

Abstract
SSX cancer/testis antigens are frequently expressed in melanoma tumors and represent attractive targets for immunotherapy, but their role in melanoma tumorigenesis has remained elusive. Here, we investigated the cellular effects of SSX2 expression. In A375 melanoma cells, SSX2 expression resulted in an increased DNA content and enlargement of cell nuclei, suggestive of replication aberrations. The cells further displayed signs of DNA damage and genomic instability, associated with p53‐mediated G1 cell cycle arrest and a late apoptotic response. These results suggest a model wherein SSX2‐mediated replication stress translates into mitotic defects and genomic instability. Arrest of cell growth and induction of DNA double‐strand breaks was also observed in MCF7 breast cancer cells in response to SSX2 expression. Additionally, MCF7 cells with ectopic SSX2 expression demonstrated typical signs of senescence (i.e. an irregular and enlarged cell shape, enhanced β‐galactosidase activity and DNA double‐strand breaks). Since replication defects, DNA damage and senescence are interconnected and well‐documented effects of oncogene expression, we tested the oncogenic potential of SSX2. Importantly, knockdown of SSX2 expression in melanoma cell lines demonstrated that SSX2 supports the growth of melanoma cells. Our results reveal two important phenotypes of ectopic SSX2 expression that may drive/support tumorigenesis: First, immediate induction of genomic instability, and second, long‐term support of tumor cell growth.
Keywords: SSX2, Cancer/testis antigen, Oncogene, Senescence, Genomic instability
Highlights
Ectopic expression of tumor antigen SSX2 induces DNA damage in cell models.
Ectopic expression of SSX2 is associated with activation of the p53‐p21 pathway and cell cycle arrest.
The outcome of ectopic SSX2 expression can be either genomic instability or senescence.
Endogenous SSX2 supports cell growth in melanoma cell lines.
Our results suggest oncogenic functions of SSX2.
Abbreviations
- CT antigen
cancer/testis antigen
- DOX
doxycycline
- DNA DSB
DNA double‐strand break
- ICC
immunocytochemistry
- IHC
immunohistochemistry
- OIS
oncogene‐induced senescence
- PRC1
Polycomb Repressive Complex 1
- SA‐β‐gal
senescence‐associated β‐galactosidase
- SSX
Synovial Sarcoma X chromosome breakpoint
1. Introduction
Cancer/testis (CT) antigens represent a group of germ cell‐specific proteins whose expression is reactivated in tumor cells (Simpson et al., 2005). Because of the immune‐privileged nature of germ cells, these proteins often elicit humoral and cellular immune responses in cancer patients (Gjerstorff et al., 2010). The unique expression pattern and immunogenic properties of CT antigens make them ideal targets for immunotherapy of multiple types of human cancer, particularly melanoma, which exhibits coordinated expression of CT antigens at high levels (Barrow et al., 2006; Sahin et al., 1998; Zendman et al., 2001). Despite their vast therapeutic potential, little is known about the role of CT antigens in melanoma tumorigenesis, and elucidation of the cellular functions of these proteins remains essential for their further development as therapeutic targets.
The family of Synovial Sarcoma X chromosome breakpoint (SSX) CT antigens was first identified as part of the chimeric SS18‐SSX fusion protein, which is generated by a translocation in more than 95% of synovial sarcomas and considered a main contributor to the development and progression of this disease (Inagaki et al., 2000). The SSX family comprises 10 highly homologous members frequently expressed in melanoma, but the biological significance of their presence in melanoma cells has not been evaluated. SSX proteins are localized in the nucleus and are believed to be transcriptional repressors. Two repressive domains are found in all SSX family members, a Krüppel‐associated box (KRAB) domain and an SSX repression domain, the latter being the most potent repressor in a GAL4‐luciferase reporter assay (Brett et al., 1997; Lim et al., 1998). SSX proteins may also have a role in regulation of gene expression through modulation of Polycomb‐mediated gene silencing, since SSX1 and SSX2 have been found to co‐localize with BMI1 and RING1B of the Polycomb Repressive Complex 1 (PRC1) in nuclear foci (Barco et al., 2009; Soulez et al., 1999), and the SSX2 part is essential for SYT‐SSX2‐mediated destabilization of BMI1 leading to reactivation of Polycomb target genes (Soulez et al., 1999).
To elucidate the role of SSX2 in tumorigenesis, we investigated the phenotypic and molecular changes associated with SSX2 expression in human melanoma and breast cancer cells and demonstrate that SSX2 possesses oncogenic potential, further highlighting the potential of this protein as a therapeutic target.
2. Materials and methods
2.1. Cell lines
A375 and MCF7 cells were obtained from American Type Culture Collection (ATCC; which uses short tandem repeat profiling for cell line authentication) and HEK293‐Flp‐In T‐REX cells were purchased from Invitrogen, Naerum, Denmark. All cell lines were kept at a low passage. A375 melanoma cells with tetracycline/doxycycline (TET/DOX)‐inducible expression of SSX2 (NM_175698) (A375‐TET‐SSX2), CRISP2 (A375‐TET‐CRISP2) and p21 (CDKN1A) (A375‐TET‐p21), and MCF7 cells with TET/DOX‐inducible SSX2 expression (MCF7‐TET‐SSX2), were generated using a modified Flp‐In system (Invitrogen). Briefly, A375 or MCF7 cells carrying a Flp recombinase recognition site were transfected with the pOG44 plasmid encoding the Flp recombinase and an expression vector with Flp recombinase recognition site carrying the genes of interest. Cells with stable integration of the expression cassette were selected with 300 μg/ml hygromycin. Three independent clones with similar growth rates were pooled to avoid undesired effects of clonal selection. Control A375 or MCF7 cells were generated by integration of an identical expression vector without an open reading frame in the same genomic site. Thus, comparing cells with TET/DOX‐induced expression of inserted genes to uninduced cells and TET/DOX‐treated isogenic control cells accounted for all effects contributed by the elements of the vector system and TET/DOX treatment. HEK293 cells (HEK293 Flp‐In T‐REX, HEK293) with TET/DOX‐inducible expression of SSX2 with an N‐terminal 3xFLAG‐tag (HEK293‐TET‐SSX2‐FLAG) were generated with the Flp‐In system (Invitrogen) according to the manufacturer's recommendations.
Melanoma cell lines were originally established by Alexei Kirkin (Kirkin et al., 1998) and kindly donated to our laboratory by Professor Mads Hald Andersen, Center for Cancer Immunotherapy (CCIT), Herlev Hospital, Denmark. Cells were grown in DMEM (A375, MCF7, HEK293) or RPMI (FM6, FM45, FM79) supplemented with 10% fetal bovine serum (FBS; Invitrogen), penicillin (100 U/ml) and streptomycin (100 mg/ml).
Thymidine‐mediated G1/S cell cycle arrest of A375 and MCF7 cells was carried out by adding 2.5 mM of thymidine to cells for 16 h. Cells were released by removing thymidine and adding 25 μM of 2′‐deoxycytidine. After 8 h, MCF7 cells were subjected to another round of thymidine arrest.
2.2. Lentiviral transfections
Lentiviral particles with SSX2‐specific shRNAs were purchased from Santa Cruz Biotech, Heidelberg, Germany (target sequences: 5′‐GUA UGA GGC UAU GAC UAA A‐3′ and 5′‐GUU AGC GUU UAC GUU GUA U‐3′). For lentiviral transfections, cells were seeded at a density of 20,000 cells/cm2 and the next day transduced (MOI = 1:5) in media with 5–8 μg/ml of polybrene. Media was changed after 16 h, and 48 h thereafter 0.1–0.4 μg/ml of puromycin was added to select stable transfectants.
2.3. Cell phenotype assays
For CellTiter blue viability assays (Promega, San Luis Obispo, CA, USA), cells were seeded in 96‐well plates at a density of 4.7 × 103 cells/cm2. After 24 h media was changed to include DOX in indicated concentrations. At indicated times, cells were stained with CellTiter‐Blue diluted in phenol red‐free MEM (Invitrogen) (1:5) and measured at 560/590 nm. For clonogenic assays, cells were seeded in 6‐ or 12‐well plates (50–100 cells/cm2) in standard media with different concentrations of DOX (where relevant). After 10–14 days, cells were stained with 0.5% crystal violet in 25% methanol. For cell cycle analysis, cells were harvested by trypsinization, fixed in 84% ethanol and incubated at 37 °C for 30 min in fresh propidium iodide/RNase solution (0.05 mg/ml propidium iodide, 3.8 mM sodium citrate, 10 mM Tris–HCl (pH 7.5), 5 mM MgCl2, 0.1 mg/ml RNaseA) at a density of 1 × 106 cells/ml propidium iodide/RNase solution. To arrest cells in mitosis before cell cycle analysis, 0.25 μM paclitaxel (Sigma–Aldrich, Brondby, Denmark) diluted in ethanol or 200 nM nocodazole (Sigma–Aldrich) in dimethyl sulfoxide was added. Staining of apoptotic cells was carried out with the Vybrant Apoptosis Assay Kit 2 (V13241; Invitrogen) essentially as specified by the manufacturer. The level of SA‐β‐galactosidase activity in cells was measured using the Senescence Detection Kit according to the manufacturers recommendations (BioVision, Milpitas, CA, USA).
2.4. Tumorigenicity in SCID mice
Female immunodeficient CB17 SCID mice (Taconic, Ry, Denmark) were maintained in pathogen‐free conditions with access to water and food ad libitum. Groups of seven mice were injected subcutaneously in the flanks with either 106 A375‐TET‐SSX2 cells or 106 control A375 cells in DMEM mixed with Matrigel in a total volume of 50 μl. DOX was administered to both groups of mice in drinking water at a concentration of 200 μg/ml. All animal experiments were performed at the Biomedical Laboratory, University of Southern Denmark and approved by The Experimental Animal Committee, The Danish Ministry of Justice.
2.5. Quantitative RT‐PCR
Total RNA was purified using Trizol (Invitrogen) for tumor samples or the RNeasy® Mini Kit from Qiagen, Copenhagen, Denmark, for cell culture samples. A RevertAid Premium Reverse Transcriptase kit (Fermentas, Slangerup, Denmark) was used for cDNA synthesis. Relative quantification of gene expression was performed with SYBR green PCR Mastermix (Applied Biosystems, Naerum, Denmark). Primers for SSX2, GAPDH and PUM1 were purchased from Qiagen. The “TissueScan RT Melanoma panel I” with cDNA from three healthy and 40 melanoma samples was purchased from OriGene, Rockville, MD, USA.
2.6. Immunostaining
Methods for Western blotting, immunocyto‐ and immunohistochemistry were described previously (Gjerstorff et al., 2006; Greve et al., 2014). The use of clinical specimens was approved by the ethical committee of Funen and Vejle County (VF20050069).
2.7. Antibodies
Anti‐panSSX (1:50 for Western blotting, sc‐28697), anti‐SSX2/SSX3 (mAb 1A4, which has been demonstrated to recognize SSX2 and SSX3, but not SSX1, SSX4 or SSX5 (Smith et al., 2011); 1:700 for immunohistochemistry (IHC) and 1:200 for immunocytochemistry (ICC);, Novus Biologicals, Cambridge, UK), anti‐cyclin A (1:200 for Western blotting, sc‐596, Santa Cruz), anti‐SSX2‐4 (mAb E3AS (dos Santos et al., 2000), 1:3000 for Western blotting), anti‐cyclin B1 (1:2000 for Western blotting, sc‐245, Santa Cruz), anti‐cyclin D1 (1:2000 for Western blotting, sc‐718, Santa Cruz), anti‐cyclin E (1:200 for Western blotting, sc‐247, Santa Cruz), anti‐p21 (1:100 for Western blotting, clone SX118, Dako, Glostrup, Denmark), anti‐p53 (1:1000 for Western blotting, MCA1703, AbD Serotec, Dusseldorf, Germany), anti‐Ki67 (1:300 for ICC, ab15580, Abcam, Oxford, UK), anti‐α‐/β‐tubulin (1:50 for ICC, #2148, Cell Signaling, Danvers, USA), anti‐γ‐H2AX (1:2000 for ICC, ab22551, Abcam), anti‐γ‐H2AX 20E3 (1:50 for fluorescence IHC; #9718, Cell Signaling), anti‐γ‐H2AX (1:50 for flow cytometry; clone 20E3, Cell Signaling), anti‐CDK1 (predicted to also react with CDK2/CDK3) (1:1000 for Western blotting; #9112, Cell Signaling), anti‐P‐CDK1‐/CDK2/CDK5‐Y15 (predicted to also react with P‐CDK3‐Y15) (1:1000 for Western blotting; #9111, Cell Signaling), anti‐P‐CHK1‐S317 (1:1000 for Western blotting; #12302, Cell Signaling), anti‐P‐CHK1‐S345 (1:1000 for Western blotting; #2341, Cell Signaling).
2.8. Confocal microscopy
An Olympus FluoView FV1000MPE confocal laser scanning microscope fitted with an Olympus objective (XLUMPLFL20xW, 20×, NA:0.95) was used at room temperature. Images were acquired sequentially with 405 nm, 488 nm and 559 nm lasers and the corresponding 400–450 nm, 477.5–522.5 nm and 575–675 nm band‐pass filters, respectively. Image acquisition of z‐stacks covering the entire cell layer depth (1 μm steps, 3× zoom) and 3D conversion was carried out with FluoView (FV‐ASW 2.1) software (Olympus, Hamburg, Germany). Three random ‘stacks’ per specimen were acquired and the number of Ki67‐positive cells and γH2AX foci per cell quantified using ImageJ software.
2.9. p53 reporter assay
Melanoma cell lines were stably transduced with lentiviral particles carrying the pGreenfire‐p53 GFP reporter plasmid (System Biosciences, Mountain View, CA, USA). In brief, HEK293T cells were co‐transfected with pGreenfire‐p53 and packaging plasmids pVSVG, pRSV‐Rev and pMDL g/p RRE (plasmids were kindly provided by the Tronolab through Addgene, Cambridge, MA, USA) and, after 72 h, supernatant containing virus was harvested and transferred to target cells. Seventy‐two hours after transduction, cells were selected with puromycin for 7 days. The GFP signal in cells treated for 16 h with 50 μM of etoposide and untreated cells was measured on an LSRII flow cytometer (BD, Albertslund, Denmark).
2.10. Statistics
The non‐parametric Mann Whitney test was chosen in the experiments involving the number of γH2AX foci where normality cannot be assumed. Student's t‐test was used in all other experiments.
2.11. EdU/BrdU flow cytometry assays
A375‐TET‐SSX2 or MCF7‐TET‐SSX2 cells were treated with 50 ng/ml DOX starting 24 h before release from G1/S arrest, or left untreated. Upon release, cells were pulsed with EdU or BrdU for 60 min and harvested at indicated times. EdU or BrdU staining was carried out according to the recommendations of the manufacturers (EdU, EdU flow cytometry assay kit, Life Technologies; BrdU, APC BrdU flow kit, BD Biosciences).
2.12. Cytometric assessment of γ‐H2AX
Cells were harvested, fixed in ice‐cold 70% ethanol for at least 2 h at −20 °C and permeabilized with PBS, 1% BSA, 0.2% Triton X‐100 (T‐BSA‐PBS). After two 5‐min washes in T‐BSA‐PBS, cells were incubated with Alexa Flour 488‐conjugated anti‐γ‐H2AX in T‐BSA‐PBS overnight at 4 °C in the dark. The cells were then washed and stained with 0.5 ml DNA staining solution [PBS, 0.5 μl FxCycle™ Far Red Stain (F10348, Life Technologies), 50 μg RNase (R4642, Sigma)] for 30 min in the dark. Flow cytometry analysis was performed on an LSRII flow cytometer (BD Biosciences). The procedure was modified from (Huang and Darzynkiewicz, 2006).
3. Results
3.1. SSX2 expression reduces cell viability and clonogenicity
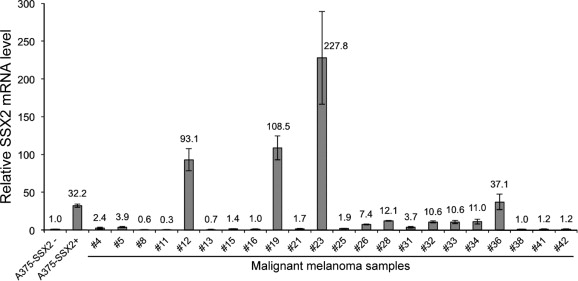
To investigate the role of SSX2 in cancer cells, we generated an A375 melanoma cell line with TET/DOX‐inducible SSX2 expression (A375‐TET‐SSX2) (Figure 1A) and an isogenic control cell line carrying an identical expression vector, but without the SSX2 open reading frame (termed A375). To validate the in vivo relevance of this cell model, we compared the DOX‐induced SSX2 expression level (A375‐TET‐SSX2 +DOX) with the level observed in tumors from melanoma patients (Supplementary Figure 1). SSX‐expression has previously been demonstrated in about 30% of melanomas (dos Santos et al., 2000). mRNA from 40 randomly selected melanomas were evaluated for SSX2 expression by quantitative PCR, and 4 of these (10%) exhibited a higher expression level of SSX2 than DOX‐treated (50 ng/ml) A375‐TET‐SSX2 cells, demonstrating that our experimental conditions reflected the clinical cancer setting.
Figure 1.
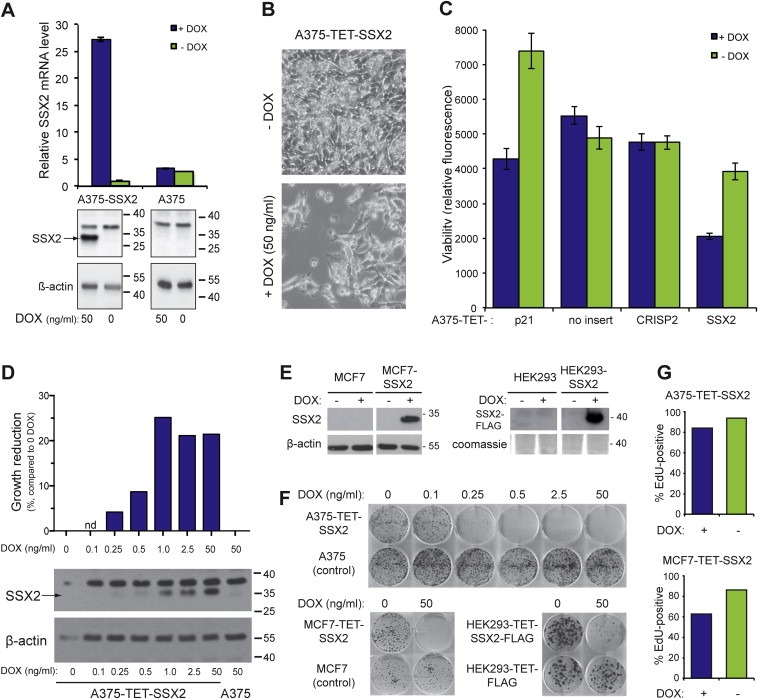
Ectopic SSX2 expression inhibits cell growth. (A) Quantitative PCR and Western blot analysis of SSX2 expression in A375‐TET‐SSX2 melanoma cells with DOX‐inducible SSX2 expression (A375‐SSX2) and control A375 cells. (B) Cell density of A375‐TET‐SSX2 cells after 96 h with or without DOX‐induction of SSX2. (C) CellTiter‐Blue assay examining the number of living (metabolic active) A375 cells after 96 h with DOX‐induced expression of SSX2, the cancer/testis antigen CRISP2 (negative control) or p21 (positive control). (D) CellTiter‐Blue viability assay (top panel, bars depict growth reduction in % compared to sample without DOX) and Western blot analysis (lower panel) of SSX2 protein levels in cells induced with increasing amounts of DOX for 48 h. (E) Western blot analysis of SSX2 expression in MCF7 and HEK293 cells with TET/DOX‐inducible SSX2 expression (MCF7‐SSX2 and HEK293‐SSX2) and control MCF7 and HEK293 cells. (F) Clonogenicity of A375‐TET‐SSX2, MCF7‐TET‐SSX2 and HEK293‐TET‐SSX2‐FLAG cells with or without SSX2 expression, relative to DOX treated controls (no SSX2 insert). Cells were stained with crystal violet after 10–14 days of growth. (G) The percentage of cycling cells was measured by EdU incorporation in A375‐TET‐SSX2 and MCF7‐TET‐SSX2 cells with or without SSX2 expression. Cells were DOX‐treated for 24 h for induction of SSX2 expression and pulsed with EdU for additional 24 h. Average of duplicates are shown. The DOX concentration used in A–C, E and G was 50 ng/ml.
Surprisingly, viability assays and growth curves showed that the SSX2‐expressing A375 cells stopped expansion after 48 h and exhibited a 47% (p < 10−5) reduction in cell viability (Figure 1B, C and Supplementary Figure 2). Using increasing amounts of DOX (0.1–50 ng/ml), we gradually increased the level of SSX2 protein induced, causing a dose‐dependent reduction in viability and clonogenicity (Figure 1D–F). Using similar TET/DOX‐regulated isogenic expression systems, SSX2 was also demonstrated to reduce the clonogenicity of MCF7 breast cancer cells (MCF‐TET‐SSX2) and HEK293 human embryonic kidney cells (HEK293‐TET‐SSX2) (Figure 1E, F), establishing that the effect was not restricted to cells of the melanocyte lineage. The reduced growth of SSX2‐expressing A375 melanoma cells was confirmed by reduction in EdU incorporation (Figure 1G) and in vivo growth studies examining the tumor‐forming capacity of A375‐TET‐SSX2 cells in immunodeficient mice (Supplementary Figure 3). Ten of 14 mice injected with control A375 melanoma cells developed large tumors, while none of the mice injected with A375‐TET‐SSX2 cells developed tumors over a period of 21 days.
3.2. SSX2 expression induces G1 checkpoint arrest in A375 cells
The growth‐inhibitory effects of SSX2 were most dramatic in A375‐TET‐SSX2 cells, and the nature of this response was investigated further. Analysis of cell cycle phase distribution after paclitaxel (Figure 2A) or nocodazole (Supplementary Figure 4) treatment to arrest cells in M‐phase suggested that A375‐TET‐SSX2 cells, after 24–48 h of SSX2 expression, exhibited either a partial G1 arrest or a bypass of the induced M‐phase arrest. The latter was ruled out by EdU incorporation in combination with paclitaxel‐induced M‐phase arrest and measurement of cells reentering G1 upon release, as SSX2 did not increase the number of cells reentering G1 (Figure 2B). To evaluate a possible G1 arrest, we synchronized cells in G1/S and EdU labeled them upon release and assessed cell cycle kinetics in cells with and without SSX2. This demonstrated that cells passed through the S‐ and M‐phases and reentered G1 at a similar rate independent of SSX2 expression. However, SSX2 induced a three‐fold increase in the number of cells stuck after reentering G1, confirming a partial G1 arrest (Figure 2C).
Figure 2.
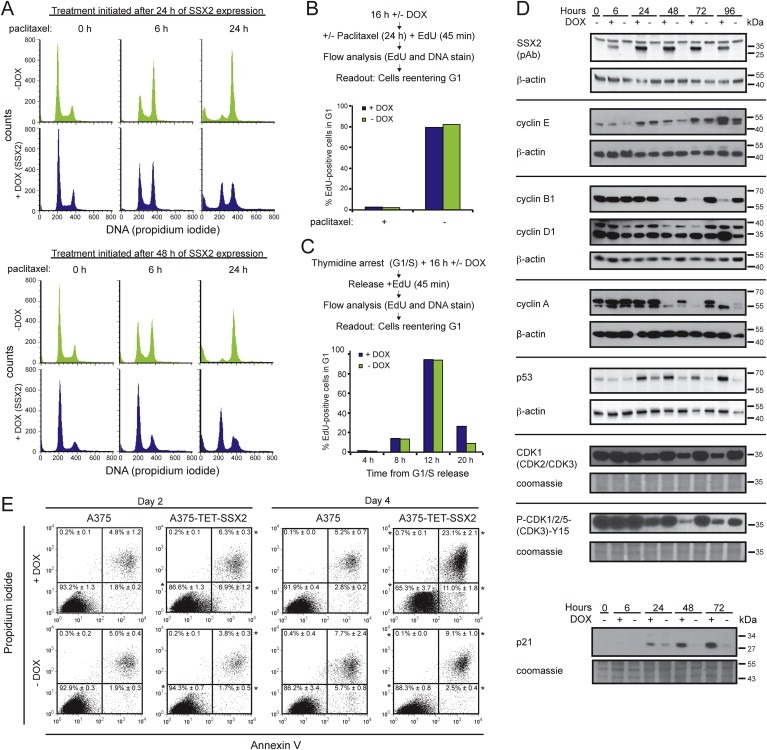
SSX2 induces G1 checkpoint arrest and apoptosis in A375 melanoma cells. (A) Cell cycle analysis of A375‐TET‐SSX2 cells after paclitaxel treatment. Paclitaxel was added either 24 or 48 h after DOX induction of SSX2 and cells harvested after 0, 6 and 24 h of paclitaxel treatment. (B) Cell cycle kinetics. A375‐TET‐SSX2 cells with or without DOX‐induced SSX2 expression were treated with paclitaxel for spindle checkpoint activation and the number of cells reentering G1 was quantified by flow cytometry after release. Average of duplicates are shown. (C) Cell cycle kinetics. A375‐TET‐SSX2 cells, with or without DOX‐induced SSX2 expression, released from thymidine induced G1/S arrest, were EdU‐labeled and cells reentering G1 was quantified by flow cytometry. Average of duplicates are shown. (D) Western blot analysis of relevant cell cycle regulators in A375‐TET‐SSX2 cells with or without DOX‐induced SSX2 expression. (E) Apoptosis and dead cell analysis with Annexin V and propidium iodide. Each plot shows accumulated data from 3 independent samples, % ± standard deviation. *+DOX significantly differently from −DOX (p ≤ 0.01). The DOX concentration used in A–E was 50 ng/ml.
Quantification of cell cycle regulators by Western blotting supported the above results (Figure 2D), as SSX2 expressing cells exhibited reduced levels of cyclin A and B, while accumulating cyclin E and D. Furthermore, SSX2 induced a clear reduction in the total level of cyclin dependent kinases (CDK1, CDK2, CDK3) and, importantly, also the active Y15‐phosphorylated form, supporting cell cycle arrest. A key regulator of the G1/S checkpoint is the p53‐p21‐RB pathway and, in agreement with this, we found increased levels of both p53 and p21. Annexin V and propidium iodide staining demonstrated an increased percentage of apoptotic and dead cells after 48–96 h of SSX2 expression (Figure 2E), which may be a direct result of p53 activation. In total, these results clearly indicate that SSX2 expression induces a p53‐p21–mediated G1 arrest in A375 cells, followed by an apoptotic response.
3.3. SSX2 expression causes replication defects and genomic instability in A375 melanoma cells
Cell cycle analysis of A375‐TET‐SSX2 cells after 4–6 days of SSX2 expression revealed a broadening and a right‐shift of both the G1 and G2/M peaks (Figure 3A), representing an increase in DNA content, which suggested induction of genomic instability in the cells. Therefore, we evaluated the level of DNA damage (DNA double‐strand breaks (DSBs), measured as the number of γ‐H2AX foci) in A375‐TET‐SSX2 cells with and without induced SSX2 expression (Figure 3B). Ki67 staining was included to distinguish proliferating from arrested cells. Indeed, we observed an overall increase in γ‐H2AX foci after 48 h of SSX2 induction, with γ‐H2AX foci present in 78% of the A375‐TET‐SSX2 +DOX cells (2.48 foci/cell) vs. 42% in A375‐TET‐SSX2 cells (0.52 foci/cell) (Figure 3C, D). After 72 h, the number of cells with a high amount of foci (>8) had increased considerably from 2 to 12% of A375‐TET‐SSX2 +DOX cells compared to an increase from 2 to 3% in A375‐TET‐SSX2 cells without induction. Ki67 staining confirmed the decreased number of proliferating SSX2‐expressing cells seen in the other assays (Figure 3E) and also showed an inverse correlation between γ‐H2AX and Ki67 staining, suggesting that DNA damage was causative for decreased proliferation (Figure 3B, arrows). The frequency of Ki67‐positive cells was also reduced in A375‐TET‐SSX2 control cells at day 3 compared to day 2, but this was due to overgrowth of these cells. DOX treatment alone did not affect Ki67 status or γ‐H2AX staining in A375 control cells (Supplementary Figure 5).
Figure 3.
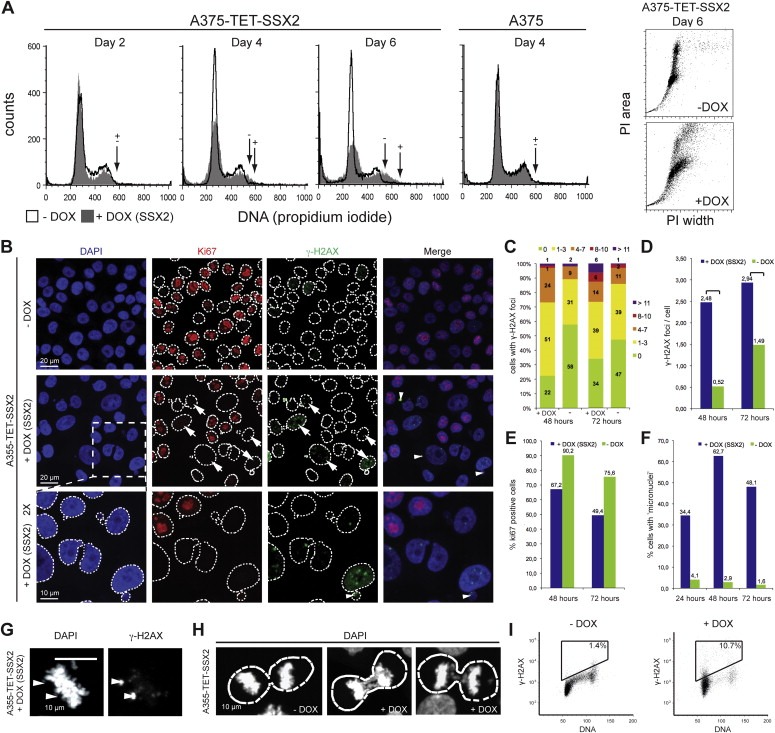
SSX2 causes genomic instability in A375 melanoma cells. (A) Cell cycle analysis showing increased DNA content in A375‐TET‐SSX2 cells with SSX2 expression. Arrows indicate the upper limit of the G2 populations. Dot plot of DNA signal width vs. area for A375‐TET‐SSX2 cells to test for cell aggregates. (B–F) A375‐TET‐SSX2 cells were grown for 48–72 h with or without 50 ng/ml DOX for SSX2 expression, fixed and stained with DAPI for nuclei visualization (blue), the proliferation marker Ki67 (Alexa‐568, red) and γ‐H2AX for identification of DNA double‐strand breaks (FITC, green). (B) Representative pictures. (C–F) Quantification of confocal images from 3 individual frames (day 2, n = 197; day 3, n = 206): (C) γ‐H2AX foci/cell were quantified in ImageJ and each cell assigned to groups with 0, 1–3, 4–7, 8–10 and >10 foci/cell, respectively. (D) Average number of γ‐H2AX foci/cell. Mann Whitney test: **, p = 0.0038; ****p < 0.0001. (E) Percentage of Ki67‐positive cells. (F) Percentage of cells with micronuclei. (G) SSX2‐expressing cell with two γ‐H2AX‐positive chromosome fragments. (H) SSX2‐expressing cells with anaphase bridges and an SXX2‐negative cell without anaphase bridges. Dotted lines mark the boundaries of the nuclei. (I) Flow cytometric analysis of γ‐H2AX/DNA stained A375‐TET‐SSX2 cells with or without 48 h of DOX‐induced SSX2 expression. The DOX concentration used in A–I was 50 ng/ml. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Interestingly, many SSX2‐expressing A375‐TET‐SSX2 cells had enlarged nuclei (Figure 3B), which can be a consequence of replication defects and inability to complete DNA synthesis (Shimizu et al., 2013; Zhu et al., 2004), thus indicating replication defects as a result of SSX2 expression. Therefore, we investigated the level of CHK1 phosphorylation at Ser317 and Ser345, which is mediated by the ATR kinase in response to replication defects/stress (Ward et al., 2004; Zhao and Piwnica‐Worms, 2001), but found no evidence of increased phosphorylation in association with SSX2 expression (Supplementary Figure 6). A high level of background CHK1 phosphorylation on Ser345 was observed in A375 cells, suggesting that this pathway was partly perturbed.
Notably, SSX2 expression resulted in a dramatic increase in the formation of micronuclei and nuclear blebs (Figure 3B, F), which are signs of ongoing genomic instability and can be caused by, for instance, replication and/or mitotic defects (Xu et al., 2011; Yasui et al., 2010). Most of the observed micronuclei showed DNA DSBs (Figure 3B, arrowheads), as did some chromosome fragments in metaphase cells (Figure 3G, arrowheads). Micronuclei comprising DNA DSBs have specifically been associated with replication stress (Xu et al., 2011). Anaphase bridges were also observed in A375‐TET‐SSX2 cells with SSX2 expression (Figure 3H), but the mitotic spindle apparatus appeared normal (Supplementary Figure 7). Cell cycle analysis in combination with γ‐H2AX staining demonstrated DNA DSBs in cells in all phases of the cell cycle, however, the majority of γ‐H2AX‐positive cells were in G1 after 48 h of SSX2 expression (Figure 3I).
In conclusion, our results suggest that SSX2 causes replication defects in A375 melanoma cells leading to anaphase bridges, genomic instability and as a result a p53‐p21‐mediated G1/S checkpoint arrest.
3.4. SSX2 expression in melanoma cells is independent of p53 status
Induction of p53 and p21 in A375 cells in response to SSX2 suggested that this pathway was essential for the observed G1 arrest and apoptotic response. In agreement, knockdown of TP53 expression in A375‐TET‐SSX2 cells using lentiviral delivery of siRNAs (Figure 4A) increased the clonogenicity of A375‐TET‐SSX2 cells with induced SSX2 expression (Figure 4B, C). This suggested that an impaired p53 response might be a prerequisite for tolerance of prolonged SSX2 expression in melanoma cells. To clarify this, we investigated the p53 response in a panel of SSX2‐positive and ‐negative melanoma cell lines using a p53 response element reporter assay (Figure 4D,E). The assay demonstrated a defective p53 response in one SSX2‐positive cell line (FM79), while two other SSX2‐positive cells lines (FM45 and FM6) showed functional p53 responses. This result suggested that SSX2 expression in melanoma cells is not dependent on p53 status, but may also reflect unidentified perturbations of p53 downstream effector molecules (e.g. p21 or RB) in SSX2‐positive cell lines, as exemplified by high viability and growth of FM6 cells with relatively high endogenous levels of p53 activity (Figure 4D,E).
Figure 4.
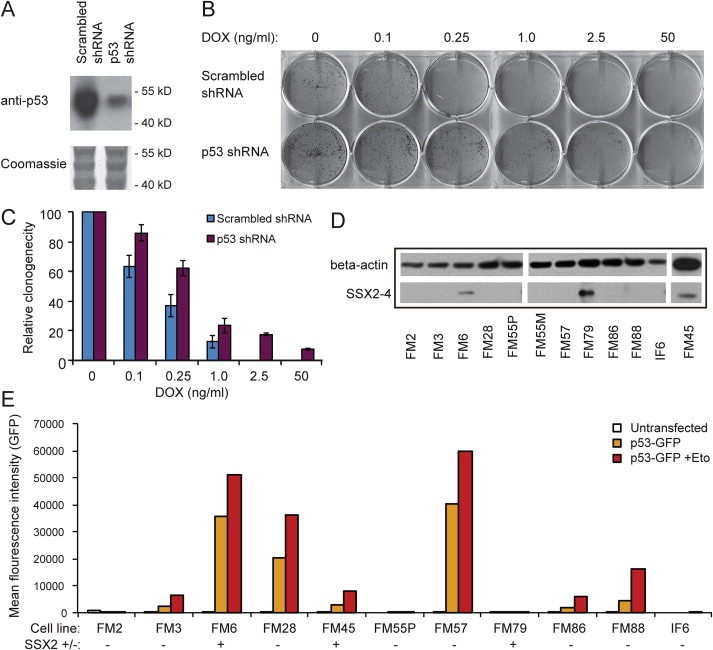
Knockdown of TP53 expression rescues A375 cells from SSX2‐mediated growth arrest. (A) A375‐TET‐SSX2 cells were stably transfected with TP53 or control scrambled shRNAs and p53 knockdown was confirmed by Western blotting. (B) The clonogenicity of A375‐TET‐SSX2 cells carrying the TP53 or scrambled shRNA was tested by seeding 100 cells/cm2 in media with increasing concentrations of DOX to induce increasing levels of SSX2 expression. Clones were stained with crystal violet after 10 days. (C) The relative clonogenicity (# of clones) of A375‐TET‐SSX2 cells carrying the TP53 or scrambled shRNA with different levels of SSX2 expression (different samples than in B) compared to cells without induction of SSX2 (error lines represent standard deviation of triplicates). (D) The SSX2 status of a panel of melanoma cell lines was assessed by Western blotting using anti‐SSX2‐4 (Mab E3AS). (E) Melanoma cell lines were stably transfected with a plasmid carrying a GFP reporter gene under the transcriptional control of a p53 response element. The basic and induced (with etoposide (50 μM); Eto) p53 activity was assayed by flow cytometry. SSX2 status (+/−) is indicated in brackets.
3.5. SSX2 expression induces a senescence‐like phenotype in MCF7 breast cancer cells
Similar to A375‐TET‐SSX2 cells, MCF7‐TET‐SSX2 cells exhibited reduced proliferation in response to SSX2 expression (Figure 1E,F), which was also corroborated by changes in cell cycle regulators (e.i. cyclin A and B1, p53, p21 and cyclin‐dependent kinases; Figure 5A) and an increased percentage of cells in G1 phase (Figure 5B). Long‐term growth (more than 7 days) of MCF7‐TET‐SSX2 cells with SSX2 expression induced a dramatic change in the morphology of the cells, which adapted an irregular and enlarged shape (Figure 5C). Similar changes were observed upon induction of SSX2 expression in HEK293‐TET‐SSX2 cells and surviving A375‐TET‐SSX2 cells, but to a lesser extent (Figure 5C). An irregular and enlarged cellular morphology is typical for cells that undergo a senescence response, which is an irreversible arrest of growth that can be induced by different types of stress, including oncogene expression (Rodier and Campisi, 2011). To further investigate if the SSX2‐expressing cells were senescent, we assayed their senescence‐associated β‐galactosidase (SA‐β‐gal) activity. MCF7‐TET‐SSX2 cultures with DOX‐induced expression of SSX2 exhibited increased SA‐β‐gal activity in a minority of cells after three days, and in the majority of cells after six days (Figure 5D). Increased SA‐β‐gal activity was also observed in some of the few surviving A375‐TET‐SSX2 cells, but not until six days of SSX2 expression. HEK293‐TET‐SSX2 cells exhibited very high background levels of SA‐β‐gal activity, and no difference was observed upon induction of SSX2 expression (Figure 5D). Immunocytochemical analysis of MCF7‐TET‐SSX2 cells 7 days after induction of SSX2 expression further substantiated their senescent phenotype, as they exhibited a clear reduction in expression of the proliferation marker Ki67 and presence of γ‐H2AX DSB foci (Figure 5E).
Figure 5.
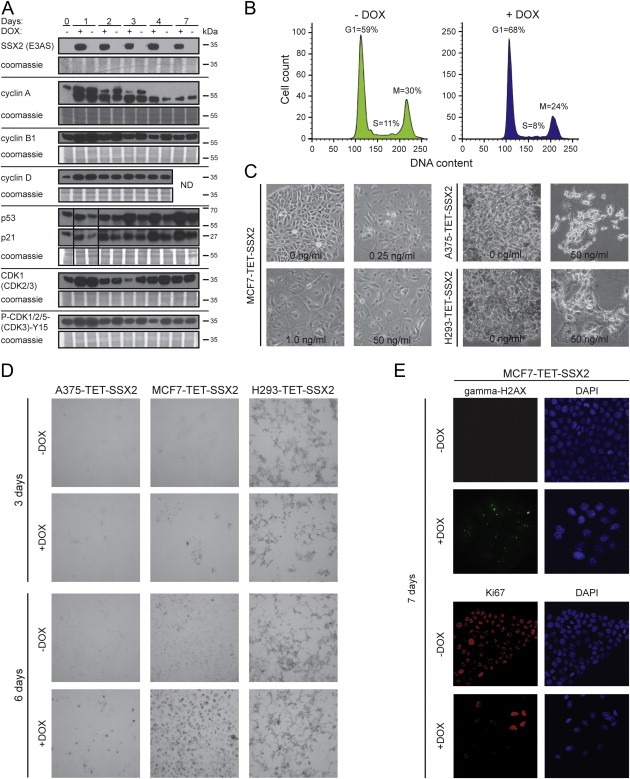
SSX2 induces a senescence‐like phenotype in MCF7 breast cancer cells. (A) Western blot analysis of relevant cell cycle regulators in MCF7‐TET‐SSX2 cells with or without DOX‐induced SSX2 expression. (B) Flow cytometric analysis of DNA content (Far Red Stain) shows an increase in the relative number of cells in G1 phase (48 h) in SSX2‐expressing MCF7‐TET‐SSX2 cells. (C) Long‐term growth of MCF7‐TET‐SSX2, A375‐TET‐SSX2 and HEK293‐TET‐SSX2 (H293‐TET‐SSX2) cells with SSX2 expression (10–14 days) induced an irregular and enlarged cells shape. DOX concentrations (induction of SSX2) are indicated. (D) Assay to detect senescence‐associated β‐galactosidase activity as represented by the ability of the cells to hydrolyze X‐gal and produce blue coloration. Cells were cultured for 3 or 6 days after induction of SSX2 expression before conducting the assay. (E) Immunocytochemical analysis of Ki67 and γ‐H2AX in MCF7 cells 7 days after induction of SSX2 expression. The DOX concentration used in A, B, D and E was 50 ng/ml.
3.6. Knockdown of SSX2 expression diminishes the growth of melanoma cells
As replication defects, genomic instability and senescence are common effects of acute oncogene expression we investigated the role of SSX2 in sustaining growth of melanoma cell lines in order to explore the oncogenic potential of SSX2. SSX2 expression was knocked down in three melanoma cell lines exhibiting endogenous SSX2 expression (i.e. FM6, FM45 and FM79) using stable lentiviral transduction (Figure 6A, B). This considerably reduced the cell number of FM45 and FM79 cells 3 and 6 days after seeding, while FM6 showed a slight increase in cell number (Figure 6C). This was corroborated by clonogenic assays, which demonstrated reduced clone‐forming capacity of FM45 and FM79 cells, and slightly increased clone‐forming capacity of FM6 cells, upon SSX2 knockdown (Figure 6D). To determine if the observed SSX2‐mediated decrease in clonogenicity of FM45 and FM79 cells was caused by an increase in the level of cell death, we performed an Annexin V apoptosis assay (Supplementary Figure 8) and found no SSX2‐related effects on the apoptotic rate of these melanoma cell lines. In total, these experiments demonstrated a context‐dependent capacity of SSX2 to support the growth of melanoma cells.
Figure 6.
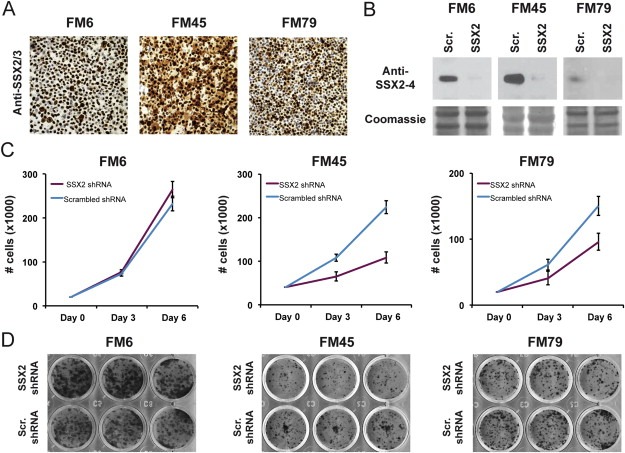
SSX2 supports the growth of melanoma cells. (A) Immunohistochemical staining of melanoma cell lines with endogenous SSX2/3 expression (FM6, FM45 and FM79) with anti‐SSX2/3 mAb 1A4. (B) The melanoma cell lines were transduced with lentivirus carrying SSX2‐specific shRNAs or scrambled control shRNA and knockdown verified by Western blotting (anti‐SSX2‐4, mAb E3AS). (C) Cells carrying SSX2‐specific or scrambled shRNAs was seeded at equal densities and the number of cells was counted after 3 and 6 days. Error bars show standard deviation of triplicates. (D) Clonogenicity of cells carrying SSX2‐specific or scrambled shRNA. Triplicates were stained with crystal violet after 10–14 days of growth.
4. Discussion
Despite the great potential of CT antigens as targets for cancer treatment, their role in melanoma, and cancer development in general, remains elusive. In this study, we investigated the phenotypic effects of SSX2 CT antigen expression and now provide the first clues to its role in tumor development.
Induced SSX2 expression was initially found to have a strong inhibitory effect on the growth of various cell types (i.e. melanoma, breast and kidney). This was most pronounced in A375 melanoma cells, where ectopic SSX2 expression was demonstrated to induce DNA DSBs and genomic instability, with clear signs of replication defects, including formation of large nuclei and increased DNA content. This led to activation of the p53‐p21 pathway and G1‐checkpoint arrest after 24–48 h, and subsequently apoptosis after 4 days. The effect of SSX2 expression in MCF7 cells was delayed compared to A375, and here SSX2 induced a senescent‐like phenotype (i.e. enlarged cell size, β‐galactosidase expression, loss of Ki67, DNA DSBs and activation of the p53‐p21 pathway) within 7 days. In contrast to A375, induction of p53 and p21 was not found until 3 days of SSX2 expression, and we detected no DNA DSB foci in MCF7 cells before 7 days of SSX2 induction (e.g. no γ‐H2AX foci after 48 h Supplementary Figure 9).
From these results, we conclude that ectopic expression of SSX2 induces DNA DSBs and activation of the p53‐p21 pathway in different types of cells, but the overall outcome varies (genomic instability vs. senescence). The likely explanation for the deviating phenotype of SSX2 expression in A375 cells compared to MCF7 cells is that the DNA damage associated with SSX2‐induced replication stress can result in genomic instability if DNA repair and/or checkpoint mechanisms are perturbed (e.g. in A375 cells). In contrast, the molecular landscape of, for example, MCF7 cells may allow maintenance of genomic integrity upon SSX2 expression and set the stage towards senescence instead of genomic instability, checkpoint activation and apoptosis. A higher ability to maintain genomic integrity in MCF7 cells, compared to A375 cells, was supported by a much higher background level of DNA DSBs in the latter.
Interestingly, it has recently been demonstrated that replication stress plays an essential role in causing genomic instability in tumors, through generation of structural chromosomal abnormalities that translate into chromosome missegregation (Burrell et al., 2013). This closely resembles the observed phenotype for SSX2 expression in A375 cells and suggests that de novo SSX2 expression in tumors may promote genomic instability through replication stress.
Within the last few years, it has become evident that cellular senescence can occur in response to oncogene expression in premalignant and malignant lesions as an important barrier to tumorigenesis (Bartek et al., 2007; Bartkova et al., 2006; Dhomen et al., 2009; Halazonetis et al., 2008; Serrano et al., 1997). Phenotypes associated with oncogene‐induced senescence (OIS) include increased DNA damage and genomic instability as a result of replication stress (Bartek et al., 2007; Di Micco et al., 2006; Halazonetis et al., 2008; Suram et al., 2012). Since the phenotypes associated with SSX2 resemble those of OIS, we investigated the effect of knockdown of endogenous SSX2 on the growth and clonogenicity of melanoma cells. This demonstrated that SSX2 supports the growth of FM45 and FM79 cells, further substantiating the role of SSX2 as an oncogene in melanoma.
SSX proteins are normally expressed at the spermatogonial stage of spermatogenesis (dos Santos et al., 2000). Spermatogonia have many similarities to cancer cells, including the ability to self‐renew and proliferate, and SSX2 may be involved in defining these phenotypes in both spermatogonia and cancer cells (Simpson et al., 2005). Interestingly, SSX2 knockdown slightly promoted the growth and clonogenicity of FM6 cells, suggesting that the role of SSX2 in supporting melanoma growth is context‐dependent and highlighting the need for further characterization of cooperating factors to enable selection of patients with SSX2‐positive tumors who might benefit from SSX2‐targeted therapy.
In conclusion, our results show that SSX2 may directly contribute to important phenotypes of cancer development (i.e. genomic instability and support of unlimited growth). This proposes SSX2 as a candidate oncogene and highlights its potential utility as a target for treatment of this disease, since the best targets are believed to support the cancer phenotypes. However, additional functional characterization is needed before the therapeutic value of SSX2 undergoes clinical trials.
Supporting information
The following are the supplementary data related to this article:
Supplementary Figure 1 Quantification of SSX2 mRNA levels in A375‐TET‐SSX2 cells with induced SSX2 expression (50 ng/ml DOX; A375‐SSX2+) and in a panel of 40 clinical malignant melanoma samples, relative to A375‐TET‐SSX2 without SSX2 expression (A375‐SSX2−). Results from three different experiments are shown as fold expression ± standard deviation relative to A375‐TET‐SSX2 controls. Tissue samples with SSX2 levels below detection limit were omitted from the histogram (22 tumor samples in total).
{kind=link}
Supplementary Figure 2 Representative growth curves for A375‐TET‐SSX2 and control A375 cells with or without 50 ng/ml DOX. DOX was added 24 h after seeding (time‐point: 0) and living (attached) cells counted daily (average of two wells shown).
{kind=link}
Supplementary Figure 3 SSX2 reduces outgrowth of A375 tumors in SCID mice. One million A375‐TET‐SSX2 cells or control A375 cells were injected subcutaneously into the flank of CB17 SCID immunodeficient mice to investigate tumor growth. All mice received DOX in drinking water to induce expression of SSX2 (in A375‐TET‐SSX2 cells only), and tumors were resected for evaluation after 22 days. All the excised tumors were examined for SSX2 expression using quantitative PCR and as expected A375‐TET‐SSX2 cells exhibited a 4.4–10.7‐fold higher expression of SSX2 than cells from control A375‐derived tumors. (a) Plot of tumor weight from two independent experiments. Bars depict mean ± SD. (b) Tumor weight and qPCR data of SSX2 expression(a) normalized to GAPDH in fold over average of parental reference samples (Ref). N/A, not available.
{kind=link}
Supplementary Figure 4 Nocodazole treatment of A375‐TET‐SSX2 cells show SSX2‐induced G1 arrest. A375‐TET‐SSX2 cells were grown for 3 days with or without DOX for induction of SSX2 expression, treated with nocodazole for 0, 6, 12 or 24 h and analyzed by PI/FACS to determine DNA content. The histograms depict the height of the PI signal (DNA) vs. counts.
{kind=link}
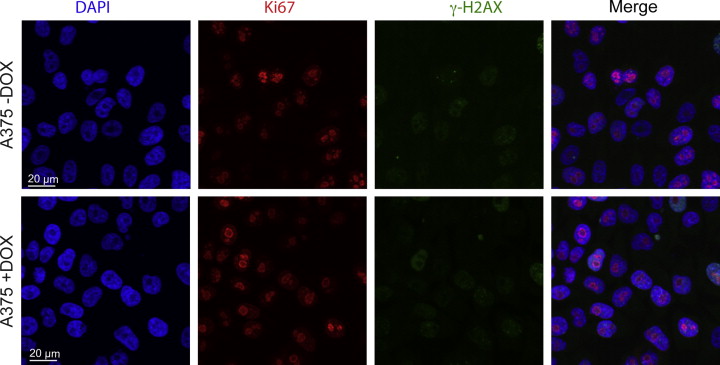
Supplementary Figure 5 DOX addition in itself does not affect γ‐H2AX levels or Ki67 proliferation status of A375 melanoma cells. Parental A375 control cells were grown with or without 50 ng/ml DOX for SSX2 expression for 3 days, fixed and stained with DAPI for nuclei visualization (blue), the proliferation marker Ki67 (Alexa‐568, red) and γ‐H2AX for localization of DNA damage foci (FITC, green).
{kind=link}
Supplementary Figure 6 Western blot analysis of CHK1 phosphorylation in A375‐TET‐SSX2 cells with or without DOX‐induced SSX2 expression (50 ng/ml). ETO = etoposide (50 μM for 4 h).
{kind=link}
Supplementary Figure 7 The mitotic spindle apparatus is normal in A375‐TET‐SSX2 cells with induced expression of SSX2. A375‐TET‐SSX2 cells were grown for 72 h with or without 50 ng/ml DOX for SSX2 expression, fixed and stained with DAPI for DNA visualization (blue), anti‐α‐/β‐tubulin (green, 1:50, #2148, Cell Signaling, Danvers, USA) and anti‐SSX2/3 mAb 1A4 (red).
{kind=link}
Supplementary Figure 8 Analysis of apoptosis in melanoma cells with and without SSX2 expression. The histograms show annexin V stainings measured by flow cytometry. Two replicates from two separate experiments (exp. 1 and 2) are shown. The dotted lines designate the control cells transfected with scrambled shRNA and the full lines cells transfected with SSX2 shRNA. Gating is indicated and the frequencies of annexin V positive cells are shown in the table below the histograms.
{kind=link}
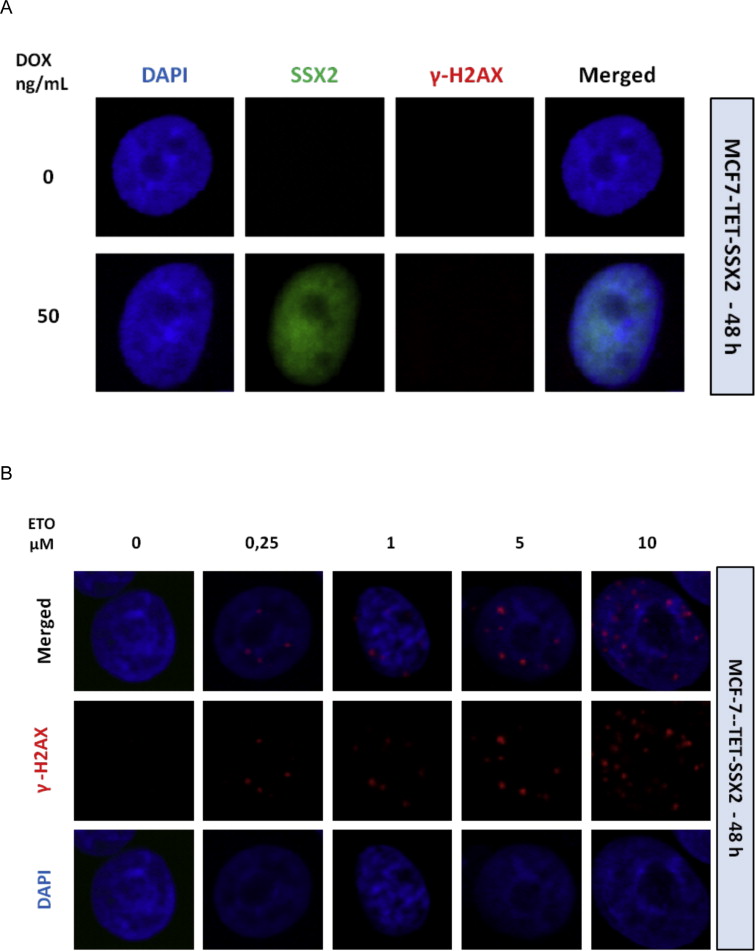
Supplementary Figure 9 SSX2 expression is not associated with increased DNA damage (γ‐H2AX foci) in MCF7 breast cancer cells after 48 h of SSX2 induction. A, SSX2 expression in MCF7‐TET‐SSX2 cells was induced with 50 ng/ml DOX and after 48 h the cells were fixed and stained for SSX2 and γ‐H2AX. B, Etoposide (ETO) treated cells were included as a positive control for γ‐H2AX foci formation.
{kind=link}
Acknowledgments
We acknowledge Thierry Boon, Jiri Lukas and Berthe M. Willumsen for critical reading of the manuscript and M. K. Occhipinti for editorial assistance. Confocal microscopy was performed at DaMBIC, (University of Southern Denmark), funded by the Danish Agency for Science Technology and Innovation. This study was supported by the Danish Research Council, The Lundbeck Foundation, the Danish Cancer Society, the Danish Cancer Research Foundation, Lundbeck Foundation Center of Excellence NanoCAN, the LeoPharma Research Foundation, Gangsted Foundation, Hørslev Foundation, the Region of Southern Denmark and Odense University Hospital Research Council.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.09.001.
Greve Katrine B.V., Lindgreen Jonas N., Terp Mikkel G., Pedersen Christina B., Schmidt Steffen, Mollenhauer Jan, Kristensen Stine B., Andersen Rikke S., Relster Mette M., Ditzel Henrik J., Gjerstorff Morten F., (2015), Ectopic expression of cancer/testis antigen SSX2 induces DNA damage and promotes genomic instability, Molecular Oncology, 9, doi: 10.1016/j.molonc.2014.09.001.
Contributor Information
Henrik J. Ditzel, Email: hditzel@health.sdu.dk
Morten F. Gjerstorff, Email: mgjerstorff@health.sdu.dk
References
- Barco, R. , Garcia, C.B. , Eid, J.E. , 2009. The synovial sarcoma-associated SYT-SSX2 oncogene antagonizes the Polycomb complex protein Bmi1. PloS One. 4, e5060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow, C. , Browning, J. , MacGregor, D. , Davis, I.D. , Sturrock, S. , Jungbluth, A.A. , Cebon, J. , 2006. Tumor antigen expression in melanoma varies according to antigen and stage. Clin. Cancer Res.: an official journal of the American Association for Cancer Research. 12, 764–771. [DOI] [PubMed] [Google Scholar]
- Bartek, J. , Bartkova, J. , Lukas, J. , 2007. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 26, 7773–7779. [DOI] [PubMed] [Google Scholar]
- Bartkova, J. , Rezaei, N. , Liontos, M. , Karakaidos, P. , Kletsas, D. , Issaeva, N. , Vassiliou, L.V. , Kolettas, E. , Niforou, K. , Zoumpourlis, V.C. , Takaoka, M. , Nakagawa, H. , Tort, F. , Fugger, K. , Johansson, F. , Sehested, M. , Andersen, C.L. , Dyrskjot, L. , Orntoft, T. , Lukas, J. , Kittas, C. , Helleday, T. , Halazonetis, T.D. , Bartek, J. , Gorgoulis, V.G. , 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 444, 633–637. [DOI] [PubMed] [Google Scholar]
- Brett, D. , Whitehouse, S. , Antonson, P. , Shipley, J. , Cooper, C. , Goodwin, G. , 1997. The SYT protein involved in the t(X;18) synovial sarcoma translocation is a transcriptional activator localised in nuclear bodies. Hum. Mol. Genet. 6, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Burrell, R.A. , McClelland, S.E. , Endesfelder, D. , Groth, P. , Weller, M.C. , Shaikh, N. , Domingo, E. , Kanu, N. , Dewhurst, S.M. , Gronroos, E. , Chew, S.K. , Rowan, A.J. , Schenk, A. , Sheffer, M. , Howell, M. , Kschischo, M. , Behrens, A. , Helleday, T. , Bartek, J. , Tomlinson, I.P. , Swanton, C. , 2013. Replication stress links structural and numerical cancer chromosomal instability. Nature. 494, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhomen, N. , Reis-Filho, J.S. , da Rocha Dias, S. , Hayward, R. , Savage, K. , Delmas, V. , Larue, L. , Pritchard, C. , Marais, R. , 2009. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 15, 294–303. [DOI] [PubMed] [Google Scholar]
- Di Micco, R. , Fumagalli, M. , Cicalese, A. , Piccinin, S. , Gasparini, P. , Luise, C. , Schurra, C. , Garre, M. , Nuciforo, P.G. , Bensimon, A. , Maestro, R. , Pelicci, P.G. , d'Adda di Fagagna, F. , 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 444, 638–642. [DOI] [PubMed] [Google Scholar]
- dos Santos, N.R. , Torensma, R. , de Vries, T.J. , Schreurs, M.W. , de Bruijn, D.R. , Kater-Baats, E. , Ruiter, D.J. , Adema, G.J. , van Muijen, G.N. , van Kessel, A.G. , 2000. Heterogeneous expression of the SSX cancer/testis antigens in human melanoma lesions and cell lines. Cancer Res. 60, 1654–1662. [PubMed] [Google Scholar]
- Gjerstorff, M.F. , Burns, J. , Ditzel, H.J. , 2010. Cancer-germline antigen vaccines and epigenetic enhancers: future strategies for cancer treatment. Expert Opin. Biol. Ther. 10, 1061–1075. [DOI] [PubMed] [Google Scholar]
- Gjerstorff, M.F. , Johansen, L.E. , Nielsen, O. , Kock, K. , Ditzel, H.J. , 2006. Restriction of GAGE protein expression to subpopulations of cancer cells is independent of genotype and may limit the use of GAGE proteins as targets for cancer immunotherapy. Br. J. Cancer. 94, 1864–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greve, K.B. , Pohl, M. , Olsen, K.E. , Nielsen, O. , Ditzel, H.J. , Gjerstorff, M.F. , 2014. SSX2-4 expression in early-stage non-small cell lung cancer. Tissue Antigens. 83, 344–349. [DOI] [PubMed] [Google Scholar]
- Halazonetis, T.D. , Gorgoulis, V.G. , Bartek, J. , 2008. An oncogene-induced DNA damage model for cancer development. Science. 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Huang, X. , Darzynkiewicz, Z. , 2006. Cytometric assessment of histone H2AX phosphorylation: a reporter of DNA damage. Methods Mol. Biol. 314, 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki, H. , Nagasaka, T. , Otsuka, T. , Sugiura, E. , Nakashima, N. , Eimoto, T. , 2000. Association of SYT-SSX fusion types with proliferative activity and prognosis in synovial sarcoma. Mod. Pathol. 13, 482–488. [DOI] [PubMed] [Google Scholar]
- Kirkin, A.F. , Dzhandzhugazyan, K. , Zeuthen, J. , 1998. Melanoma-associated antigens recognized by cytotoxic T lymphocytes. APMIS. 106, 665–679. [DOI] [PubMed] [Google Scholar]
- Lim, F.L. , Soulez, M. , Koczan, D. , Thiesen, H.J. , Knight, J.C. , 1998. A KRAB-related domain and a novel transcription repression domain in proteins encoded by SSX genes that are disrupted in human sarcomas. Oncogene. 17, 2013–2018. [DOI] [PubMed] [Google Scholar]
- Rodier, F. , Campisi, J. , 2011. Four faces of cellular senescence. J. Cell Biol. 192, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin, U. , Tureci, O. , Chen, Y.T. , Seitz, G. , Villena-Heinsen, C. , Old, L.J. , Pfreundschuh, M. , 1998. Expression of multiple cancer/testis (CT) antigens in breast cancer and melanoma: basis for polyvalent CT vaccine strategies. Int. J. Cancer. 78, 387–389. [DOI] [PubMed] [Google Scholar]
- Serrano, M. , Lin, A.W. , McCurrach, M.E. , Beach, D. , Lowe, S.W. , 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Shimizu, N. , Nakajima, N.I. , Tsunematsu, T. , Ogawa, I. , Kawai, H. , Hirayama, R. , Fujimori, A. , Yamada, A. , Okayasu, R. , Ishimaru, N. , Takata, T. , Kudo, Y. , 2013. Selective enhancing effect of early mitotic inhibitor 1 (Emi1) depletion on the sensitivity of doxorubicin or X-ray treatment in human cancer cells. J. Biol. Chem. 288, 17238–17252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, A.J. , Caballero, O.L. , Jungbluth, A. , Chen, Y.T. , Old, L.J. , 2005. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer. 5, 615–625. [DOI] [PubMed] [Google Scholar]
- Smith, H.A. , Cronk, R.J. , Lang, J.M. , McNeel, D.G. , 2011. Expression and immunotherapeutic targeting of the SSX family of cancer-testis antigens in prostate cancer. Cancer Res. 71, 6785–6795. [DOI] [PubMed] [Google Scholar]
- Soulez, M. , Saurin, A.J. , Freemont, P.S. , Knight, J.C. , 1999. SSX and the synovial-sarcoma-specific chimaeric protein SYT-SSX co-localize with the human Polycomb group complex. Oncogene. 18, 2739–2746. [DOI] [PubMed] [Google Scholar]
- Suram, A. , Kaplunov, J. , Patel, P.L. , Ruan, H. , Cerutti, A. , Boccardi, V. , Fumagalli, M. , Di Micco, R. , Mirani, N. , Gurung, R.L. , Hande, M.P. , d'Adda di Fagagna, F. , Herbig, U. , 2012. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 31, 2839–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, I.M. , Minn, K. , Chen, J. , 2004. UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J. Biol. Chem. 279, 9677–9680. [DOI] [PubMed] [Google Scholar]
- Xu, B. , Sun, Z. , Liu, Z. , Guo, H. , Liu, Q. , Jiang, H. , Zou, Y. , Gong, Y. , Tischfield, J.A. , Shao, C. , 2011. Replication stress induces micronuclei comprising of aggregated DNA double-strand breaks. PloS One. 6, e18618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui, M. , Koyama, N. , Koizumi, T. , Senda-Murata, K. , Takashima, Y. , Hayashi, M. , Sugimoto, K. , Honma, M. , 2010. Live cell imaging of micronucleus formation and development. Mutat. Res. 692, 12–18. [DOI] [PubMed] [Google Scholar]
- Zendman, A.J. , de Wit, N.J. , van Kraats, A.A. , Weidle, U.H. , Ruiter, D.J. , van Muijen, G.N. , 2001. Expression profile of genes coding for melanoma differentiation antigens and cancer/testis antigens in metastatic lesions of human cutaneous melanoma. Melanoma Res. 11, 451–459. [DOI] [PubMed] [Google Scholar]
- Zhao, H. , Piwnica-Worms, H. , 2001. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 21, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, W. , Chen, Y. , Dutta, A. , 2004. Rereplication by depletion of geminin is seen regardless of p53 status and activates a G2/M checkpoint. Mol. Cell. Biol. 24, 7140–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary Figure 1 Quantification of SSX2 mRNA levels in A375‐TET‐SSX2 cells with induced SSX2 expression (50 ng/ml DOX; A375‐SSX2+) and in a panel of 40 clinical malignant melanoma samples, relative to A375‐TET‐SSX2 without SSX2 expression (A375‐SSX2−). Results from three different experiments are shown as fold expression ± standard deviation relative to A375‐TET‐SSX2 controls. Tissue samples with SSX2 levels below detection limit were omitted from the histogram (22 tumor samples in total).
Supplementary Figure 2 Representative growth curves for A375‐TET‐SSX2 and control A375 cells with or without 50 ng/ml DOX. DOX was added 24 h after seeding (time‐point: 0) and living (attached) cells counted daily (average of two wells shown).
Supplementary Figure 3 SSX2 reduces outgrowth of A375 tumors in SCID mice. One million A375‐TET‐SSX2 cells or control A375 cells were injected subcutaneously into the flank of CB17 SCID immunodeficient mice to investigate tumor growth. All mice received DOX in drinking water to induce expression of SSX2 (in A375‐TET‐SSX2 cells only), and tumors were resected for evaluation after 22 days. All the excised tumors were examined for SSX2 expression using quantitative PCR and as expected A375‐TET‐SSX2 cells exhibited a 4.4–10.7‐fold higher expression of SSX2 than cells from control A375‐derived tumors. (a) Plot of tumor weight from two independent experiments. Bars depict mean ± SD. (b) Tumor weight and qPCR data of SSX2 expression(a) normalized to GAPDH in fold over average of parental reference samples (Ref). N/A, not available.
Supplementary Figure 4 Nocodazole treatment of A375‐TET‐SSX2 cells show SSX2‐induced G1 arrest. A375‐TET‐SSX2 cells were grown for 3 days with or without DOX for induction of SSX2 expression, treated with nocodazole for 0, 6, 12 or 24 h and analyzed by PI/FACS to determine DNA content. The histograms depict the height of the PI signal (DNA) vs. counts.
Supplementary Figure 5 DOX addition in itself does not affect γ‐H2AX levels or Ki67 proliferation status of A375 melanoma cells. Parental A375 control cells were grown with or without 50 ng/ml DOX for SSX2 expression for 3 days, fixed and stained with DAPI for nuclei visualization (blue), the proliferation marker Ki67 (Alexa‐568, red) and γ‐H2AX for localization of DNA damage foci (FITC, green).
Supplementary Figure 6 Western blot analysis of CHK1 phosphorylation in A375‐TET‐SSX2 cells with or without DOX‐induced SSX2 expression (50 ng/ml). ETO = etoposide (50 μM for 4 h).
Supplementary Figure 7 The mitotic spindle apparatus is normal in A375‐TET‐SSX2 cells with induced expression of SSX2. A375‐TET‐SSX2 cells were grown for 72 h with or without 50 ng/ml DOX for SSX2 expression, fixed and stained with DAPI for DNA visualization (blue), anti‐α‐/β‐tubulin (green, 1:50, #2148, Cell Signaling, Danvers, USA) and anti‐SSX2/3 mAb 1A4 (red).
Supplementary Figure 8 Analysis of apoptosis in melanoma cells with and without SSX2 expression. The histograms show annexin V stainings measured by flow cytometry. Two replicates from two separate experiments (exp. 1 and 2) are shown. The dotted lines designate the control cells transfected with scrambled shRNA and the full lines cells transfected with SSX2 shRNA. Gating is indicated and the frequencies of annexin V positive cells are shown in the table below the histograms.
Supplementary Figure 9 SSX2 expression is not associated with increased DNA damage (γ‐H2AX foci) in MCF7 breast cancer cells after 48 h of SSX2 induction. A, SSX2 expression in MCF7‐TET‐SSX2 cells was induced with 50 ng/ml DOX and after 48 h the cells were fixed and stained for SSX2 and γ‐H2AX. B, Etoposide (ETO) treated cells were included as a positive control for γ‐H2AX foci formation.