together with determined relative stereochemistry of the products from UHPLC-MS experiments allow determination of the absolute stereochemistry of each product formed. As can be seen from the Figure S3, oxygen addition to C11 of the pentadienyl radical inside the COX active site is highly stereospecific occurring exclusively from the pro-R side. This result correlates well with literature data on stereoselectivity of the COX oxygenation reactions.35 On the other hand the stereoselectivity of the oxygen addition to C15 is very poor. Reaction of 8S,9R-EET with the COX-1 enzyme is highly regio- and stereo-selective producing almost exclusively 8S,9R,11R-EHET, while hydroxylation of 8R,9S-EET results in the formation of 8R,9S,11R-EHET as a major product together with smaller but similar amounts of the 8R,9S,15R-EHET and 8R,9S,15S-EHET isomers. Similarly, enzymatic hydroxylation by COX-2 of 8S,9R-EET is more regioselective compared to 8R,9S-EET, however, unlike the reaction with the COX-1 enzyme, formation of the noticeable amounts of 8S,9R,15R-EHET and 8S,9R,15S-EHET was observed in this case.
Conclusions
In summary, we synthesized a mixture of all eight 11- and 15-EHETs through biomimetic free radical autoxidation of (±)-8,9-EET. An optimized UHPLC method has been developed for the separation of the diastereomeric mixtures of 8,9,11-EHET and 8,9,15-EHET. To determine relative stereochemistry of the EHET products within each chromatographic peak a short and easy synthetic approach toward 8R,9S,11R-EHET, 8S,9R,11R-EHET and 8R,9S,15R-EHET was developed. Analysis of the COX hydroxylation products obtained from enantiomerically pure 8,9-EETs with optimized LC-MS/MS method revealed that while hydroxylation at C11 is highly stereoselective, poor stereoselctivity is observed for the hydroxylation at C15 center. Both COX-1 and COX-2 enzymes have preference for hydroxylation of 8,9-EETs at C11 position with reaction being more regioselective for 8S,9R-EET enantiomer. The identity of the biological and autoxidation products formed from 8,9-EET and synthetic EHETs was unambiguously supported by comparison of the retention times, fragmentation patterns and co-elution experiments. The biological roles of EHETs are currently under investigation in our laboratory.36
Supplementary Material
Acknowledgments
We acknowledge support from the National Institute of Environmental Health Sciences R01 ES0027010; NIEHS Superfund Program P42 ES004699; and CounterACT Program, National Institutes of Health Office of the Director, and the National Institute of Neurological Disorders and Stroke, Grant Number U54 NS079202. T. Cajka is supported by the NIH grants U24 DK097154 and P20 HL113452. A. Rand is a fellow of NIH/NIEHS T32 CA108459. K.S.S.L. is partially supported by the NIH grant R00 ES024806. We greatly appreciate instrument funding through the NIH grant S10-RR031630.
Footnotes
† Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Brash AR. J Biol Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 2.Smith WL, Garavito RM, DeWitt DL. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 3.Zeldin DC. J Biol Chem. 2001;276:36059–62. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 4.Morisseau C, Hammock BD. Annu Rev Pharmacol Toxicol. 2013;53:37–58. doi: 10.1146/annurev-pharmtox-011112-140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Medhora M, Daniels J, Mundey K, Fisslthaler B, Busse R, Jacobs ER, Harder DR. Am J Physiol Heart Circ Physiol. 2003;284:H215–24. doi: 10.1152/ajpheart.01118.2001. [DOI] [PubMed] [Google Scholar]
- 6.Cheranov SY, Karpurapu M, Wang D, Zhang B, Venema RC, Rao GN. Blood. 2008;111:5581–91. doi: 10.1182/blood-2007-11-126680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleming I. Prostaglandins Other Lipid Mediat. 2007;82:60–7. doi: 10.1016/j.prostaglandins.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC. Cancer Metastasis Rev. 2011;30:525–40. doi: 10.1007/s10555-011-9315-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pozzi A, Macias-Perez I, Abair T, Wei S, Su Y, Zent R, Falck JR, Capdevila JH. J Biol Chem. 2005;280:27138–46. doi: 10.1074/jbc.M501730200. [DOI] [PubMed] [Google Scholar]
- 10.Newman JW, Morisseau C, Hammock BD. Prog Lipid Res. 2005;44:1–51. doi: 10.1016/j.plipres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 11.Teder T, Boeglin WE, Brash AR. J Lipid Res. 2014;55:2587–96. doi: 10.1194/jlr.M054072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moghaddam M, Motoba K, Borhan B, Pinot F, Hammock BD. Biochim Biophys Acta. 1996;1290:327–39. doi: 10.1016/0304-4165(96)00037-2. [DOI] [PubMed] [Google Scholar]
- 13.Quéré V Le, Plée-Gautier E, Potin P, Madec S, Salaün J-P. J Lipid Res. 2004;45:1446–58. doi: 10.1194/jlr.M300463-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Halarnkar PP, Nourooz-Zadeh J, Kuwano E, Jones AD, Hammock BD. Arch Biochem Biophys. 1992;294:586–593. doi: 10.1016/0003-9861(92)90729-g. [DOI] [PubMed] [Google Scholar]
- 15.Falck JR, Reddy LM, Byun K, Campbell WB, Yi XY. Bioorg Med Chem Lett. 2007;17:2634–8. doi: 10.1016/j.bmcl.2007.01.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cowart LA, Wei S, Hsu MH, Johnson EF, Krishna MU, Falck JR, Capdevila JH. J Biol Chem. 2002;277:35105–12. doi: 10.1074/jbc.M201575200. [DOI] [PubMed] [Google Scholar]
- 17.Carroll MA, Balazy M, Margiotta P, Falck JR, McGiff JC. J Biol Chem. 1993;268:12260–12266. [PubMed] [Google Scholar]
- 18.Homma T, Zhang JY, Shimizu T, Prakash C, Blair IA, Harris RC. Biochem Biophys Res Commun. 1993;191:282–8. doi: 10.1006/bbrc.1993.1214. [DOI] [PubMed] [Google Scholar]
- 19.Moreland KT, Procknow JD, Sprague RS, Iverson JL, Lonigro AJ, Stephenson AH. J Pharmacol Exp Ther. 2007;321:446–454. doi: 10.1124/jpet.106.107904. [DOI] [PubMed] [Google Scholar]
- 20.Zhang JY, Prakash C, Yamashita K, Blair IA. Biochem Biophys Res Commun. 1992;183:138–143. doi: 10.1016/0006-291x(92)91619-2. [DOI] [PubMed] [Google Scholar]
- 21.Zhang G, Panigrahy D, Hwang SH, Yang J, Mahakian LM, Wettersten HI, Liu JY, Wang Y, Ingham ES, Tam S, Kieran MW, Weiss RH, Ferrara KW, Hammock BD. Proc Natl Acad Sci U S A. 2014;111:11127–32. doi: 10.1073/pnas.1410432111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pratt DA, Tallman KA, Porter NA. Acc Chem Res. 2011;44:458–67. doi: 10.1021/ar200024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin H, Xu L, Porter NA. Chem Rev. 2011;111:5944–72. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- 24.Punta C, Rector CL, Porter NA. Chem Res Toxicol. 2005;18:349–56. doi: 10.1021/tx049685x. [DOI] [PubMed] [Google Scholar]
- 25.Brown CA, Ahuja VK. J Chem Soc Chem Commun. 1973;553–554 [Google Scholar]
- 26.Mihelich ED, Daniels K, Eickhoff DJ. J Am Chem Soc. 1981;103:7690–7692. [Google Scholar]
- 27.Jeffery T. Tetrahedron. 1996;52:10113–10130. [Google Scholar]
- 28.Trost BM, Ball ZT, Jöge T. J Am Chem Soc. 2002;124:7922–7923. doi: 10.1021/ja026457l. [DOI] [PubMed] [Google Scholar]
- 29.Fürstner A, Radkowski K. Chem Commun. 2002;93:2182–2183. doi: 10.1039/b207169j. [DOI] [PubMed] [Google Scholar]
- 30.Frihed TG, Furstner A. Bull Chem Soc Jpn. 2016;89:135–160. [Google Scholar]
- 31.Gallagher WP, Terstiege I, Maleczka RE. J Am Chem Soc. 2001;123:3194–3204. doi: 10.1021/ja0035295. [DOI] [PubMed] [Google Scholar]
- 32.Darwish A, Lang A, Kim T, Chong JM. Org Lett. 2008;10:861–864. doi: 10.1021/ol702982x. [DOI] [PubMed] [Google Scholar]
- 33.Allred GD, Liebeskind LS. J Am Chem Soc. 1996;118:2748–2749. [Google Scholar]
- 34.Bischop M, Doum V, Nordschild née Rieche A, Pietruszka J, Sandkuhl D. Synthesis (Stuttg) 2010;2010:527–537. [Google Scholar]
- 35.Schneider C, Pratt DA, Porter NA, Brash AR. Chem Biol. 2007;14:473–488. doi: 10.1016/j.chembiol.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rand AA, Barnych B, Morisseau C, Cajka T, Lee KSS, Panigrahy D, Hammock BD. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1616893114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.