

Abstract
Targeted toxin‐based therapeutics are hindered by poor intracellular uptake, limited stability and non‐specific immune stimulation. To address these problems, ligand‐targeted toxins in combination with low dose saponin mixtures have been adapted and tested in vivo in the past, however, undefined saponin raw mixtures are not suitable for use in clinical development. In the present work we therefore used a targeted toxin (Sap3‐EGF, i.e. saporin fused to epidermal growth factor) in combination with a structurally defined isolated saponin m/z 1861 (SO‐1861). In vitro evaluation confirmed a 6900‐fold enhancement in the cytotoxic efficacy of Sap3‐EGF against TSA‐EGFR target cells. The required dose of the targeted toxin was appreciably reduced and there was a highly synergistic effect observed. An ex vivo hemolysis assay showed no or very less hemolysis up to 10 μg/mL of SO‐1861. In the acute toxicity studies SO‐1861 was found to be non‐toxic up to a dose of 100 μg/treatment. The enzymes aspartate aminotransferase, alanine aminotransferase, and glutamate dehydrogenase did not show any statistically significant liver damage, which was further confirmed by histological examination. Additionally, creatinine was also similar to the control group thus ruling out damage to kidney. In vivo studies in a syngeneic BALB/c tumor model characterized by EGFR overexpression were done by applying 30 μg SO‐1861 and 0.1 μg Sap3‐EGF per treatment. A more than 90% reduction (p < 0.05) in the average tumor volume was observed by this combined therapy.
Keywords: Targeted toxin, Synergistic toxicity enhancement, Saponaria saponins, Ligand-mediated therapeutics, Saporin, Epidermal growth factor
Highlights
-
►
A 6900‐fold enhancement in the in‐vitro efficacy of targeted toxins.
-
►
First report on usage of a purified saponin in‐vivo as enhancer of targeted toxin‐based tumor therapy.
-
►
Reduced toxin dose with higher efficacy provides evidence for better translational research.
-
►
Basis for testing in heterogenous tumor models and for numerous plant‐derived toxins.
1. Introduction
Cancer remains to be a major cause of death world‐wide and accounted for nearly 12–14% of recorded deaths in 2008 (Jemal et al., 2011). With advancements in diagnostic techniques the detection of tumors at an earlier stage is plausible. Simultaneously, there has been an appreciable progression in the field of molecular targeted therapies in general and more so in case of tumor treatment (Ashley et al., 2011; Vallera et al., 2010). Some of these modalities are in clinical practice as well. The problems that continue to restrict success of targeted therapies are the stratifications in the type of tumors with different levels of receptor expressions. Expression of tumor‐associated cell surface antigens in off‐target cells is another obstacle, which leads to unpredictable grades of unwanted toxicity in these cells (Ashley et al., 2011).
Targeted toxins (TTs) comprise a toxin moiety fused with antibodies, growth factors or cytokines that bind to a target cell and induce cell death (Polito et al., 2011). The fusion protein Sap3‐EGF (in some publications simply abbreviated as SE) is one such TT, which contains the plant protein toxin Saporin (Sap3) and epidermal growth factor (EGF) (Fuchs et al., 2007). This TT was found to be highly specific for EGFR overexpressing cells. Two main problems appeared with the application of this TT in vivo. Firstly, there was a substantial immune response against the toxin moiety and secondly, the cytosolic uptake of this TT was insufficient so that high systemic concentrations of the TT were required for significant anti‐tumor efficacy resulting in undesired adverse effects. These problems were partly solved by introducing a molecular adapter with cell penetrating properties into the TT (Fuchs et al., 2007) and finally circumvented by using saponins as a synergistic enhancer of the site‐specific toxicity of Sap3‐EGF.
In previous studies we have been able to demonstrate that the combination of Sap3‐EGF with a commercial saponin composite or an isolated pure saponin from the commercial mixture resulted in a reduction of the tumor volume of more than 94% (Bachran et al., 2010); however, saponins used in these experiments were a non‐standardized complex mixture, which is inapplicable for clinical studies and in the meanwhile commercially unavailable (Weng et al., 2010). Therefore, it was envisaged to isolate structurally defined saponins from natural sources which can potentially enhance the in vivo effectiveness of TTs. Sap3, a type I ribosome inactivating protein (RIP), is isolated from the seeds of Saponaria officinalis L., a plant that is also a rich source of triterpenoid saponins. It is hypothesized that the co‐existence of a highly toxic protein together with saponins, which are synergistic enhancers of the protein's toxicity, is an evolutionary advantage for numerous plants from the family Caryophyllaceae (Thakur et al., 2011a).
The three R's, i.e. replacement, refinement and reduction, were the guiding principle (Russell and Burch, 1959) to utilize a purified single saponin in combination with Sap3‐EGF, both well characterized, to treat tumors. In the present work we refined the process of saponin isolation and applied the highly efficacious saponin SO‐1861, resulting in a reduced dosage of Sap3‐EGF compared to the conventional treatment of EGFR overexpressing tumors with TTs.
2. Materials and methods
2.1. Purification and isolation of SO‐1861
The roots of S. officinalis L. (Galke, Gittelde, Germany) were coarsely powdered using a mechanical grinder. Extraction was carried out by percolation in 90% methanol, wherein 50 g drug powder was continuously stirred in 200 mL of 90% methanol. After 24 h the extract was filtered and concentrated in vacuo. Acetone was added to the extract in a 1:1 ratio and the precipitate was collected past centrifugation. This precipitate was further washed three times with acetone and subjected to dialysis (molecular mass cutoff 1000 Da) against deionized water for 72 h with 5 changes of dialyzing medium (Weng et al., 2010). The dialyzed acetone precipitate was lyophilized (Heto, Germany) and the dried extract was thereafter subjected to isolation using a preparative high performance liquid chromatography (Shimadzu, Japan) using an UltraSep ES Pharm RP18E column. The mobile phase used was a gradient of methanol (A) and water + 0.05% trifluoroacetic acid (B) at a flow rate of 1.5 mL/min. The program was 0–30 min 60% A–100% A, 30–45 min 30% A, 45–50 min 60% A. For further purification a semi‐preparative HPLC was performed using acetonitrile: water + 0.05% trifluoroacetic acid gradient of 70–30% acetonitrile in 40 min at a flow rate of 0.5 mL/min.
2.2. Characterization of SO‐1861
The purity of isolated saponins was preliminarily determined by thin layer chromatographic analysis. The mobile phase was n‐butanol:acetic acid:water (4:1:5 v/v). The mass of the isolated saponins was determined by electrospray ionization time‐of‐flight mass spectrometry (ESI‐TOF‐MS) (Agilent, Santa Clara, USA) in a negative ion mode to reduce the protonation. Determination of the aglycone and other structural features was done by H1 and C13 NMR analysis. The electrophoretic mobility of SO‐1861 was determined according to the reported protocol.
2.3. In vitro hemolytic activity of SO‐1861
SO‐1861 was tested for its in vitro hemolytic activity. Whole human blood was collected in EDTA vials and spun‐down at 900g (4 °C) for 10 min, the pellet was washed twice and thereafter carefully resuspended. A 4% (v/v) solution of the RBC in 0.9% NaCl was prepared and 150 μL of it was pipetted into a Nunc immuno module (U16 Maxisorp, Nalge Nunc International, Denmark). To this RBC solution 50 μL of the saponin sample was added (concentration range 500 μg/mL to 15.63 μg/mL) in triplicate. The plate was incubated at 37 °C for 30 min. Thereafter, the plate was centrifuged at 800g for 10 min. The supernatant (100 μL) was collected and transferred to a flat bottomed 96‐well plate (Sarstedt, Newton, USA). The absorbance was measured at 405 nm. A negative control (0.9% (w/v) NaCl) (0% hemolysis) and a positive control (0.1% Triton‐X‐100) (100% hemolysis) were included in the experiment to calculate the degree of hemolysis.
2.4. Protein expression and purification
The methods for expression and purification of Sap3‐EGF (also called SE) are defined in Weng et al. (2012). The protein Sap3‐EGF comprises an N‐terminal 6xHis‐tag, Saporin and human EGF as ligand. Plasmid DNA was transformed into the Escherichia coli strain, Rosetta DE3 pLysS (Novagen). Transformed cells were grown overnight in Luria Broth medium (400 mL) supplemented with ampicillin (50 μg/mL). The overnight culture was further split into 4 portions with each 100 mL transferred to 300 mL fresh LB medium and the culture was grown to A550 = 0.6–0.8. Isopropyl β‐d‐thiogalactopyranoside was added to a final concentration of 1 mM and the culture further incubated for 3 h. Thereafter the cells were centrifuged (10 min, 4 °C, 4000g) and the culture pellets were resuspended in phosphate‐buffered saline (PBS), pH 7.0, and stored at −20 °C.
To purify Sap3‐EGF, the resuspended cells were thawed, sonicated on ice five times for 20 s (Branson Sonopuls, microtip SH213G, duty cycle 20%) with 20‐s‐breaks, and centrifuged at 16,100g for 30 min at 4 °C. The supernatant was subjected to affinity chromatography on a pre‐equilibrated nickel–nitrilotriacetic acid column. The toxins were eluted using successively 50, 70 and 250 mM imidazole buffer. After elution the fractions were analyzed by SDS‐PAGE (12%). The fractions containing Sap3‐EGF were concentrated using Amicon centrifugal filter devices (30 kDa, Millipore, Eschborn, Germany) and imidazole was removed by buffer exchange against PBS via PD10 columns (GE Healthcare, Munich, Germany). The protein concentration was determined by a commercial protein assay kit (Advance protein assay Kit, Cytoskeleton, Inc., USA).
2.5. Endotoxin removal
For removal of endotoxins 400 μL of Sap3‐EGF was diluted to 2000 μL by addition of sterile Dulbecco's PBS in a sterile pyrogen‐free tube. To this solution 20 μL of Triton‐X‐114 (Sigma, Germany) was added and stirred for 30 min at 4 °C. Thereafter, the mixture was transferred to a pre‐heated water bath (37 °C) for 7 min. The solution was transferred to a pyrogen‐free Eppendorf tube and centrifuged at 19,900g for 10 min at 25 °C. The supernatant was collected and the complete process was repeated once more. The amount of protein was determined using BCA assay (Pierce Biotechnology, Rockford, USA).
2.6. Enzymatic activity
The N‐glycosidase activity of Sap3‐EGF was determined using an adenine release assay as described by Weng et al. (2012) without any further modifications.
2.7. Cell culture and cytotoxicity studies
Swiss mouse embryo NIH‐3T3 cells were obtained from the German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany; HER‐14 cells were a kind gift by Prof. E. J. van Zoelen, Department of Cell Biology, University of Nijmegen, The Netherlands. The mammary adenocarcinoma cell line (TSA) was a kind gift from Dr. S Bulfone‐Paus (Forschungszentrum Borstel, Borstel, Germany). Transfection of EGFR to the TSA cells is already reported in Fuchs et al. (2007) and these transfected cells were used for the induction of tumors in BALB/c mice.
Cytotoxicity studies were essentially carried out as already described in previous publications (Thakur et al., 2012) using an MTT assay. Evaluation of the cytotoxicity of SO‐1861 alone was carried out to determine the no‐observed‐effect levels (NOEL values). Furthermore, a real‐time evaluation of SO‐1861 in combination with Sap3‐EGF was determined in TSA‐EGFR cells by an impedance‐based measurement as described in Thakur et al. (2012). In brief, 4000 cells were seeded on an Adcon Plate (Bionas, Germany) and allowed to proliferate for 24 h. Thereafter, Sap3‐EGF (0.003–10 nM) with or without SO‐1861 (2 μg/mL) was added and the effects of compounds were monitored in real‐time.
2.8. In vivo experiments
All animal experiments were carried out in compliance with the German Law and the recommendations of the ethical committee (approval G 0260/10 from LAGeSo, Berlin, Germany). The in vivo experiments were performed in 6–8‐week‐old BALB/c mice (18–20 g) (Charles Rivers, Germany). The mice were housed in plexi‐glass cages under constant day–night cycle (12 h each) and had free access to pellet feed and water. A period of 1 week was rendered to the mice for acclimatization. All the animals were monitored routinely for well‐being during the entire course of the experiment.
The PBS used for all animal experiments was Dulbecco's PBS without Ca and Mg (PAA, Austria). The acute toxicity study for SO‐1861 was carried out at 5 different dosages (15, 30, 60, 100 and 200 μg/treatment) in 100 μL of PBS. The mice (3 in each group) were observed routinely for the first two days (6 h and 24 h interval) and thereafter, every day. The animals were visually monitored and their body weights were recorded on a regular basis. The blood was collected (∼150 μL) from the sub‐mandibular vein of all the mice (only twice fortnightly for each individual) and different blood parameters (see Results) were determined photometrically by standard procedures of clinical chemistry on a Modular P analyzer (Roche Diagnostics, Mannheim, Germany) using system reagents supplied by the manufacturer. After 28 days the mice were sacrificed by an isoflurane overdose and their organs collected for histopathological examination.
The induction of the tumor is described in Bachran et al. (2010). In brief, 1.25 × 105 TSA‐EGFR cells were injected in the right flank in 100 μL PBS on day 0. On day 5, when a solid tumor was palpable in all the mice, the 18 animals were distributed randomly to two groups and the therapy was started. The treated group comprised 10 mice, which received per medication 30 μg of SO‐1861 (s.c. into the neck) in 100 μL PBS and 1 h later 0.1 μg Sap3‐EGF (s.c. in the right flank) again in 100 μL PBS. The control (placebo) group (8 mice) was treated with PBS alone in the neck and right flank. In total there were 8 administrations per mouse done on days 5, 8, 12, 15, 19, 22, 25 and 28. The growth of the tumor was monitored on a daily basis using a digital caliper and the final tumor volume was determined past excision.
2.9. Histopathological examination
The organs and tumors were collected after sacrificing the mice, fixed in formalin and embedded in paraffin blocks. Thin 3‐μm‐sections were cut by a sliding microtome, and the organs were stained with hematoxylin/eosin (HE). The sections of liver, spleen and kidney were examined for apoptosis, necrosis, evidences of inflammation, tumor manifestation and other histological aspects, which may indicate a severe adverse effect of the combination therapy. Mild hepatic injury was defined as cytoplasmic degeneration of hepatocytes and necrosis of not more than four single cells per 4 mm2, moderate liver injury as hydropic change of hepatocytes and necrosis of more than four but not more than ten single hepatocytes per 4 mm2, severe hepatic failure as necrosis of more than ten hepatocytes per 4 mm2 to extended necrosis of hepatocytes.
2.10. Statistical analyses
Data is reported as mean ± SEM. Instat (version 2.01) (Statistical Service Centre, University of Reading, UK) was used for the statistical analysis. All data was tested by Dunnet's test comparing all the groups versus control; p < 0.05 was considered significant.
3. Results
3.1. Isolation and characterization of saponins and Sap3‐EGF
In the present work we applied a new modified isolation procedure for saponins. They were extracted from the powdered roots of common soapwort by methanol percolation and concentrated by acetone precipitation followed by dialysis. The developed isolation process was less tedious and led to a higher yield, i.e. 15% (w/w) of the crude extract compared to typically 6–8% for other methods. ESI‐MS‐TOF analysis of the dialyzed acetone precipitate confirmed the presence of only high molecular weight saponins. A two‐step preparative HPLC rendered the isolation of five different saponins. One of them referred to as SO‐1861 was obtained with highest purity; it was an amorphous solid and had a molecular formula C83H13O46 (m/z 1861.7679). Thus, it appears that a pentose in the previously reported structure of Saponarioside A (Jia et al., 1998) is replaced with a hexose at the sugar chain in C‐28 position. The aglycone is anticipated to be a quillaic acid as already illustrated in S. officinalis rhizomes (Henry et al., 1981). This is a novel structure, which was obtained in appreciable purity as evident from the mass spectra in [M − H]− mode (Supplementary Figure 1). The structure was further verified by the H1 and C13 NMR spectroscopy. Thin layer chromatographic analysis of the isolated saponin showed only one spot in both 1D and 2D mode thus validating its purity (Supplementary Figure 2). The saponin had a relative electrophoretic mobility of 0.59 related to bromophenol blue set to 1.0 (Supplementary Figure 3).
Sap3‐EGF was expressed in E. coli and the protein purified via nickel–nitrilotriacetic acid chromatography. The total amount of Sap3‐EGF was found to be 0.35 mg/mL and the process of endotoxin removal did not cause any major changes in the final concentration. No degradation products were observable on the SDS‐PAGE (Supplementary Figure 4).
3.2. Self‐cytotoxicity of SO‐1861 and enhancement of the cytotoxicity of Sap3‐EGF
Preliminary evaluations in TSA‐EGFR cells showed a cell lytic effect of SO‐1861 at concentrations greater than 10 μg/mL (not shown). Since the major aim of purifying saponins in this study was to apply them in combination with TTs to synergistically enhance the toxicity of the TT against TSA‐EGFR target cells, we first investigated the self‐cytotoxicity of SO‐1861 in cell culture (Figure 1A). There was absolutely no effect of SO‐1861 up to 4 μg/mL and only a very slight effect up to 10 μg/mL. We therefore decided to apply 2 μg/mL of SO‐1861 for the combined treatment to be on the safe side. In addition, an impedance‐based assay confirmed a non‐perturbing effect on membranes of SO‐1861 up to 10 μg/mL (Thakur et al., 2012). The combination of Sap3‐EGF and SO‐1861 was highly toxic in a synergistic fashion, whereby a more than 50% cell death was observed at a Sap3‐EGF concentration of 0.0001 nM (IC50 0.13 pM) and higher while no toxic effects were observed at that concentration for Sap3‐EGF alone (IC50 0.9 nM) (Figure 1B).
Figure 1.
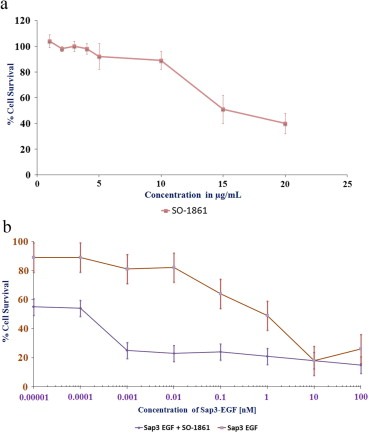
(A) Self‐toxicity of SO‐1861 in TSA‐EGFR cells. The cells were incubated with different concentrations of SO‐1861 for 48 h. The cell proliferation was thereafter determined using an MTT assay. The data points reflect the mean of three experiments conducted in triplicate. (B) Cytotoxic effects of Sap3‐EGF on TSA‐EGFR cells with (solid line) or without (dot‐dashed line) SO‐1861. The cells (2000 cells/well) were seeded and allowed to proliferate for 24‐h, thereafter SO‐1861 was added at a concentration of 2 μg/mL to half the wells, and medium to the rest of the wells; after 5 min Sap3‐EGF was added in different concentrations within the range of 0.00001–100 nM. Cell viability was thereafter determined using an MTT assay. All the experiments were performed in triplicate and the reported results are a mean of 3 experiments.
In case of the impedance‐based cytotoxicity determination assay shown in Figure 2A and B the following results were obtained. The cells proliferated well up to 24 h of seeding of the cell as demonstrated by a continuous increase of impedance (Figure 2A). Thereafter, the plate was taken out of the plate station and the compounds SO‐1861 (2 μg/mL) and Sap3‐EGF (0.01–3.0 nM) were added, and the impedance was reset to zero. While the cells treated either only with Sap3‐EGF or only with SO‐1861 continued to proliferate as indicated by positive impedance values, the cells treated with the combination of Sap3‐EGF and SO‐1861 were already reduced by more than 50% within the first 8 h after addition of the compounds demonstrating the synergistic effect of this combination.
Figure 2.
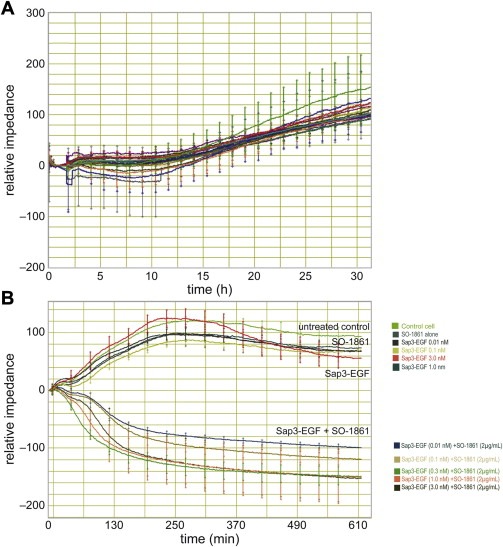
Real‐time monitoring of the cell viability using an impedance‐based assay. (A) The impedance of the cells after seeding and settling was set to zero. After a lag phase of 12 h, the cells started into a continuous growth phase. About 31 h after seeding, the cells were removed from the measurement to add the drugs. (B) To consider the slight differences during growth seen in (A), the impedance was set to zero again after starting of the treatment. Thus, positive relative impedance values indicate further growth after treatment while negative values illustrate cell death. The time scale was also set to zero at that time point. The uppermost curve represents untreated cells, the curve directly beneath depicts the cells treated with 2 μg/mL SO‐1861 only. The remaining curves of the upper bundle represent cells treated with increasing concentrations of Sap3‐EGF (0.01–3.0 nM). The lower arrays of curves displays the cells treated with the same concentrations of Sap3‐EGF, but in addition with SO‐1861 which are further defined by the color legends. Note the very rapid effect of the combined treatment within 6 h.
3.3. Ex vivo hemolytic activity
The hemolytic activity of SO‐1861 against freshly isolated human erythrocytes was determined photometrically by monitoring the released hemoglobin upon incubation with different concentrations of saponins (Figure 3). This was compared with Saponinum album (SA), the crude saponin mixture, which was previously used as enhancer of TTs (Bachran et al., 2010). There was no hemolysis observed up to a concentration of 10 μg/mL. SO‐1861 which was marginally more hemolytic when compared to SA.
Figure 3.
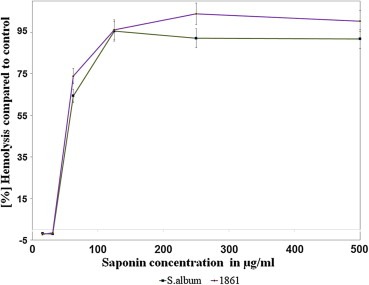
Evaluation of the degree of hemolysis in freshly prepared human erythrocytes determined by spectroscopic measurement of the released hemoglobin. There was no observed hemolysis up to 10 μg/mL of SO‐1861, thereafter the percentage of hemolysis incremented drastically at higher concentrations.
3.4. Acute toxicity in BALB/c mice
All the animals treated with an acute dose of 200 μg SO‐1861 per application died on the second day of the experiment as detailed in the survival curve (Figure 4). None of the animals from 100 μg/treatment or below died during the entire observation period of 28 days. Blood parameters were compared for all the treated groups to the placebo (buffer) treated control. Although there was a slight increase at some individuals in serum creatinine levels as well as in aspartate aminotransferase, alanine aminotransferase and glutamate dehydrogenase activity in case of 60 μg and 100 μg/treatment, this was still well below a moderately toxic effect (as evidenced per histopathological assessment) and there was no substantial increment in the average of these parameters compared to placebo‐treated control animals (Figure 5).
Figure 4.
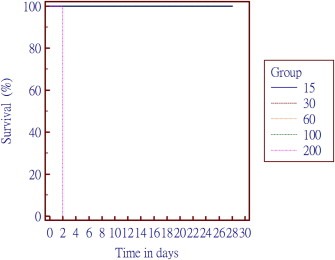
Survival curve for the acute toxicity of SO‐1861. Different dosages tested were 15, 30, 60, 100 and 200 μg per treatment. All the animals in the group treated with 200 μg died on day 2 of experiment, while no further mortality was observed in any other group.
Figure 5.
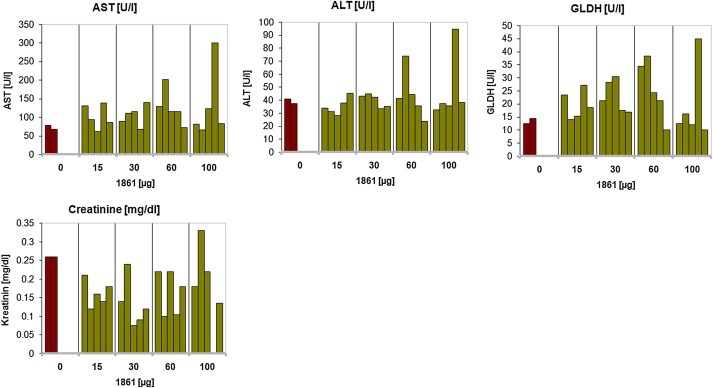
Different hematological parameters evaluated in SO‐1861‐treated mice during the evaluation of the acute toxicity: aspartate serum transferase (AST), alanine aminotransferase (ALT), creatinine and glutamate dehydrogenase (GLDH). Apart from the values being slightly higher in one mice each in the 60 and 100 μg/treatment group, no such effects were observed in other animals of the treated group in comparison to controls. The p values calculated for the different parameters by performing one‐way ANOVA using Dunnett's test are 0.84 (AST), 0.88 (ALT), 0.32 (creatinine), and 0.69 (GLDH) indicating no significant effect.
Histopathological examination of the liver indicated mild alterations in the majority of animals, which were treated with 15 μg–100 μg SO‐1861. Less than 10% of these mice showed moderate injuries of the liver. In contrast, severe damage of the liver was often induced in animals receiving 200 μg SO‐1861. The kidneys and spleens of all mice were not damaged by any SO‐1861 application; however, the spleens showed lymphatic hyperplasia indicating that injected reagents might be immunogenic.
3.5. Tumor growth inhibitory effects
Based on the hemolysis assay and acute toxicity studies, the two lower dosages of saponins (15 and 30 μg) were considered the safest for performing the in vivo experimentations in tumor bearing BALB/c mice. A combination therapy of the saponin mixture Saponinum album together with SA2E, another Sap3‐EGF‐based TT that differ from the fusion protein used in the present study by additional functional domains, has shown tremendous potential in previous studies. The major problem associated with this study was the use of a non‐purified and completely uncharacterized saponin mixture unsuitable for clinical studies. There were also some problems associated with the stability of the fusion protein SA2E as well. The reason to use SA2E in previous studies was the evident advantage of a molecular adapter that enhances the endosomal escape of Sap3. This led to some reduction in side‐effects which were slightly more pronounced in case of Sap3‐EGF. Since the enhancement of the endosomal escape is highly supported by purified saponins, it facilitated us to apply toxins at a much lower dose than the one exhibiting side‐effects and toxicity and to use the more stable Sap3‐EGF (compared to SA2E).
On 29th day of treatment, the average tumor volume in placebo‐treated mice was significantly (p < 0.023) larger than in the mice treated with the combination of 30 μg SO‐1861 and 0.1 μg Sap3‐EGF. The average reduction in the tumor volume was more than 90% in treated mice compared to control mice and 8 of 10 mice had a complete remission (1 of 8 in the controls) indicating a strong dependency of the therapy on the combined treatment (Figure 6).
Figure 6.
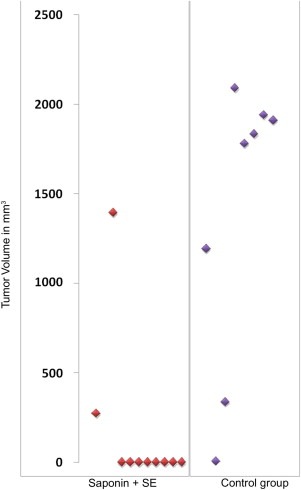
Distribution of the tumor volume in all the animals in placebo‐ and verum‐treated mice. The most effective combination known was used: 0.1 μg Sap‐EGF (SE) and 30 μg SO‐1861 per treatment. A total of 8 therapies were given within the duration of 28 days. Each data point is a representative of the tumor volume determined for each individual animal on day 29 past sacrificing and excision of the tumor.
4. Discussion
TTs are gaining an increasing prominence in the disease management of cancer. Improved methods of protein engineering have resulted in a number of innovative ideas to overcome the major problems associated with the use of protein drugs including systemic immune response, off‐target effects and insufficient cytosolic uptake by cancer cells (Fuchs and Bachran, 2011; Madhumathi and Verma, 2012). Published strategies comprise the use of fully humanized targeted toxins (Psarras et al., 2000; Stahnke et al., 2008), the introduction of deimmunized toxins (Cizeau et al., 2009; Liu et al., 2012), the design of bispecific antibodies (Oh et al., 2012) and methods that result in the increase of endosomal escape including photochemical internalization (Selbo et al., 2012), cell penetrating peptides (Hetzel et al., 2008; Yuan et al., 2012) and enhancer substances such as monensin or saponin (Fuchs et al., 2009; Shaik et al., 2001). A large number of targeted toxins are undergoing different phases of clinical trials (Madhumathi and Verma, 2012). Type I RIPs are toxins that have the advantage not to possess a natural cell binding domain, which makes them ideal candidates to fuse them with a cancer cell targeting moiety; however, since these TTs are generally doomed to get stuck due to their inability to efficiently escape the endo/lysosomal compartment (Puri et al., 2012), the role of saponins in promoting the endosomal escape appears to be highly beneficial (Thakur et al., 2011a). In the present study, we used Sap3‐EGF as a targeted toxin. S. officinalis (common soapwort) is the source of the type I RIP saporin (Lombardi et al., 2010b) that has already been tested in a high number of targeted toxins and in several clinical trials (Polito et al., 2011). Furthermore, a lot of work has been done to improve the expression of saporin‐based toxins including codon‐usage optimization and stabilization by PDZ domains (Lombardi et al., 2010a). The EGFR is an accepted target for anti‐cancer drugs, and corresponding monoclonal antibodies are approved by the U.S. Food and Drug Administration (Panitumumab, Cetuximab) or are under clinical investigation (Zalutumumab) (Yan et al., 2011). Moreover, new imaging techniques allow to evaluate the response to treatment as shown for murine xenograft tumor models by far red imaging after application of an anti‐EGFR‐based targeted toxin (Pardo et al., 2012). In addition to saporin, S. officinalis is also a rich source of triterpenoid saponins. Interestingly, the combination of a saporin‐based TT and saponins from the plant family Caryophyllaceae are reported for synergistic toxic effects. Therefore, it was considered worthwhile to isolate and identify a purified saponin with desired structural features for use with saporin‐based TTs (Weng et al., 2012). It is also established from previous work that the saponin of interest should have a glucuronic acid at C‐3 position and possess an aglycone being preferably a triterpenoid (gypsogenin or quillaic acid), and must have a bidesmosidic structure (Fuchs and Bachran, 2009). Ideal saponins should also have a relatively high electrophoretic mobility (Thakur et al., 2011b). All these structural attributes are fulfilled by SO‐1861, thus validating it as the most suitable purified saponin for testing in a tumor bearing mice model.
Since saponins are relatively rapidly distributed in vivo and excreted mainly in the urine, it is anticipated that the cytotoxic effects on tumor cells should be exhibited in quick succession past the administration of the saponin and TT (Bachran et al., 2010). Impedance‐based real‐time monitoring of TSA‐EGFR cells further confirmed this in vitro when the cells were exposed to the combination of TT and SO‐1861 (Figure 2). These cells were further used for induction of syngeneic solid tumors in BALB/c mice. The results of the present study clearly demonstrate a high level of synergy and reduced side‐effects in the treatment of tumors. The amount of 0.1 μg Sap3‐EGF required to achieve a more than 90% tumor reduction is only 2% of the amount (5 μg) necessary to get a regression of only 33% with Sap3‐EGF in the absence of saponins (Fuchs et al., 2007). This effect was substantially better than those achieved by cell penetrating peptides used for the targeted delivery of gelonin (Yuan et al., 2012), by photochemical internalization applied to EGF‐saporin (Selbo et al., 2012), and by long‐circulating monensin nanoparticles to enhance the cytotoxicity of a ricin‐based immunotoxin (Shaik et al., 2001). Nevertheless, the latter approach is notable since the use of nanoparticles can be used to target the saponins. Although target cell specificity is maintained due to the targeting moiety of the TT, it is important to highlight the lack of saponin specificity of the present approach since it is administered without a target recognition component. Therefore, it would be worthwhile to make tumor‐specific saponin delivery systems to further increase the specificity of the treatment and bringing down off‐target effects to a minimum. Another possibility to improve the therapy with Sap3‐EGF is the use of bispecific ligand‐directed toxins as demonstrated for deimmunized Pseudomonas exotoxin using either a single chain EGF/interleukin‐4 ligand or a fusion of EGF and the amino terminal fragment of urokinase‐type plasminogen activator (Oh et al., 2012; Waldron et al., 2012).
In the present study, we applied a syngeneic tumor model that allows us to use immune competent mice and to consider the immune response. Nonetheless, an aspect of interest would be the testing of this therapeutic combination in a heterogeneous nu/nu mouse tumor model with a human‐derived tumor as in case of disseminated human pancreatic cancer treated with an anti‐EGFR TT (Bruell et al., 2005). In any case, the combination approach presented here provides an exciting avenue for further evaluation of different targeted toxins composed of plant‐based type I RIP's. SO‐1861 appears to be an ideal candidate for testing in such combination therapy with other antibody directed bacterial, plant or humanized toxins.
Conflict of interest
None to declare.
Supporting information
Supplementary data
Acknowledgments
MT would like to acknowledge Alexander von Humboldt Foundation for the postdoctoral research fellowship. Funding from German Research Foundation (DFG grants FU 408/6‐1, TH1810/1‐1, and WE 4784/1‐1) is also acknowledged.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2012.12.004.
Thakur Mayank, Mergel Katharina, Weng Alexander, von Mallinckrodt Benedicta, Gilabert-Oriol Roger, Dürkop Horst, Melzig Matthias F., Fuchs Hendrik, (2013), Targeted tumor therapy by epidermal growth factor appended toxin and purified saponin: An evaluation of toxicity and therapeutic potential in syngeneic tumor bearing mice, Molecular Oncology, 7, doi: 10.1016/j.molonc.2012.12.004.
Contributor Information
Mayank Thakur, Email: mayank.thakur@charite.de.
Hendrik Fuchs, Email: hendrik.fuchs@charite.de.
References
- Ashley, C.E. , Carnes, E.C. , Phillips, G.K. , Padilla, D. , Durfee, P.N. , Brown, P.A. , Hanna, T.N. , Liu, J. , Phillips, B. , Carter, M.B. , Carroll, N.J. , Jiang, X. , Dunphy, D.R. , Willman, C.L. , Petsev, D.N. , Evans, D.G. , Parikh, A.N. , Chackerian, B. , Wharton, W. , Peabody, D.S. , Brinker, C.J. , 2011. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat. Mater. 10, 476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachran, C. , Weng, A. , Bachran, D. , Riese, S.B. , Schellmann, N. , Melzig, M.F. , Fuchs, H. , 2010. The distribution of saponins in vivo affects their synergy with chimeric toxins against tumours expressing human epidermal growth factor receptors in mice. Br. J. Pharmacol. 159, 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruell, D. , Bruns, C.J. , Yezhelyev, M. , Huhn, M. , Muller, J. , Ischenko, I. , Fischer, R. , Finnern, R. , Jauch, K.W. , Barth, S. , 2005. Recombinant anti-EGFR immunotoxin 425(scFv)-ETA' demonstrates anti-tumor activity against disseminated human pancreatic cancer in nude mice. Int. J. Mol. Med. 15, 305–313. [PubMed] [Google Scholar]
- Cizeau, J. , Grenkow, D.M. , Brown, J.G. , Entwistle, J. , MacDonald, G.C. , 2009. Engineering and biological characterization of VB6-845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J. Immunother. 32, 574–584. [DOI] [PubMed] [Google Scholar]
- Fuchs, H. , Bachran, C. , 2009. Targeted tumor therapies at a glance. Curr. Drug Targets 10, 89–93. [DOI] [PubMed] [Google Scholar]
- Fuchs, H. , Bachran, C. , 2011. Design of targeted protein toxins. In Kratz F., Senter P., Steinhagen H.(Eds.), Drug Delivery in Oncology – from Basic Research to Cancer Therapy Wiley-VCH; Weinheim, Germany: 1443–1487. [Google Scholar]
- Fuchs, H. , Bachran, C. , Li, T. , Heisler, I. , Durkop, H. , Sutherland, M. , 2007. A cleavable molecular adapter reduces side effects and concomitantly enhances efficacy in tumor treatment by targeted toxins in mice. J. Control Release 117, 342–350. [DOI] [PubMed] [Google Scholar]
- Fuchs, H. , Bachran, D. , Panjideh, H. , Schellmann, N. , Weng, A. , Melzig, M.F. , Sutherland, M. , Bachran, C. , 2009. Saponins as tool for improved targeted tumor therapies. Curr. Drug Targets 10, 140–151. [DOI] [PubMed] [Google Scholar]
- Henry, M. , Brion, J.D. , Guignard, J.L. , 1981. Plant Med. Phytol. 15, 192–200. [Google Scholar]
- Hetzel, C. , Bachran, C. , Fischer, R. , Fuchs, H. , Barth, S. , Stocker, M. , 2008. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 31, 370–376. [DOI] [PubMed] [Google Scholar]
- Jemal, A. , Bray, F. , Center, M.M. , Ferlay, J. , Ward, E. , Forman, D. , 2011. Global cancer statistics. CA Cancer J. Clin. 61, 69–90. [DOI] [PubMed] [Google Scholar]
- Jia, Z. , Koike, K. , Nikaido, T. , 1998. Major triterpenoid saponins from Saponaria officinalis. J. Nat. Prod. 61, 1368–1373. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Onda, M. , Lee, B. , Kreitman, R.J. , Hassan, R. , Xiang, L. , Pastan, I. , 2012. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl. Acad. Sci. U S A 109, 11782–11787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi, A. , Bursomanno, S. , Lopardo, T. , Traini, R. , Colombatti, M. , Ippoliti, R. , Flavell, D.J. , Flavell, S.U. , Ceriotti, A. , Fabbrini, M.S. , 2010. Pichia pastoris as a host for secretion of toxic saporin chimeras. FASEB J. 24, 253–265. [DOI] [PubMed] [Google Scholar]
- Lombardi, A. , Marshall, R.S. , Savino, C. , Fabbrini, M.S. , Ceriotti, A. , 2010. Type I ribosome-inactivating proteins from Saponaria officinalis. In Lord J.M., Hartley M.R.(Eds.), Toxic Plant Proteins, Plant Cell Monographs Springer Verlag; Heidelberg: 55–78. [Google Scholar]
- Madhumathi, J. , Verma, R.S. , 2012. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 15, 300–309. [DOI] [PubMed] [Google Scholar]
- Oh, S. , Todhunter, D.A. , Panoskaltsis-Mortari, A. , Buchsbaum, D.J. , Toma, S. , Vallera, D.A. , 2012. A deimmunized bispecific ligand-directed toxin that shows an impressive anti-pancreatic cancer effect in a systemic nude mouse orthotopic model. Pancreas 41, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo, A. , Stocker, M. , Kampmeier, F. , Melmer, G. , Fischer, R. , Thepen, T. , Barth, S. , 2012. In vivo imaging of immunotoxin treatment using Katushka-transfected A-431 cells in a murine xenograft tumour model. Cancer Immunol. Immunother. 61, 1617–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polito, L. , Bortolotti, M. , Pedrazzi, M. , Bolognesi, A. , 2011. Immunotoxins and other conjugates containing saporin-s6 for cancer therapy. Toxins (Basel) 3, 697–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psarras, K. , Ueda, M. , Tanabe, M. , Kitajima, M. , Aiso, S. , Komatsu, S. , Seno, M. , 2000. Targeting activated lymphocytes with an entirely human immunotoxin analogue: human pancreatic RNase1-human IL-2 fusion. Cytokine 12, 786–790. [DOI] [PubMed] [Google Scholar]
- Puri, M. , Kaur, I. , Perugini, M.A. , Gupta, R.C. , 2012. Ribosome-inactivating proteins: current status and biomedical applications. Drug Discov. Today 17, 774–783. [DOI] [PubMed] [Google Scholar]
- Russell, W.M.S. , Burch, R.L. , 1959. The Principles of Humane Experimental Technique Methuen; London: [Google Scholar]
- Selbo, P.K. , Weyergang, A. , Eng, M.S. , Bostad, M. , Maelandsmo, G.M. , Hogset, A. , Berg, K. , 2012. Strongly amphiphilic photosensitizers are not substrates of the cancer stem cell marker ABCG2 and provides specific and efficient light-triggered drug delivery of an EGFR-targeted cytotoxic drug. J. Control Release 159, 197–203. [DOI] [PubMed] [Google Scholar]
- Shaik, M.S. , Ikediobi, O. , Turnage, V.D. , McSween, J. , Kanikkannan, N. , Singh, M. , 2001. Long-circulating monensin nanoparticles for the potentiation of immunotoxin and anticancer drugs. J. Pharm. Pharmacol. 53, 617–627. [DOI] [PubMed] [Google Scholar]
- Stahnke, B. , Thepen, T. , Stocker, M. , Rosinke, R. , Jost, E. , Fischer, R. , Tur, M.K. , Barth, S. , 2008. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 7, 2924–2932. [DOI] [PubMed] [Google Scholar]
- Thakur, M. , Melzig, M.F. , Fuchs, H. , Weng, A. , 2011. Chemistry and pharmacology of saponins: special focus on cytotoxic properties. Bot.: Targets Ther. 2011, 19–29. [Google Scholar]
- Thakur, M. , Weng, A. , Bachran, D. , Riese, S.B. , Bottger, S. , Melzig, M.F. , Fuchs, H. , 2011. Electrophoretic isolation of saponin fractions from Saponinum album and their evaluation in synergistically enhancing the receptor-specific cytotoxicity of targeted toxins. Electrophoresis 32, 3085–3089. [DOI] [PubMed] [Google Scholar]
- Thakur, M. , Mergel, K. , Weng, A. , Frech, S. , Gilabert-Oriol, R. , Bachran, D. , Melzig, M.F. , Fuchs, H. , 2012. Real time monitoring of the cell viability during treatment with tumor-targeted toxins and saponins using impedance measurement. Biosens. Bioelectron. [DOI] [PubMed] [Google Scholar]
- Vallera, D.A. , Oh, S. , Chen, H. , Shu, Y. , Frankel, A.E. , 2010. Bioengineering a unique deimmunized bispecific targeted toxin that simultaneously recognizes human CD22 and CD19 receptors in a mouse model of B-cell metastases. Mol. Cancer Ther. 9, 1872–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldron, N.N. , Oh, S. , Vallera, D.A. , 2012. Bispecific targeting of EGFR and uPAR in a mouse model of head and neck squamous cell carcinoma. Oral Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng, A. , Jenett-Siems, K. , Schmieder, P. , Bachran, D. , Bachran, C. , Gorick, C. , Thakur, M. , Fuchs, H. , Melzig, M.F. , 2010. A convenient method for saponin isolation in tumour therapy. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878, 713–718. [DOI] [PubMed] [Google Scholar]
- Weng, A. , Thakur, M. , Beceren-Braun, F. , Bachran, D. , Bachran, C. , Riese, S.B. , Jenett-Siems, K. , Gilabert-Oriol, R. , Melzig, M.F. , Fuchs, H. , 2012. The toxin component of targeted anti-tumor toxins determines their efficacy increase by saponins. Mol. Oncol. 6, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, L. , Rosen, N. , Arteaga, C. , 2011. Targeted cancer therapies. Chin. J. Cancer 30, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, X. , Lin, X. , Manorek, G. , Howell, S.B. , 2012. Challenges associated with the targeted delivery of gelonin to claudin-expressing cancer cells with the use of activatable cell penetrating peptides to enhance potency. BMC Cancer 11, 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data