

Abstract
The following fictional case is intended as a learning tool within the Pathology Competencies for Medical Education (PCME), a set of national standards for teaching pathology. These are divided into three basic competencies: Disease Mechanisms and Processes, Organ System Pathology, and Diagnostic Medicine and Therapeutic Pathology. For additional information, and a full list of learning objectives for all three competencies, see http://journals.sagepub.com/doi/10.1177/2374289517715040.
Keywords: pathology competencies, organ system pathology, autosomal recessive, cystic disorders, congenital, kidney, oligohydramnios
Primary Objective
Objective UTK4.1: Inherited Renal Disorders
Compare autosomal dominant and autosomal recessive polycystic kidney disease in terms of pathological anatomy, molecular pathogenesis, and clinical presentation.
Competency 2: Organ System Pathology; Topic UTK: Kidney; Learning Goal 4: Congenital Disorders of the Kidney.
Patient Presentation
A 25-year-old G1 woman with no prenatal care presents to her physician with absent fetal movement. A diagnosis of in utero fetal demise is made. The third trimester infant shown was delivered (Figure 1). Significant oligohydramnios was noted at delivery.
Figure 1.
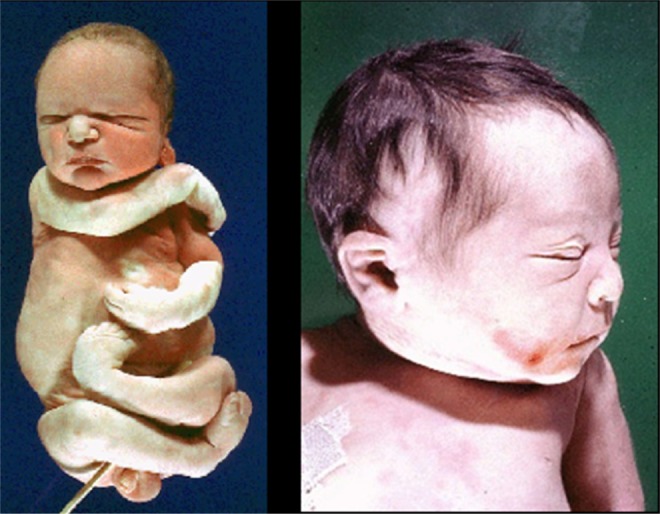
Infant with oligohydramnios (Potter) sequence. The left image shows a third trimester infant with flexion of the upper and lower extremities and spade-like hands and feet with abnormal facies. The right image shows the characteristic facial features of beaked nose, low-set flattened ears, prominent epicanthal folds, and receding chin.
Diagnostic Findings
An autopsy performed on the stillborn infant showed marked enlargement of the kidneys (combined weight of 105 g vs expected of 20.8 g) and the following changes on gross and microscopic examination in addition to pulmonary hypoplasia (combined lung weight of 20 g vs expected of 43 g) (Figure 2).
Figure 2.
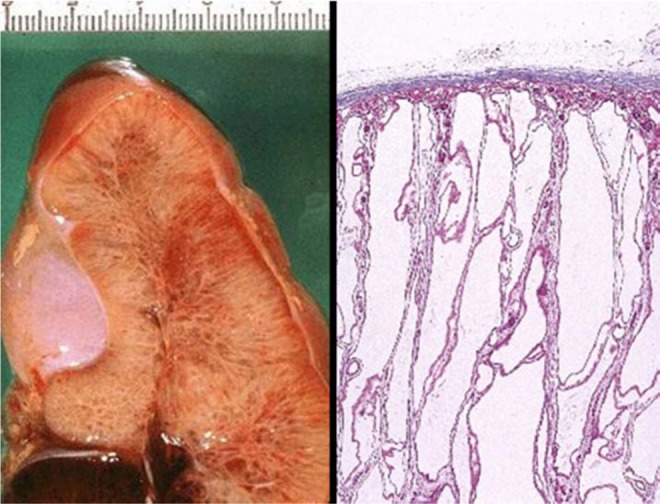
Gross appearance of kidney demonstrating cystic dilation of collecting ducts. Left: Medium power H&E stain of renal parenchyma illustrating cystic dilation of collecting ducts. Right: The long axis of the cysts is perpendicular to the connective tissue capsule. Normal glomeruli can be seen between collecting ducts.
Questions/Discussion Points
What is the Differential Diagnosis for Oligohydramnios?
The infant shows the classic features of oligohydramnios sequence, also called Potter sequence. This sequence includes limb deformities, such as clubbed feet and hip deformities, craniofacial anomalies, such as flattened ears and nose, hypoplastic mandible and epicanthal malformations, and pulmonary hypoplasia.1 The differential diagnosis of oligohydramnios consists of bilateral renal agenesis, autosomal recessive polycystic kidney disease (ARPKD), bilateral cystic renal dysplasia, obstructive uropathy, and chronic loss of amniotic fluid.2
Describe the Gross and Microscopic Pathologic Features Observed in the Kidneys at Autopsy
Grossly, the kidneys are massively enlarged with a reniform appearance. On sectioning, the kidneys show cystic dilation of the collecting ducts demonstrating a radial pattern. On microscopic examination, the collecting ducts are cystically dilated with the long axis of the cyst perpendicular to the capsule. Glomeruli and tubules may be seen between the collecting ducts.2
What is the Diagnosis Based on the Autopsy Findings?
The gross and histologic appearance of the kidneys is consistent with ARPKD.
Outline the Etiology and Pathogenesis of Autosomal Recessive Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease has an estimated incidence between 1:10 000 and 1:40 000.3 Neither race nor gender has been linked to increased prevalence.4 Different clinical presentations are observed. Fetal demise or death within the first year of life occurs in 30% of patients. Those who survive through infancy go on to develop progressive renal failure and liver disease leading to portal hypertension.5
Autosomal recessive polycystic kidney disease is due to a mutation in the PKHD1 gene on chromosome 6 that codes for the protein fibrocystin.2 There have been more than 750 PKHD1 mutations identified, the most common mutation of which is a missense mutation on exon 3 that accounts for more than 20% of cases.6 Most cases are familial, but de novo mutations are possible and account for 2 to 5% of cases.7 Diagnostic DNA testing is available for some of the most common causative mutations but is often not needed due to characteristic phenotypic and histologic findings. Genetic testing can be useful when the diagnosis is considered prenatally.8
Fibrocystin is a protein highly expressed in the primary cilia lining the collecting ducts of the kidney, the bile ducts, and pancreatic ducts. Abnormal expression of fibrocystin leads to defective intracellular signaling of epithelial cells during development of the kidney and biliary tract leading to cystic dilation of the collecting ducts and ductal plate malformations in the biliary tree. Defective remodeling of the ductal plate during development leading to dilation of the bile ducts and accompanied by increased periductal fibrosis may eventually lead to portal hypertension.8
Pulmonary hypoplasia is a complication of ARPKD. Several factors are involved. Decreased amniotic fluid affects the expansion of the lungs in utero. External compression of the fetus by the uterine wall secondary to oligohydramnios and the enlargement of the kidneys pushing up on the diaphragm decrease the thoracic cavity volume contributing to the pulmonary hypoplasia.
Contrast Autosomal Recessive Polycystic Kidney Disease With Autosomal Dominant Polycystic Kidney Disease
In contrast to ARPKD, autosomal dominant polycystic kidney (ADPKD) is most frequently due to a mutation in the PKD1 gene (chromosome 16) or the PKD2 gene (chromosome 4) that codes for the proteins polycystin-1 and polycystin-2 (Table 1). These proteins are also involved with ciliary function and intercellular signaling during development. Primary cilia are thought to have both chemoreceptor and mechanoreceptor functions and play a role in cell growth. Autosomal dominant polycystic kidney disease is observed in 1/400 to 1/1000 live births and accounts for about 10% of renal transplants. Most patients are asymptomatic early in life and present in middle age with chronic renal failure. Manifestations include hematuria, proteinuria, and hypertension. A small percentage of cases are associated with intracranial berry aneurysms as well as mitral valve prolapse and hepatic cysts. Grossly, the kidneys are enlarged and may weigh up to 4 kg (normal kidney weight 120-170 g). On sectioning, cysts up to 3 to 4 cm in diameter are haphazardly distributed throughout the renal parenchyma. On microscopic examination, all parts of the nephron may be cystically dilated. Normal renal parenchyma may be seen between cysts.2
Table 1.
Comparison of ARPKD with ADPKD.
| Feature | ARPKD | ADPKD |
|---|---|---|
| Incidence | 1:10 000 to 1:40 000 | 1:400 to 1:1 000 |
| Involved gene(s) | PKHD1 on chromosome 6 | PKD1 on chromosome 16 PKD2 on chromosome 4 |
| Protein defects | Fibrocystin | Polycystin-1 Polycystin-2 |
| Histologic appearance | Diffuse cystic dilation of the collecting ducts with the long axis of the cyst perpendicular to the capsule | Haphazardly arranged, cystic dilation of all parts of the involved nephron with normal renal parenchyma interspersed |
| Age at symptom onset | Infancy | Middle-aged adulthood |
| Complications | In utero fetal demise Neonatal respiratory distress Renal failure Liver failure | Renal failure Rupture of berry aneurysms |
Abbreviations: ARPKD, Autosomal recessive polycystic kidney disease; ADPKD, Autosomal dominant polycystic kidney disease.
Teaching Points
Oligohydramnios (Potter) sequence is defined as the presence of clubbed feet, craniofacial anomalies, and pulmonary hypoplasia in the setting of a below normal volume of amniotic fluid1:
Autosomal recessive polycystic kidney disease should be considered in the differential diagnosis for any patient presenting with oligohydramnios.2
In ARPKD, the characteristic histologic appearance of the kidneys includes large, cystic dilations that are oriented with the long axis perpendicular to the renal capsule and represent dilations of the collecting ducts.2
Thirty percent of affected patients die prenatally or within the first year of life. Patients surviving past the first year of life may go on to develop progressive renal failure and liver disease.8
Autosomal recessive polycystic kidney disease is due to abnormal expression of the PKHD gene on chromosome 6 that codes for fibrocystin. Abnormal fibrocystin results in defective intracellular signaling in utero, leading to the development of cystic dilations of the renal collecting ducts and biliary tree.8
Autosomal dominant polycystic kidney disease is due to a mutation in PKD1 gene on chromosome 16 or PKD2 gene on chromosome 4. These genes code for polycystin-1 and polycystin-2, respectively, which are also involved in intracellular signaling in utero, as well as ciliary function.2
Unlike ARPKD, ADPKD presents in middle age with hematuria, proteinuria, and hypertension, leading eventually to renal failure. Other conditions associated with ADPKD include intracranial berry aneurysms, mitral valve prolapse, and hepatic cysts.2
The histologic appearance of ADPKD differs from ARPKD in that cystic areas are scattered throughout the renal parenchyma, without uniform orientation, and all parts of the nephron may be involved, instead of only the collecting duct as in ARPKD.2
Acknowledgment
Images were obtained during the scope of federal employment for Dr. Conran.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Shastry SM, Kolte SS, Sanaagapati PR. Potter’s sequence. J Clin Neonatol. 2012;1(3):157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alpers CE, Chang A. The kidney In: Kumar V, Abbas AK, Aster JC, eds. Robbins and Cotran Pathologic Basis of Disease. 9th ed Philadelphia, PA: Elsevier; 2015:897–957. [Google Scholar]
- 3. Büscher R, Büscher AK, Weber S, et al. Clinical manifestations of autosomal recessive polycystic kidney disease (ARPKD): kidney-related and non-kidney-related phenotypes. Pediatr Nephrol. 2014;29:1915–1925. [DOI] [PubMed] [Google Scholar]
- 4. Patil A, Sweeney WE, Jr, Avner ED, et al. Childhood polycystic kidney disease In: Li X, ed. Polycystic Kidney Disease [Internet]. Brisbane, Australia: Codon Publications; 2015. Chap 2 https://www.ncbi.nlm.nih.gov/books/NBK373381/. Accessed March 7, 2017. doi:10.15586/codon.pkd.2015.ch2. [PubMed] [Google Scholar]
- 5. Parfrey P. Autosomal-recessive polycystic kidney disease. Kid Int. 2005;67:1638–1648. [DOI] [PubMed] [Google Scholar]
- 6. Department of Human Genetics. RWTH Aachen University. Mutation Database Autosomal Recessive Polycystic Kidney Disease. www.humgen.rwth-aachen.de. Accessed March 7, 2017.
- 7. Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol. 2015;30:15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sweeney WE, Avner ED . Polycystic kidney disease, autosomal recessive In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993-2017. https://www.ncbi.nlm.nih.gov/books/NBK1326/. Accessed March 7, 2017. [PubMed] [Google Scholar]