

Abstract
Nasopharyngeal carcinoma (NPC) is a cancer that arises from the epithelium of the nasopharynx. Celastrol is a triterpene from traditional Chinese medicine, which demonstrates anti-proliferative activity in several cancer cell lines. However, the effect of celastrol on human NPC and the underlying mechanisms are not yet elucidated. The present study investigated whether celastrol induced apoptosis in human NPC cells, and the underlying molecular mechanisms were explored. Celastrol decreased the viability of HONE-1 and NPC-039 cells in a dose-dependent manner, and induced G1 and G2/M phase cell cycle arrest. The level of cleaved caspases-3, −8, and −9 and poly (ADP-ribose) polymerase 1 increased in cells treated with celastrol. There was an increase in active Bcl-2-like 11 isoform S, Bcl-2-associated X, Bcl-2 antagonist/killer and truncated BH3-interacting death antagonist, and the levels of the anti-apoptotic Bcl-2 and Bcl-2-like 1 decreased. Celastrol induced an increase in Fas, Fas-associated via death domain, TNF receptor superfamily members (TNRSF) 1A and 10B, and TNFRSF1A associated via death domain, and induced a dose-dependent reduction in mitochondrial membrane potential. Celastrol inhibited activation of mitogen-activated protein kinase (MAPK) 1/3 and 14, and induced MAPK 8/9 activation. The results indicated that celastrol induced apoptosis through the death receptor and the mitochondrial pathway in human NPC cells, and is a promising candidate in the development of drugs against NPC.
Keywords: nasopharyngeal carcinoma, celastrol, apoptosis, death receptor, mitochondria
Introduction
Nasopharyngeal carcinoma (NPC) is a cancer that arises from the epithelium of nasopharynx. This cancer is uncommon in Europe and America, and its incidence is higher in Africa and Southeast Asia (1). Its unique epidemiology, pathogenesis and association with the Epstein-Barr virus make it distinct from other types of head and neck cancer. Although NPC is reported to respond well to radiation therapy, chemotherapy also has a function in the current treatment protocol (2). For cases that have spread beyond the nasopharynx (i.e., cancer stages II, III, IVA and IVB), the addition of chemotherapy is common. Typically, platinum based agents, including cisplatin or carboplatin are used to treat NPC alongside fluorouracil (3). However, these medications also harm normal cells, causing side effects that include a decrease in white blood cells, anemia, kidney toxicity and nausea.
Apoptosis, the process of programmed cell death, is a crucial self-defense mechanism for the human body to counteract cancerous cells. A number of existing chemotherapeutic agents aim to initiate the apoptosis cascade for an anti-tumor effect (4,5). However, as the disease progresses, certain cancer cell populations become resistant to chemotherapeutic agents and the efficacy of chemotherapy gradually deteriorates. Therefore, developing novel chemotherapeutic agents against NPC is crucial.
Tripterygium wilfordii, also known as thunder god vine, is an herb traditionally used in Asia to treat various diseases, including tissue inflammation, arthritis, certain rheumatoid diseases, and neurodegenerative diseases (6). Previous studies have revealed that celastrol, a bioactive isolate from the root of T. wilfordii, has potential as a cancer treatment. The chemical structure of celastrol is depicted in Fig. 1. Several studies have produced promising results using celastrol against various cancer cell lines in vitro, including gastric cancer (7), prostate cancer (8), hepatocellular carcinoma (9), breast cancer (10), esophageal cancer (11), lung cancer (12) and osteosarcoma (13). However, the therapeutic effects of celastrol on nasopharyngeal cancer have yet to been studied thoroughly. As natural products have been increasingly utilized in chemotherapy, the present study aimed to assess the cytotoxic effect of celastrol on NPC cells, identify the mechanism for the effect, and therefore, evaluate the potential for the utilization of this traditional herb as a novel agent against NPC.
Figure 1.
Chemical structure of celastrol.
Materials and methods
Reagents
Celastrol was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) as ≥98% powder. Stock solutions of celastrol were prepared with concentrations of 1, 2 and 4 mM in DMSO, and stored at −20°C. The final concentration of DMSO for all treatments was consistently <0.1%. MTT, U0126, SB203580 and DAPI reagents were obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell culture
The human NPC cell lines HONE-1 and NPC-039 were purchased from American Type Culture Collection (Manassas, VA, USA), and cultured in Gibco RPMI-1640 Medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA), 0.1 mM non-essential amino acids, 1 mM glutamine, 1% penicillin/streptomycin, 1.5 g/l sodium bicarbonate, and 1 mM sodium pyruvate (Sigma-Aldrich; Merck KGaA). The cell cultures were maintained at 37°C in a humidified atmosphere of 5% CO2.
In vitro cytotoxicity MTT assay
The influence of celastrol on cell growth was assayed by the MTT method. A total of 5×104 HONE-1 and NPC-039 cells were seeded in each well of 24-well plates. Celastrol (0, 1, 2 or 4 µM) was then added to the culture media and incubated at 37°C for 24 h. Following celastrol treatment, MTT was added to each well to a 0.5 mg/ml final concentration, and the cells were incubated for a further 4 h. The viable cell number was directly proportional to the production of formazan following solubilization with isopropanol, as detected by the optical density at 595 nm.
Cell cycle analysis
To determine the effect of celastrol on the cell cycle, 1×106 cells were cultured in 1 ml serum-free RPMI-1640 medium for 18 h to induce starvation, and then incubated with 1, 2 or 4 µM celastrol at 37°C for 24 h. Following the harvesting of cells by centrifugation (300 × g at room temperature for 5 min), washing in PBS and fixing with 70% ethanol overnight, they were incubated for 30 min in the dark at room temperature, with propidium iodide (PI) buffer (4 g/ml PI, 1% Triton X-100, 0.5 mg/ml RNase A (Affymetrix, Inc., Santa Clara, CA, USA) in PBS). The cells were filtered with a Falcon 40 µm nylon cell strainer (Corning Incorporated, Corning, NY, USA). The cell cycle distribution of 3,000 cells was analyzed by Muse Cell Analyzer flow cytometry (EMD Millipore, Billerica, MA, USA) and analysis data by Muse Cell Soft V1.4.0.0 Analyzer Assays (EMD Millipore).
Annexin V/PI double staining
To detect apoptosis in cells following celastrol exposure, a Muse Annexin V and Dead Cell Assay kit was used to quantify the number of cells at each stage of cell death (EMD Millipore). Briefly, 1×105 cells were suspended in 100 µl Muse reagent. Following incubation for 20 min at room temperature, the cell suspension was gently mixed and analyzed by the Muse Cell Analyzer flow cytometer (EMD Millipore). The data was processed with Muse Cell Soft V1.4.0.0 Analyzer Assays (EMD Millipore).
DAPI staining
Cells (3×105) that had been incubated with 0, 1, 2 or 4 µM celastrol at 37°C for 24 h were fixed with 4% paraformaldehyde for 15 min at room temperature, then at 4°C overnight. The plates were washed twice with PBS and the cell nuclei from the plates were stained with 100 ng/ml DAPI for 15 min in the dark. Following 3 washes with tap water, the cells were examined under a fluorescence microscope. Cells that appeared to exhibit a rough surface and darkly-stained nuclei with fragmented chromosomes were considered to be apoptotic.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay buffer (EMD Millipore) containing protease inhibitor cocktail and phosphatase inhibitor cocktail (EMD Millipore). Cells were harvested by centrifugation (12,000 × g, 4°C, 10 min). Protein concentration was determined by the Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Inc.). Cell lysates (total 20 µg) were separated on a 10 or 15% polyacrylamide gel and transferred onto a PVDF membrane (EMD Millipore). The blot was subsequently incubated with 3–5% skimmed milk in PBS at room temperature for 1 h to block non-specific binding, and probed with the following antibodies: Cleaved caspase-3 (cat. no. 9664; dilution, 1:1,000); cleaved caspase-8 (cat. no. 9496; dilution, 1:1,000); cleaved caspase-9 (cat. no. 9505; dilution, 1:1,000); poly (ADP-ribose) polymerase (PARP) (cat. no. 556494; dilution, 1:1,000); cyclin A (cat. no. 4656; 1:1,000); cyclin B (cat. no. 12231; 1:1,000); cyclin-dependent kinase (CDK)1 (cat. no. 9116; dilution, 1:1,000); CDK2 (cat. no. 2546; dilution, 1:1,000); CDK inhibitor 1A (p21Cip; cat. no, 2947; dilution, 1:1,000); CDK inhibitor 1B (p27Kip; cat. no. 3686; dilution, 1:1,000); Bcl-2 (cat. no. 2870; dilution, 1:1,000); B-cell lymphoma-extra-large (Bcl-xL; cat. no. 2764; dilution, 1:1,000); Bcl-2 associated X, apoptosis regulator (Bax; cat. no. 5023; dilution, 1:1,000); Bcl-2 antagonist/killer 1 (Bak; cat. no. 12105; dilution, 1:1,000); Bcl-2-like 11 (Bim; cat. no. 2933; dilution, 1:1,000); BH3-interacting death agonist (Bid; cat. no. 2002; dilution, 1:1,000); Fas (cat. no. 4233; dilution, 1:1,000), Fas-associated via death domain (FADD; cat. no. 2782; dilution, 1:1,000); TNFRSF1A-associated via death domain (TRADD; cat. no. 3684; dilution, 1:1,000); TNF receptor superfamily member 1A (TNF-R1; cat. no. 3736; dilution, 1:1,000); TNF receptor superfamily member 10b (DR5; cat. no. 8074; dilution, 1:1,000); protein kinase B (AKT; cat. no. 4298; dilution, 1:1,000); phosphorylated (p-)AKT (cat. no. 4060; dilution, 1:1,000); mitogen-activated protein kinase 14 (p38; cat. no. 9212; dilution, 1:1,000); p-p38 (cat. no. 9211; dilution, 1:1,000); extracellular signal-regulated kinase 1/2 (ERK1/2; cat. no. 4695; dilution, 1:1,000); p-ERK1/2 (cat. no. 4370; dilution, 1:1,000); c-Jun N-terminal kinase 1/2 (JNK1/2; cat. no. 9258; dilution, 1:1,000); p-JNK1/2 (cat. no. 4668; dilution, 1:1,000); and β-actin (cat. no. NB600-501; dilution, 1:5,000). Membranes were incubated with primary antibodies overnight at 4°C, and then with an appropriate peroxidase-conjugated secondary antibody at room temperature for 1 h [anti-mouse Ig; dilution, 1:3,000 (cat. no. 7076); anti-rabbit IgG; dilution, 1:3,000 (cat. no. 7074)]. Following the final wash, the immunoreactive signal was detected with an enhanced chemiluminescence detection system (WBKLS0500; EMD Millipore), and the relative density was quantitated with gel documentation and analysis (AlphaImager 2000; Alpha Innotech Corporation, San Leandro, CA, USA).
Mitochondrial membrane potential (Δψm) measurement
The reduction in Δψm was detected with a Muse Mitopotential Assay kit (EMD Millipore). Briefly, 1×105 cells which had been treated with celastrol were washed with PBS. The cells were suspended in Muse MitoPotential working solution and incubated at 37°C for 20 min. Following incubation, 5 µl Muse 7-AAD was added, and incubated at room temperature for 5 min. The reaction volume was thoroughly mixed, run on Muse Cell Analyzer flow cytometry (EMD Millipore), and analyzed by Muse Cell Soft V1.4.0.0 Analyzer Assays (EMD Millipore).
Statistical analysis
All values included represent the mean ± standard deviation of three repetitions per procedure. Statistical analyses of >3 groups were performed with a one-way analysis of variance test followed by Tukey's post hoc test. Comparisons between two groups were performed with a Student's t-test. Statistical tests were performed using SigmaStat 2.0 software (Systat Software, Inc., San Jose, CA, USA). P<0.05 was considered to represent a statistically significant difference.
Results
Celastrol has cytotoxic effects on NPC cell lines
To evaluate the effects of celastrol on cell viability, HONE-1 and NPC-039 cells were treated with celastrol for varying concentrations and durations, and viability was assessed with an MTT assay. The result demonstrated that celastrol decreased the viability of HONE-1 and NPC-039 cells, compared with untreated cells, in a dose-(Fig. 2A) and time-(Fig. 2B) dependent manner.
Figure 2.

Celastrol significantly reduced the viability of HONE-1 and NPC-039 nasopharyngeal carcinoma cells in an (A) dose- and (B) time-dependent manner, as detected with an MTT assay. *P<0.05 vs. 0 µM control.
Celastrol induces cell cycle arrest and apoptosis in human NPC cell lines
To determine whether the inhibitory effect on cell viability produced by celastrol was associated with the induction of apoptosis, HONE-1 and NPC-039 cells were treated with 0, 1, 2 or 4 µM of celastrol for 24 h and analyzed for cell cycle distribution analysis using flow cytometry. There was an accumulation of sub-G1 phase cells in HONE-1 cells (Fig. 3A). In NPC-039 cells, the treatment induced an increase of cells in the Sub-G1 and G2/M phases compared with the untreated control, in a dose dependent manner (Fig. 3A). The appearance of a sub-G1 population indicated apoptotic cells. These results suggested that the reduction in cell viability associated with celastrol may involve the induction of G2/M phase arrest and apoptosis. To further verify whether celastrol-induced cell death was associated with apoptosis, Annexin V/PI double staining was performed to quantify the level of apoptosis. A dose-dependent increase in apoptotic cells following 24 h of celastrol treatment was demonstrated for HONE-1 and NPC-039 cells (Fig. 3B). In addition, DAPI staining was applied to observe changes in nuclear morphology. In the two cell types, the condensed and fragmented nuclei in the treated cells was smaller compared with the control, which was observed following 24 h of 4 µM celastrol treatment (Fig. 3C).
Figure 3.

Celastrol induces cell cycle arrest and apoptosis in HONE-1 and NPC-039 nasopharyngeal carcinoma cells. HONE-1 and NPC-039 cells were treated with celastrol at doses of 1, 2 or 4 µM for 24 h. (A) The cell cycle distribution was analyzed with flow cytometry. (B) Annexin-V/propidium iodide staining followed by flow cytometry analysis revealed a significant increase in the rate of apoptosis. (C) Characteristic ‘blebbing’ morphology of DAPI-stained nuclei was observed in a dose-dependent manner (magnification, ×100).
Celastrol inhibits the cyclin-CDK checkpoint and induces activation of caspase-3, −8 and −9 in HONE-1 cells
To clarify whether caspase activation occurred in celastrol-induced apoptosis, apoptosis-associated molecules were examined with western blot analysis. Following treatment with celastrol at doses of 1–4 µM for 24 h, the levels of cleaved fragments of caspases-3, −8, and −9 and PARP increased in HONE-1 cells vs. untreated cells (Fig. 4A). A celastrol treatment of 4 µM for 24 h significantly increased the level of cleaved caspases-3, −8, −9 and PARP, by 52, 72, 30 and 42%, respectively, compared with the control (Fig. 4B). In addition, celastrol treatment significantly decreased the level of cyclin A and B and CDK1 and 2, and increased p21Cip and p27Kip levels, compared with the control (Fig. 4C and D).
Figure 4.

Effect of celastrol on cell cycle-associated proteins, and caspase and PARP cleavage. Cells were treated with celastrol, at doses of 1, 2 or 4 µM, for 24 h. (A) Western blot analysis detected an increase in cleaved caspase-3, −8, −9 and PARP levels. (B) The western blot was quantified, with detected protein levels normalized to β-actin using a densitometer. (C) Western blot analysis was used to detect the expression level change in cyclin A, cyclin B, CDK1, CDK2, p21Cip and p27Kip (D) The western blot was quantified, with detected protein levels normalized to β-actin with a densitometer. *P<0.05 vs. 0 µM control. PARP, poly (ADP-ribose) polymerase 1; CDK, cyclin-dependent kinase; p21Cip, cyclin-dependent kinase inhibitor 1A; p27Kip, cyclin-dependent kinase inhibitor 1B.
Bcl-2 family expression is altered in celastrol-treatment HONE-1 cells
Bcl-2 family proteins are associated with the regulation of apoptosis, therefore, western blot analysis was performed to determine whether Bcl-2 family members were involved in the process of apoptosis that was previously observed. The expression levels of the pro-apoptotic proteins Bax, Bak, t-Bid and BimS were increased, whereas the anti-apoptotic Bcl-2 and Bcl-xL decreased (Fig. 5A). Celastrol treatment at 4 µM for 24 h significantly increased the expression levels of Bax, Bak, t-Bid and BimS by 24, 135, 93 and 23% respectively, compared with the control (Fig. 5B). The 4 µM celastrol treatment also significantly decreased the expression levels of Bcl-2 and Bcl-xL by 30 and 35%, respectively, compared with the control (Fig. 5B).
Figure 5.
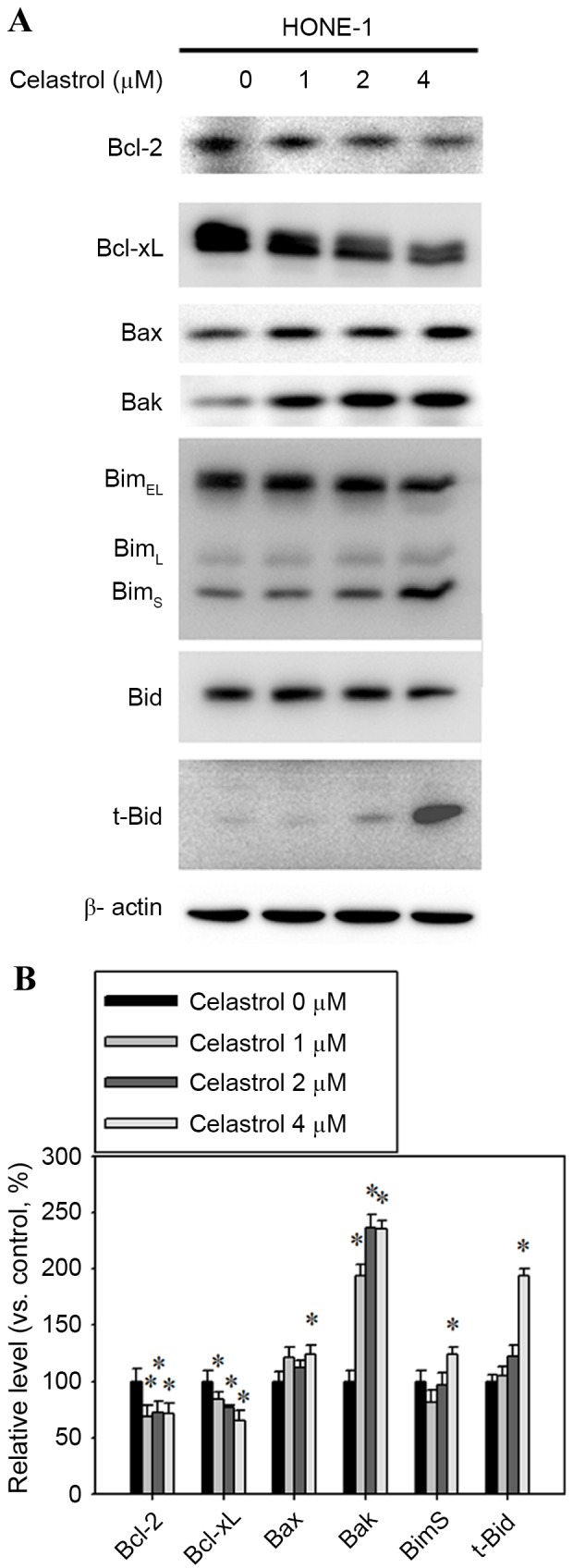
Effect of celastrol on the expression of Bcl-2, Bcl-xL, Bax, Bak, Bim and t-Bid. Cells were treated with celastrol, at doses of 1, 2 or 4 µM, for 24 h. (A) Western blot analysis detected the change in the expression level of Bcl-2, Bcl-xL, Bax, Bak, Bim and t-Bid. (B) The western blot was quantified, with detected protein levels normalized to β-actin with a densitometer. *P<0.05 vs. 0 µM control. Bcl-xL, B-cell lymphoma-extra large; Bax, Bcl-2 associated X, apoptosis regulator; Bak, Bcl-2 antagonist/killer 1; Bid, BH3-interacting death agonist; t-, truncated; BimEL, Bcl-2-like 11 isoform EL; BimL, Bcl-2-like 11 isoform L; BimS, Bcl-2-like 11 isoform S.
Celastrol-induced apoptosis is specifically mediated by the mitochondrial and Fas-mediated pathways
To determine the molecular mechanism by which celastrol induced the apoptosis of HONE-1 cells, the mitochondrial membrane potential and the protein levels of death receptor proteins were assessed. The mitochondrial membrane potential was reduced in celastrol-treated HONE-1 cells (Fig. 6A). In addition, celastrol also increased Fas, FADD, TRADD, TNF-R1 and DR5 expression, as assessed with a western blot analysis (Fig. 6B). A 24 h, 4 µM celastrol treatment significantly increased the expression levels of Fas, FADD, TRADD, TNF-R1 and DR5 by 39, 51, 103, 126 and 88%, respectively, compared with the control (Fig. 6C).
Figure 6.

Celastrol induces cell apoptosis by the death receptor and mitochondrial pathways. Cells were treated with celastrol, at doses of 1, 2 or 4 µM, for 24 h. (A) The mitochondrial membrane potential was analyzed with flow cytometry. (B) Western blot analysis detected the change in the expression level of Fas, FADD, TRADD, TNF-R1 and DR5. (C) The western blot was quantified, with detected protein levels normalized to β-actin using a densitometer. *P<0.05 vs. 0 µM control. FADD, Fas associated via death domain; TRADD, TNFRSF1A associated via death domain; TNF-R1, TNF receptor superfamily member 1A; DR5, TNF receptor superfamily member 10B.
Celastrol increases JNK1/2 activation and decreases ERK1/2 and p38 activation
The mitogen-activated protein kinase (MAPK) signaling pathway is involved in the induction of apoptosis by chemotherapeutic drugs (14). Western blot analysis was used to assess whether MAPKs were activated following celastrol treatment of HONE-1 cells. The relative phosphorylation level of JNK1/2 was increased and the phosphorylation of p38 and ERK1/2 was decreased, but AKT phosphorylation was not significantly altered (Fig. 7A). Celastrol treatment significantly increased the extent of JNK1/2 phosphorylation; phosphorylation was increased by 34% compared with the control (Fig. 7B). In addition, celastrol treatment significantly decreased the levels of ERK1/2 and p38 phosphorylation by 52 and 82%, respectively, compared with the control (Fig. 7B). Celastrol combined with an ERK1/2 inhibitor (U0126) or p38 inhibitor (SB203580) increased the rate of apoptosis in HONE-1 cells, compared with celastrol alone (Fig. 7C).
Figure 7.

Effect of celastrol on the expression of mitogen-activated protein kinase pathway components. Cells were treated with celastrol, at doses of 1, 2 or 4 µM, for 24 h. (A) Western blot analysis detected the change in AKT, p38, ERK1/2 and JNK1/2 phosphorylation. (B) The western blot was quantified, with detected protein levels normalized to β-actin with a densitometer. (C) Cells were treated with a combination of celastrol (1 µM) and U0126 (20 µM) or SB203580 (20 µM) for 24 h. The number of apoptotic cells was detected with flow cytometry. *P<0.05 vs. 0 µM control. #P<0.05 vs. celastrol-only group. AKT, protein kinase B; p38, mitogen-activated protein kinase 14; ERK1/2, extracellular signal-regulated kinase 1/2; JNK1/2, c-Jun N-terminal kinase 1/2.
Discussion
Celastrol is a bioactive isolate from T. wilfordii. Multiple in vitro studies have demonstrated that celastrol exhibits promising results against several types of cancer, including lung cancer, esophageal cancer, osteosarcoma, prostate cancer, breast cancer, hepatocellular carcinoma, and gastric cancer (7–9). However, the effect of celastrol on NPC cells remains uncertain. The purpose of the present study was to investigate the effect of celastrol on human NPC cells, and to identify the mechanisms underlying the effect. The findings revealed that celastrol may inhibit cancer cell viability, delay cell cycle progression and induce apoptosis. The viability of NPC cells was decreased in a dose-dependent manner. Apoptosis was induced in NPC-039 and HONE-1 cells following 24 h of celastrol treatment, and the extent of apoptosis was dose-dependent.
Cell cycle arrest, which is induced by a number of novel chemotherapeutic medications, is a potential strategy to hinder the proliferation of cancer cells (13). During cell cycle arrest, cells may initiate self-repairing mechanisms or enter the apoptosis cascade. A number of studies have previously identified that celastrol may induce cell cycle arrest. A study by Peng et al (15) demonstrated that celastrol arrested the human monocytic leukemia cell line U937 in G0/G1. Another study, by Rajendran et al (16), revealed that celastrol causes the accumulation of human hepatocellular carcinoma cells in the sub-G1 phase of the cell cycle. Furthermore, G2/M arrest has been identified in human cervical carcinoma and prostate cancer cells (15,17). The present study demonstrated that celastrol treatment caused the accumulation of HONE-1 and NPC-039 cells in the sub-G1 and G2/M phases. Therefore, celastrol may cause cell cycle arrest and prompt apoptosis in NPC cells.
Celastrol treatment (4 µM) significantly decreased the expression levels of Bcl-2 and Bcl-xL compared with the control (Fig. 5B). Members of the Bcl-2 family regulate the intrinsic mitochondrial mediated apoptotic pathway (18). The Bcl-2 family includes pro-apoptotic and anti-apoptotic members. Pro-apoptotic Bcl-2-like proteins, including Bax, promote apoptosis by opening the mitochondrial voltage-dependent anion channel to cause permeabilization of the mitochondrial outer membrane and the release of other pro-apoptotic factors (19). In the present study, celastrol treatment increased Fas, FADD, TRADD, TNF-R1 and DR5 expression and reduced the mitochondrial membrane potential (Fig. 6).
Caspases are also involved in mediating apoptotic processes. Western blot analysis revealed that the administration of celastrol led to increased expression levels of cleaved caspase-9, −8 and −3 and the cleavage of PARP in HONE-1 cells. A previous study has identified that celastrol treatment resulted in a significant activation of p38 and ERK and a marginal activation of stress-activated protein kinases/JNK in a dose- and time-dependent manner (17). Zhu et al (20) hypothesized that DR4 and DR5 are involved in the sensitization of celastrol-treated cells to TRAIL/Apo-2L-induced apoptosis, in a p38-independent manner. In addition, Choi et al (21) observed that celastrol reduced the extent of rotenone-induced generation of reactive oxygen species and mitochondrial membrane potential loss by inhibiting p38 activation in the SH-SY5Y human neuroblastoma cell line. In the present study, celastrol treatment significantly decreased the phosphorylation of p38 and ERK1/2, however, the level of JNK1/2 phosphorylation was increased (Fig. 7).
In conclusion, the results of the present study demonstrated that celastrol inhibited the viability of NPC cells, caused G2/M cycle arrest and the accumulation of cells in the sub-G1 phase, and induced apoptosis. Therefore, it is possible to hypothesize that celastrol may be a promising candidate in the development of drugs against NPC.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Al-Sarraf M, LeBlanc M, Giri PG, Fu KK, Cooper J, Vuong T, Forastiere AA, Adams G, Sakr WA, Schuller DE, Ensley JF. Chemoradiotherapy versus radiotherapy in patients with advanced nasopharyngeal cancer: Phase III randomized Intergroup study 0099. J Clin Oncol. 1998;16:1310–1317. doi: 10.1200/JCO.1998.16.4.1310. [DOI] [PubMed] [Google Scholar]
- 3.Bensouda Y, Kaikani W, Ahbeddou N, Rahhali R, Jabri M, Mrabti H, Boussen H, Errihani H. Treatment for metastatic nasopharyngeal carcinoma. Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128:79–85. doi: 10.1016/j.anorl.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 5.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: A link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/S0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 6.Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:1341–1357. doi: 10.1016/S0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 7.Ji N, Li J, Wei Z, Kong F, Jin H, Chen X, Li Y, Deng Y. Effect of celastrol on growth inhibition of prostate cancer cells through the regulation of hERG channel in vitro. Biomed Res Int. 2015;2015:308475. doi: 10.1155/2015/308475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH, Chun KH. Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep. 2014;47:697–702. doi: 10.5483/BMBRep.2014.47.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li PP, He W, Yuan PF, Song SS, Lu JT, Wei W. Celastrol induces mitochondria-mediated apoptosis in hepatocellular carcinoma Bel-7402 cells. Am J Chin Med. 2015;43:137–148. doi: 10.1142/S0192415X15500093. [DOI] [PubMed] [Google Scholar]
- 10.Shrivastava S, Jeengar MK, Reddy VS, Reddy GB, Naidu VG. Anticancer effect of celastrol on human triple negative breast cancer: Possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3K/Akt pathways. Exp Mol Pathol. 2015;98:313–327. doi: 10.1016/j.yexmp.2015.03.031. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Wu CL. Anti-metastasis of celastrol on esophageal cancer cells and its mechanism. Sheng Li Xue Bao. 2015;67:341–347. (In Chinese) [PubMed] [Google Scholar]
- 12.Xu J, Wu CL, Huang J. Effect of celastrol in inhibiting metastasis of lung cancer cells by influencing Akt signaling pathway and expressing integrins. Zhongguo Zhong Yao Za Zhi. 2015;40:1129–1133. (In Chinese) [PubMed] [Google Scholar]
- 13.Yu X, Zhou X, Fu C, Wang Q, Nie T, Zou F, Guo R, Liu H, Zhang B, Dai M. Celastrol induces apoptosis of human osteosarcoma cells via the mitochondrial apoptotic pathway. Oncol Rep. 2015;34:1129–1136. doi: 10.3892/or.2015.4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen T, Wong YS. Selenocystine induces S-phase arrest and apoptosis in human breast adenocarcinoma MCF-7 cells by modulating ERK and Akt phosphorylation. J Agric Food Chem. 2008;56:10574–10581. doi: 10.1021/jf802125t. [DOI] [PubMed] [Google Scholar]
- 15.Peng B, Xu L, Cao F, Wei T, Yang C, Uzan G, Zhang D. HSP90 inhibitor, celastrol, arrests human monocytic leukemia cell U937 at G0/G1 in thiol-containing agents reversible way. Mol Cancer. 2010;9:79. doi: 10.1186/1476-4598-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajendran P, Li F, Shanmugam MK, Kannaiyan R, Goh JN, Wong KF, Wang W, Khin E, Tergaonkar V, Kumar AP, et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev Res (Phila) 2012;5:631–643. doi: 10.1158/1940-6207.CAPR-11-0420. [DOI] [PubMed] [Google Scholar]
- 17.Wang WB, Feng LX, Yue QX, Wu WY, Guan SH, Jiang BH, Yang M, Liu X, Guo DA. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J Cell Physiol. 2012;227:2196–2206. doi: 10.1002/jcp.22956. [DOI] [PubMed] [Google Scholar]
- 18.Anilkumar U, Prehn JH. Anti-apoptotic BCL-2 family proteins in acute neural injury. Front Cell Neurosci. 2014;8:281. doi: 10.3389/fncel.2014.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Primikyri A, Chatziathanasiadou MV, Karali E, Kostaras E, Mantzaris MD, Hatzimichael E, Shin JS, Chi SW, Briasoulis E, Kolettas E, et al. Direct binding of Bcl-2 family proteins by quercetin triggers its pro-apoptotic activity. ACS Chem Biol. 2014;9:2737–2741. doi: 10.1021/cb500259e. [DOI] [PubMed] [Google Scholar]
- 20.Zhu H, Liu XW, Ding WJ, Xu DQ, Zhao YC, Lu W, He QJ, Yang B. Up-regulation of death receptor 4 and 5 by celastrol enhances the anti-cancer activity of TRAIL/Apo-2L. Cancer Lett. 2010;297:155–164. doi: 10.1016/j.canlet.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 21.Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S, Kim SJ. Celastrol from ‘Thunder God Vine’ protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson's disease. Neurochem Res. 2014;39:84–96. doi: 10.1007/s11064-013-1193-y. [DOI] [PubMed] [Google Scholar]