

Abstract
Background
Chorismate mutases of the AroQ homology class are widespread in the Bacteria and the Archaea. Many of these exist as domains that are fused with other aromatic-pathway catalytic domains. Among the monofunctional AroQ proteins, that from Erwinia herbicola was previously shown to have a cleavable signal peptide and located in the periplasmic compartment. Whether or not this might be unique to E. herbicola was unknown.
Results
The gene coding for the AroQ protein was cloned from Salmonella typhimurium, and the AroQ protein purified from both S. typhimurium and Pseudomonas aeruginosa was shown to have a periplasmic location. The periplasmic chorismate mutases (denoted *AroQ) are shown to be a distinct subclass of AroQ, being about twice the size of cytoplasmic AroQ proteins. The increased size is due to a carboxy-terminal extension of unknown function. In addition, a so-far novel aromatic aminotransferase was shown to be present in the periplasm of P. aeruginosa.
Conclusions
Our analysis has detected a number of additional *aroQ genes. The joint presence of *AroQ, cyclohexadienyl dehydratase and aromatic aminotransferase in the periplasmic compartment of P. aeruginosa comprises a complete chorismate-to-phenylalanine pathway and accounts for the "hidden overflow pathway" to phenylalanine described previously.
Background
The enzyme chorismate mutase performs the initial catalytic step of divergence to L-phenylalanine and L-tyrosine in aromatic biosynthesis. This reaction has long intrigued biochemists as the sole example of an enzyme-catalyzed pericyclic (Claisen) rearrangement [1]. The existence of two (or maybe three) different homology groups of chorismate mutases (Table 1) points to separate evolutionary origins. Such independent evolutionary origins of analogous enzymes that mediate the same function are becoming increasingly apparent [2].
Table 1.
Gene families for cyclohexadienyl mutase
| Class I* | Class II* | |||
| Subclass | Function | Acronym† | Function | Acronym† |
| Iα | Chorismate | AroQ | Chorismate | AroH |
| mutase | mutase | |||
| Isochorismate | PchB | |||
| mutase | ||||
| 4-Amino, | PapB | |||
| 4-deoxy- | ||||
| chorismate | ||||
| mutase | ||||
| Iβ | Chorismate | AroR‡ | ||
| mutase | ||||
*Class I and Class II specify independent homology groups (analogs). †Chorismate mutases are shown in bold. ‡In view of the closely related tertiary structures of the Saccharomyces cerevisiae chorismate mutase and the E. coli AroQ domain of AroQPheA [9], the placement of AroR within class I is consistent with the suggestion of MacBeath et al. [8].
Assigning a name to an enzyme would be vastly more simple if all homologs belonging to a given protein family possessed identical substrate specificities. At one extreme, however, completely independent homology groups can possess the same substrate specificity; that is, a given function may have evolved more than once. At the other extreme, a single protein family may possess member enzymes that have different substrate specificities. In fact, distinct and dispersed subsets of proteins having the same substrate specificity may exist within a single protein family whose members possess an admixture of different substrate specificities. The latter case is exemplified by the α, β and γ subfamilies of aromatic aminotransferases within the aminotransferases of family I [3]. Nevertheless, the differing substrate specificities within the catalytic repertoire of a particular protein family are generally a reflection of similar reaction mechanisms. Therefore, it should usually be feasible to name a protein family (or superfamily) at a hierarchical level that encompasses all substrate specificities of its member proteins. Chorismate mutase, isochorismate mutase [4] and 4-amino, 4-deoxychorismate mutase [5], which all catalyze similar reactions, are common members of a group that we have called cyclohexadienyl mutase (Table 1). In this vein, class I cyclohexadienyl mutases possess chorismate mutase members that belong to either family Iα (AroQ) or family Iβ (AroR). A third group of chorismate mutases (AroH) so far constitute the sole members of a separate homology group, the class II cyclohexadienyl mutases.
Chorismate mutases encoded by both aroQ and aroH are represented among prokaryotes. The distribution of AroH is quite limited, known at this time to be present only in some Bacillus species (B. subtilis, B. halodurans, and B. stearothermophilus), cyanobacteria (Synechocystis sp., Synechococcus sp., Nostoc punctiforme, Anabaena sp., and Prochlorococcus marinus), Streptomyces coelicolor, and Thermomonospora fusca. On the other hand, AroQ is widely distributed. Multiple paralogs of aroQ are commonly present in a single organism. Genes encoding AroQ and AroH coexist in some representatives of Bacillus. The AroQ proteins are all-helix-bundle domains [6], whereas the AroH proteins possess a trimeric pseudo α/β-barrel structure [7]. The AroR proteins (known only in eukaryotes) are structurally similar to AroQ, deploying a four-helix-bundle structure around the active site. The AroR and AroQ proteins also share a colinear arrangement of amphiphilic patterns, as well as conserved spacing between patches of closely neighboring patterns in the primary sequence. Thus, AroR can be viewed as a version of AroQ that has inserted loops related to allosteric regulation. The latter rationale is the basis for classification of AroQ and AroR into the same homology group ([8] and references therein) even though homology is barely detectable at the sequence level. Both AroQ and AroH are small (20 kDa) proteins. AroR is much larger and exhibits complex allostery, being activated by L-tryptophan and inhibited by L-tyrosine in the case of yeast [9] or being activated by L-tryptophan and inhibited by both L-tyrosine and L-phenylalanine in the case of higher plants [10].
The aroQ gene has been an extremely popular partner for fusion with other aromatic-pathway genes, as illustrated in Table 2. In Archaeoglobus, aroQ encodes the central domain of a tripartite fusion protein, which is flanked at the amino end by TyrA and at the carboxyl end by PheA. The most widely distributed aroQ fusion is with pheA, being present in most of the gamma and beta proteobacteria. The aroQ•pheA fusion also exists in Thermotoga maritima and Clostridium difficile, apparently having arisen as independent fusion events (G.X. and R.A.J., unpublished data). All enteric bacteria and their nearest phylogenetic neighbors possess an aroQ•tyrA fusion. Clostridium acetobutylicum possesses a so-far unique fusion of aroQ with aroD, the gene encoding shikimate dehydrogenase. Many years ago, B. subtilis was shown to have fused domains specifying chorismate mutase (aroQ) and 3-deoxy-D-arabino-heptulosonate 7-P (DAHP) synthase (aroA). The AroQ domain was shown to function not only as a catalytic domain for chorismate mutase, but also as the allosteric domain for the fused AroA domain. This fusion thus provided a physical basis for the allosteric pattern of control called sequential feedback inhibition [11]. This aroQ•aroA fusion also occurs in Staphylococcus and in Deinococcus. Although Porphyromonas gingivalis also possesses fused domains of aroQ and aroA, the fusion joint is at the 5' end of aroQ instead of at the 3' end.
Table 2.
aroQ subtypes
| Designation | Nature of gene product | Prior gene/ |
| protein names | ||
| aroQ | Monofunctional protein | aroQf/CM-F |
| *aroQ | Periplasmic/secreted | aroQf/CM-F |
| •aroQ or aroQ• | Fused with another domain | |
| Fusion examples | ||
| tyrA•aroQ•pheA | Archaeoglobus fulgidus | |
| aroQ•pheA | Most Proteobacteria | AroQp† |
| aroQ•tyrA | Enteric bacteria | AroQt† |
| aroQ•aroD | Clostridium acetobutylicum | |
| aroQ•aroA | Deinococcus, Bacillus, | AroQd |
| Staphylococcus | ||
| aroA•aroQ | Porphyromonas gingivalis |
†The chorismate mutase domain (AroQp or AroQt) has also been designated CM-P or CM-T, respectively, in the older literature ([45] and see [22]). The rationale for terminology to facilitate genomic comparisons, including the convention of using a bullet to separate the domains of fusion proteins, was presented in Gu et al. [46].
Among the monofunctional chorismate mutases, that present in Erwinia herbicola possesses a cleavable signal peptide and accordingly is translocated into the periplasm. E. herbicola chorismate mutase was shown [12] to be not only periplasmic but also located at the poles of the cells, similar to cyclohexadienyl dehydratase of Pseudomonas aeruginosa [13]. In this study we have examined the extent to which monofunctional chorismate mutases are periplasmic or cytoplasmic. A chorismate mutase of Salmonella typhimurium is shown to be periplasmic. A periplasmic chorismate mutase in P. aeruginosa is shown to be part of an complete three-step pathway for periplasmic conversion of chorismate to phenylalanine.
Figure 1 shows the biochemical pathway context of chorismate mutase with respect to phenylalanine and tyrosine biosynthesis and provides an overview of differences between S. typhimurium and P. aeruginosa. It also shows the nomenclature used. In this era of comparative genomics, it is no longer tenable to use acronyms for genes and their gene products that differ from organism to organism. The common-pathway enzymes through chorismate synthase are named in order, that is [AroA] catalyzes the first pathway reaction, [AroB] the second, and so on. In cases where different homolog classes are known to exist, these analog groups are designated with Roman numerals, for example, AroAI or AroAII. Different subgroups within a homology class are usually distinguished with α, β, and γ designations. Three of the common-pathway steps are represented by independently evolved analog classes of enzyme. S. typhimurium has three paralogs of DAHP synthase belonging to the AroAIα family, whereas P. aeruginosa possesses two paralogs of AroAIα enzyme and two paralogs of class II DAHP synthase (AroAII) [14]. S. typhimurium utilizes a single dehydroquinase that belongs to class I (AroCI), whereas P. aeruginosa deploys two paralogs of class II (AroCII). Both S. typhimurium and P. aeruginosa utilize class I shikimate kinase (AroEI), as do all bacteria [15]. However, S. typhimurium possesses two paralogs that are characteristic of E. coli and its closest relatives and encoded by genes labeled as aroK and aroL in the literature.
Figure 1.
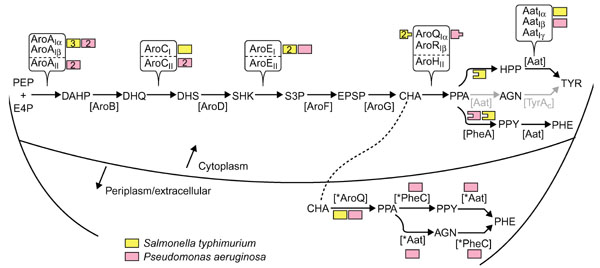
Biochemical pathway for phenylalanine and tyrosine biosynthesis. Small-molecule abbreviations: AGN, L-arogenate; CHA, chorismate; DAHP, 2-keto-3-deoxy-D-arabino-heptulosonate 7-P; DHQ, dehydroquinate; DHS, dehydroshikimate; E4P, erythrose-4-P; EPSP, enolpyruvylshikimate 3-P; HPP, 4-hydroxyphenylpyruvate; PEP, phosphoenolpyruvate; PHE, phenylalanine; PPA, prephenate; PPY, phenylpyruvate; SHK, shikimate; S3P, shikimate-3-P; TYR, tyrosine. Enzyme acronyms: [Aat], aromatic aminotransferase; [AroA], DAHP synthase; [AroB] DHQ synthase; [AroC], dehydroquinase; [AroD], SHK dehydrogenase; [AroE], SHK kinase; [AroF], EPSP synthase; [AroG], CHA synthase; [AroQ/AroR/AroH], CHA mutase; [PheA], prephenate dehydratase; [TyrAc], prephenate (cyclohexadienyl) dehydrogenase; [*Aat], periplasmic aromatic aminotransferase; [*AroQ], periplasmic chorismate mutase; [*PheC], periplasmic cyclohexadienyl dehydratase. (In P. aeruginosa, the only organism so far shown to have *Aat, it belongs to the *AatIβ grouping.) The flow route that progresses from PPA → AGN → TYR is shown in grey because this is a minor route in both P. aeruginosa and S. typhimurium. Different homology groups known so far that correspond with a given catalytic step are distinguished by Roman numeral subscripts, for example AroAI or AroAII. Subgroups within a homology group carry a further identifier, for example AroAIα or AroAIβ . As far as possible, these identifiers have been chosen to accommodate (within the constraints of logical naming) established precedents of naming. For example, the two homology groups of AroA were originally distinguished as class I or class II [50], and α and β subgroups for family-I DAHP synthases were defined recently [24]. AroCI and AroCII have been distinguished as biosynthetic or catabolic dehydroquinase, respectively, but this is a misnomer as many genomes use AroCII for aromatic amino acid biosynthesis (G.X. and R.A.J., unpublished data). Thus far, AroEI is used exclusively in Bacteria, and AroEII exclusively in Archaea [14]. The AroQIα group of chorismate mutases has been so named to reflect previous reference to them as AroQ [22, 46]. Likewise, AroRIβreflects previous reference to it as AroR [32]. The α, β and γ subgrouping identifiers for Aat reflect the subgroupings within family-I aminotransferases defined by Jensen and Gu [3]. Color-coding is used to illustrate the substantial difference between P.aeruginosa and S. typhimurium in deployment of different homolog or analog proteins (numbers within boxes indicate the number of paralogs), chorismate mutase fusions (peg-and-hole matches) and periplasm-localized enzymes.
Chorismate mutase is the focal point of this paper, and Figure 1 illustrates that the cytoplasm of S. typhimurium has two paralogs of the Iα family of chorismate mutases denoted AroQ. One is fused to TyrA and the other to PheA, as indicated by the peg-and-hole depiction in the figure. A third AroQ paralog of the Iα group is present in the periplasm (*AroQ). P. aeruginosa is similar to S. typhimurium in having an AroQ•PheA fusion in the cytoplasm as well as the periplasmic AroQ species (*AroQ), but differs in lacking the AroQ•TyrA fusion. Multidomain proteins whose domains have separable counterparts elsewhere must be appropriately named to facilitate comparisons (see Table 2).
Results
Chorismate-to-phenylalanine pathway in the P. aeruginosa periplasm
A modified magnesium shock procedure was used to separate periplasmic and cytoplasmic fractions of P. aeruginosa cultures grown in minimal medium. The established periplasmic enzyme *PheC [13, 16] was used as a periplasmic protein marker and shikimate dehydrogenase was used as a cytoplasmic protein marker. It was preferable to use prephenate rather than L-arogenate as a substrate in the assay of *PheC activity, not only because the assay for L-arogenate dehydratase is tedious [17] but also because prephenate is much less expensive. However, *PheC cannot be monitored as prephenate dehydratase in the presence of cytoplasmic P-protein because the PheA domain of that protein is prephenate dehydratase. To avoid this problem, the aroQ•pheA-deficient mutant PAT1051 was used. All the prephenate dehydratase activity in PAT1051 is derived from *PheC, which thus can be used as a convenient marker for the quality of the separation of the cellular compartments.
*PheC can also be selectively assayed without interference from PheA in wild-type PAO1. In the wild-type, the prephenate dehydratase activity of the P-protein is completely inhibited by phenylalanine, whereas *PheC is completely insensitive. Thus, one can calculate the fraction of total activity contributed by each of the two enzymes by comparing activities measured in the presence and absence of L-phenylalanine. From Table 3, a calculated 91% of *PheC was found in the periplasmic fraction of PAT1051, compared to 93% of *PheC from PAO1.
Table 3.
The presence of chorismate mutase and aromatic aminotransferase in the periplasmic fraction of Pseudomonas aeruginosa
| Fractions | Enzymes | Total enzyme activities† | ||
| PA01 | PAT1051 | |||
| Periplasmic fraction | DT | Total PDT‡ | 1154 | 1154 |
| *PheC§ (marker) | 1154 | 1154 | ||
| PheA¶ | 0 | 0 | ||
| *AroQ | 184 | 81 | ||
| AAT | 181 | 244 | ||
| SDH (marker) | 13 | 38 | ||
| Cytoplasmic fraction | DT | Total PDT‡ | 1373 | 71 |
| *PheC§ (marker) | 50 | 60 | ||
| PheA¶ | 1323 | 11 | ||
| AroQ• | 605 | 22 | ||
| AAT | 862 | 2528 | ||
| SDH (marker) | 375 | 478 | ||
| Whole-cell extract | DT | Total PDT‡ | 2706 | 617 |
| *PheC§ | 1220 | 604 | ||
| PheA¶ | 1486 | 13 | ||
| *AroQ + AroQ• | 734 | 88 | ||
| AAT | 905 | 3714 | ||
| SDH | 402 | 512 | ||
†Total enzyme activities are expressed as units (nmol of product permin) per 150 ml of culture. ‡Prephenate dehydratase activity assayed in the absence of L-phenylalanine. §Prephenate dehydratase activity assayed in the presence of 1 mM L-phenylalanine. ¶Calculated as 'Total PDT' minus '*PheC'. DT,dehydratase; PDT, prephenate dehydratase; *PheC, cyclohexadienyl dehydratase; PheA, prephenate dehydratase domain of AroQPheA; AroQ, chorismate mutase domain of aroQPheA; *AroQ, periplasmic chorismate mutase; AAT, aromatic aminotransferase; SDH, shikimate dehydrogenase.
*AroQ was found in the periplasm in P. aeruginosa PAO1. None of the periplasmic chorismate mutase activity was attributed to AroQ•PheA because no PheA-domain activity was detected (Table 3). Hence, the chorismate mutase activity in the periplasm must be exclusively *AroQ. Identification of this activity as *AroQ, and not AroQ•PheA, was verified by elution of a single peak of chorismate mutase in the wash fraction recovered after Mono Q chromatography of the periplasmic fraction. This is the established elution behavior of *AroQ [18]. Of the total aromatic aminotransferase activity, 10–20% was found in the periplasm.
Purification of *AroQ and *AatIβ from P. aeruginosa periplasm
*AroQ was purified from the periplasmic fraction of PAO1. Ninety-seven percent of shikimate dehydrogenase was in the cytoplasm of PAO1, compared to 93% from PAT1051. These data affirm a technically good separation of the periplasmic and cytoplasmic fractions. The purification steps are summarized in Table 4. After the last purification step, one distinctive band with a relative molecular mass (Mr) of around 22,000 was revealed by SDS-PAGE after staining with Coomassie blue. The Mr estimated by gel filtration was around 38,000, suggesting a native homodimer structure. Amino-terminal amino-acid sequencing gave a sequence QPLSPALQQL (in the single-letter amino-acid notation), which corresponds to a sequence beginning at residue 24 of gene PA5184 of the completed P. aeruginosa genome [19].
Table 4.
Purification of *AroQ from Pseudomonas aeruginosa
| Purification | Total protein | Specific | Purification | Yield |
| (mg) | activity* | (fold) | (%) | |
| Shock fluid | 240 | 17.7 | 1 | 100 |
| Hydroxylapatite | 39.9 | 39.2 | 2.2 | 36.8 |
| Mono Q (pH 7.0) | 10.5 | 53.1 | 3 | 13.1 |
| Mono Q (pH 8.4) | 1.7 | 116.5 | 6.6 | 4.7 |
| Superdex 75 | 0.44 | 487.5 | 27.5 | 5.0 |
*Specific activity expressed as nmol/min/mg.
Aromatic aminotransferase (*AatIβ) was purified from the periplasmic fraction of PAO1. The protein fractions with enzymatic activity that eluted from the gradient of a hydroxylapatite column were chromatographed on a mono Q column and recovered as single peaks. The leading peak fractions yielded one major band and several minor bands after SDS-PAGE. Amino-terminal amino-acid sequencing of the major band gave GSGATSAAGK, which corresponds to the sequence beginning with residue 29 of the deduced protein encoded by PA2531 in the completed P. aeruginosa genome [19]. In addition, one of the minor bands exhibited the sequence AGKPQETFLD, which corresponds to the sequence of the protein encoded by gene PA2531 beginning at residue 36. Thus, a favored and a minor cleavage site seem to exist for the signal peptide. The Mr estimated by SDS-PAGE staining with Coomassie blue was about 37,000. The Mr estimated by gel filtration was 73,000, indicating the presence of a homodimer.
Isolation of S. typhimurium *AroQ
E. coli BL21(pJB-1) containing *aroQ from S. typhimurium was grown to late-exponential growth in 1 liter of LB medium containing kanamycin. After cell harvest by centrifugation, 1.5 g of wet cells were subjected to osmotic shock [20]. After centrifugation, the supernatant was collected and designated as the shocked periplasmic fraction. An equal portion of wet cells, treated identically but not subjected to osmotic shock, was designated as the non-shock periplasmic fraction. Crude extracts were also prepared from each cell-pellet fraction.
The shocked periplasmic fraction contained 26,100 units (1 unit = 1 nmol of prephenate produced per minute) of chorismate mutase activity. As the prephenate dehydratase and prephenate dehydrogenase activities associated with the cytoplasmic AroQ•PheA and AroQ•TyrA proteins were not detected in this fraction, contamination with cytoplasmic species of AroQ was less than 1%. The additional assay for shikimate dehydrogenase as a marker for cytoplasmic contamination also showed less than 1% of total activity to be in the shocked periplasmic fraction. On the other hand, 97% of the total alkaline phosphatase (periplasmic marker) was recovered in the shocked periplasmic fraction. Although cross-reactivity was relatively poor, the presence of S. typhimurium *AroQ in periplasmic fractions and its absence in cytoplasmic fractions was demonstrated by western blotting using antibody obtained against E. herbicola AroQ [12].
The S. typhimurium *AroQ was purified using the protocol of Xia et al. [12] to give a final preparation having a specific activity of 59.1 units/mg, and an estimated 184-fold purification with 19% yield. SDS-PAGE showed a major band and one minor band. The sequence of the ten amino acids at the amino terminus of the major band (DSVTSVSLGA) matched perfectly with the deduced amino-acid sequence, beginning at the 21st residue, consistent with the predicted cleavage indicated in Figure 2.
Figure 2.
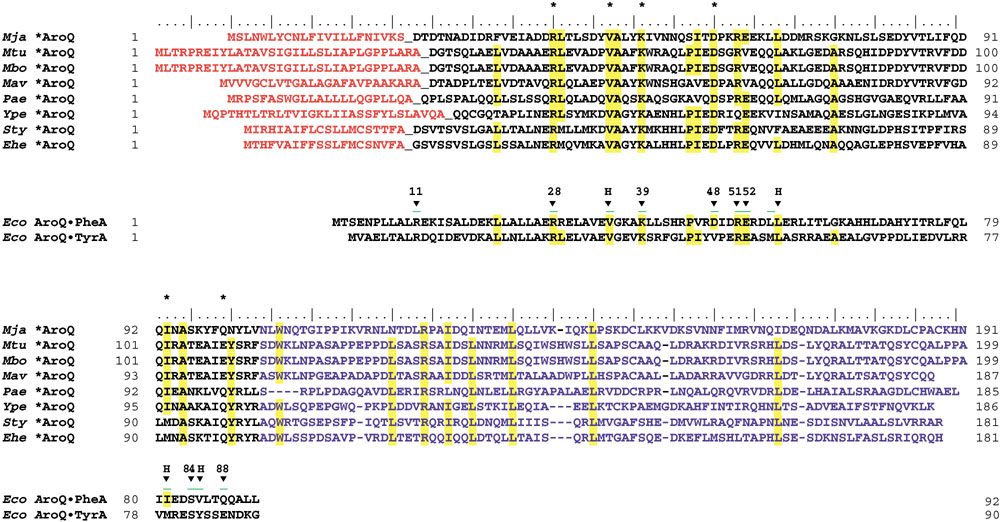
Multiple sequence alignment of the *AroQ protein family in relationship with the known three-dimensional structure of the AroQ domain of AroQPheA from E. coli. (The AroQ domain of AroQTyrA from E. coli is also shown.) Acronyms for organisms are as given in Table 7. The cleavable signal peptides of *AroQ proteins are shown in orange. The carboxy-terminal extensions of *AroQ proteins, which have no counterpart in other AroQ protein domains, are shown in blue. Residues located at the active site of the AroQ domain of E. coli AroQPheA as demonstrated by X-ray crystallography, are marked with arrowheads and assigned the E. coli AroQPheA residue numbers. Arrowheads marked with H are hydrophobic residues, and residue 88 is always Q or E. Conserved residues are shown in yellow. Active-site residues of AroQPheA from E. coli that are conserved in all eight *AroQ sequences are marked with an asterisk.
Enzymological characterization of periplasmic chorismate mutases
Purified enzymes from P. aeruginosa, S. typhimurium and E. herbicola were compared with respect to kinetic parameters under identical assay conditions (Table 5). Each enzyme followed Michaelis-Menten kinetics. Parameters of Km and kcat were similar to one another, as well as to other chorismate mutases characterized in the literature. Table 5 also shows a comparison of isoelectric points and molecular masses for all of the periplasmic chorismate mutases.
Table 5.
Kinetic constants, pI and Mw of *AroQ enzymes
| pI/Mr | ||||
| Organism | Km (μM) | kcat (s-1) | Uncleaved | Cleaved |
| Pseudomonas | 98 | 6.4 | 8.77 / 20,417 | 8.04 / 17,939 |
| aeruginosa | ||||
| Salmonella | 142 | 8.9 | 7.02 / 20,646 | 6.58 / 18,392 |
| typhimurium | ||||
| Erwinia | 169 | 9.7 | 6.28 / 20,299 | 6.24 / 18,017 |
| herbicola | ||||
| Yersinia | 6.43 / 20,761 | 5.66 / 17,504 | ||
| pestis | ||||
| Mycobacterium | 5.40 / 21,945 | 5.03 / 18,475 | ||
| tuberculosis | ||||
| Meloidogyne | 6.58 / 22,087 | 5.90 / 19,460 | ||
| javanica | ||||
Reaction kinetics were monitored at 32°C using 40–75 nM enzyme purified in this study (P. aeruginosa and S. typhimurium) or previously (E. herbicola [12]). Bovine serum albumin was present at 0.1 mg/ml. Initial rates were used to calculate the steady-state kinetic parameters of Km and kcat. pI/Mr valueswere determined by use of the compute pI/Mw tool at ExPASy [47].
Discussion
New periplasmic subgroup of AroQ
When the periplasmic chorismate mutase from E. herbicola was initially cloned and sequenced, it was a unique case of an AroQ protein having a cleavable signal peptide and a carboxy-terminal extension. The current repertoire of additional AroQ proteins that can be gleaned from the contemporary databases on the criterion of these two features numbers eight. One of them has been located in the esophageal glands of the phytoparasitic nematode Meloidogyne javanica. Presumably, this AroQ protein is secreted into plant cells as part of the parasitic process [21]. The erratic phylogenetic distribution of *AroQ may have a variety of explanations that include massive gene loss, horizontal transfer or independent evolution of a signal peptide and carboxy-terminal extension.
Figure 2 shows a multiple sequence alignment which highlights conserved amino-acid residues in the *AroQ subfamily in relationship to the homologous residues that have been shown by X-ray crystallography [6] to be located in the active site of the AroQ domain of E. coli AroQ•PheA. *AroQ proteins are twice the size of the basic AroQ catalytic domain. The carboxy-terminal half of each *AroQ protein is of similar length but not highly conserved, and it has no known catalytic or regulatory function. When used as a query sequence, it returns no significant hits. One reasonable speculation is that as *AroQ maybe localized at the polar cap(s) of the cell, this region might accomplish some sort of protein-protein or protein-membrane interaction that might facilitate its spatial location at the polar regions of the periplasmic milieu.
Substrate specificities of AroQ proteins
Chorismate mutase catalyzes a facile rearrangement reaction that can progress to a significant extent under nonenzymatic conditions. The aroQ genes are easily missed during genome annotation because they are small and do not show strong sequence conservation. We have identified many initially dubious AroQ hits to BLAST queries by alignment of amino-acid residues known to be critical for catalysis.
Figure 3 illustrates several chemical rearrangements that closely parallel the chorismate mutase reaction. Zamir et al. [4] have demonstrated the isochorismate mutase reaction in higher plants, and Xie et al. [22] presented a rationale in support of the possibility that PchB from P. aeruginosa catalyzes the isochorismate mutase reaction as a step in salicylate biosynthesis [23]. 4-Amino 4-deoxy-chorismate mutase catalyzes a step in antibiotic (pristinamycin) biosynthesis [5].
Figure 3.
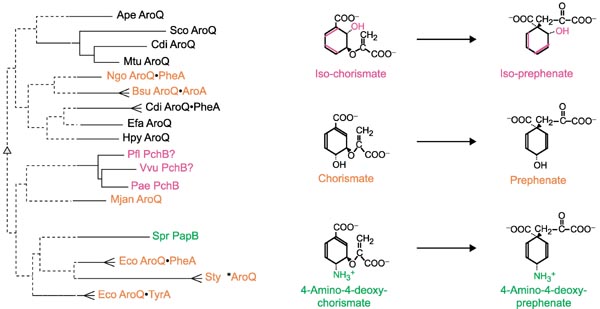
Substrate specificity within the Iα family of cyclohexadienyl mutase. The substrates and products of chorismate mutase (middle), isochorismate mutase (top) and 4-amino-4-deoxy-chorismate mutase (bottom) are shown at the right. Enzymes from species shown on the left that are known (but see text for PchB) to catalyze the reactions shown on the right are color coded to correspond. Chorismate mutase proteins or domains from selected organisms (see Figure 4 legend for organism abbreviations) are shown as a phylogram tree. The tree shown was obtained with the Phylip program using the input of a Multalign multiple alignment [51]. Bootstrap values over 50% (500 iterations) determine the placement of solid lines, which show significant clustering relationships.
There is precedent for the finding that within a homolog grouping whose members have differing substrate specificities, proteins sharing a substrate specificity do not necessarily cluster together; for example, B. subtilis AroA and close relatives are clustered more specifically with KdsA sequences than with E. coli AroA and its close relatives [24]. The phylogram tree at the left in Figure 3 illustrates this for the AroQ protein family. The current limitations of homology detection are such that the monofunctional AroQ proteins from Aeropyrum pernix, Enterococcus faecalis, Helicobacter pylori and Methanococcus jannaschii cannot be shown to be closer to one another or to any other AroQ proteins/domains than they are to PapB or PchB. This is because bootstrap values only support the branching nodes to the level shown in bold. Thus, even though the computational program used placed A. pernix AroQ with the valid grouping of Streptomyces coelicolor AroQ, C. difficile AroQ, and Mycobacterium tuberculosis AroQ, the A. pernix AroQ sequence could in fact be closer to any other group. Thus, one can recognize distinct and valid groupings (for example, the *AroQ periplasmic chorismate mutases), but not necessarily the exact relationship of these groupings with one another.
The AroQ family of chorismate mutases
Figure 4 shows a phylogram tree of the entire set of AroQ protein or AroQ domain sequences available from Bacteria and Archaea. A number of cohesive clusters are evident. One of these is the periplasmic group of monofunctional AroQ proteins. The cytoplasmic monofunctional AroQ proteins include five-member, three-member, and two-member groups in addition to four other sequences isolated so far. AroQ domains that are fused with TyrA form a cohesive homology grouping within enteric bacteria and their closest relatives. These same organisms also possess AroQ•PheA bifunctional proteins whose AroQ domains also form a cohesive homology grouping. Other AroQ•PheA fusions are widespread, and the sequences of their AroQ domains are highly divergent. Only the AroQ•PheA proteins from P. aeruginosa/ P. stutzeri, from Neisseria gonorrhoeae/N. meningitidis and from Archaeoglobus fulgidus/Thermotoga maritima possess AroQ domains that cluster with bootstrap values greater than 50%. Analysis of interdomain linker regions and codon usage suggests that the aroQ and pheA genes have fused independently on many occasions (G.X. and R.A.J., manuscript in preparation). The three cases of AroQ•AroA fusions fall into a single cluster supported by a bootstrap value of 61%.
Figure 4.
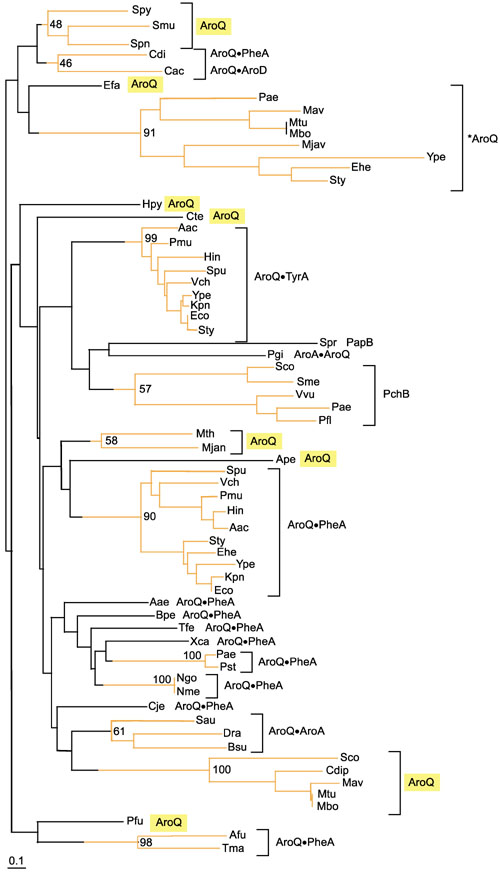
Relationship between chorismate mutase domains belonging to the Iα family of cyclohexadienyl mutases. The relationships are shown as a phylogram tree computed with the neighbor-joining method using the Phylo-Win program and multiple alignment input from the ClustalW program. Bootstrap percentages (for 500 iterations) are shown at nodes that define clusters deemed to be significant (above 50%). Accordingly, that portion of the tree where branching order is significant is shown in orange. AroQ, monofunctional chorismate mutase; *AroQ, periplasmic chorismate mutase. A bullet indicates a fusion joint. Thus, AroQPheA indicates an AroQ domain fused at its carboxyl terminus with the amino-terminal portion of a PheA domain. PapB and PchB carry out reactions analogous to chorismate mutase as illustrated in Figure 3. The divergent AroQ protein groupings are highlighted in yellow. Abbreviations for organisms: Actinobacillus actinomycetemcomitans (Aac), Aeropyrum pernix (Ape), Archaeoglobus fulgidus (Afu), Bacillus subtilis (Bsu), Bordetella pertussis (Bpe), Campylobacterium jejeuni (Cje), Chlorobium tepidum (Cte), Clostridium acetobutylicum (Cac), Clostridium difficile (Cdi), Enterococcus faecalis (Efa), Erwinia herbicola (Ehe), Escherichia coli (Eco), Haemophilus influenzae (Hin), Helicobacter pylori (Hpy), Klebsiella pneumoniae (Kpn), Methanobacterium thermoautotrophicum (Mth), Methanococcus jannaschii (Mjan), Mycobacterium avis (Mav), Meloidogyne javanica (Mjav), Mycobacterium bovis (Mbo), Mycobacterium tuberculosis (Mtu), Neisseria gonorrhoeae (Ngo), Neisseria meningitidis (Nme), Pasteurella multocida (Pmu), Porphyromonas gingivalis (Pgi), Pseudomonas aeruginosa (Pae), Pseudomonas fluorescens (Pfl), Pseudomonas stutzeri (Pst), Salmonella typhimurium (Sty), Shewanella putrefaciens (Spu), Sinorhizobium meliloti (Sme), Staphylococcus aureus (Sau), Streptomyces coelicolor (Sco), Streptococcus mutans (Smu), Streptococcus pneumoniae (Spn), Streptococcus pyogenes (Spy), Streptomyces coelicolor (Sco), Streptomyces pristinaespiralis (Spr), Thermotoga maritima (Tma), Thiobacillus ferrioxidans (Tfe), Vibrio cholerae (Vch), Vibrio vulnificans (Vvu), Xanthomonas campestris (Xca), and Yersinia pestis (Ype). Within the ten-member cluster of AroQPheA proteins present in enteric bacteria and their closest relatives, the S. putrefaciens (Spu) protein possesses a third domain (AroQPheAAroA). However, the AroA domain is altered at many otherwise-conserved residues and presumably lacks catalytic function (pseudogene).
In the five-member group labeled PchB in Figure 4, the P. aeruginosa protein has been shown to participate in salicylate synthesis and the proposed reaction (catalyzed by isochorismate mutase) has been discussed in the previous section. The P. fluorescens protein undoubtedly has the same role. This substrate specificity does not necessarily apply to the remaining members. In P. aeruginosa, pchA (isochorismate synthase) and pchB are translationally coupled, and cis-expression is obligatory for activity [23]. In Vibrio vulnificus, pchB is closely linked to venB (isochorismatase) [25]. V. cholerae does not exhibit a pchB homolog upstream of the gene encoding isochorismatase (vibF) [26]. Perhaps the clustering of PchB proteins together reflects their interaction with proteins able to bind isochorismate.
The periplasmic pathway of phenylalanine biosynthesis
It has been known for some time that phenylalanine-pathway redundancy exists in P. aeruginosa. The enzymological basis for this became apparent with the recognition of the presence of an 'extra' species of chorismate mutase (CM-F) and cyclohexadienyl dehydratase in addition to the E. coli-like enzymes [27]. This redundancy explained the "reluctant auxotrophy" of P. aeruginosa [27], as well as the "hidden overflow pathway" to phenylalanine [28]. The latter phenomenon is one whereby early-pathway regulatory mutations that increase intracellular chorismate concentrations result in exclusive metabolite flow toward L-phenylalanine because cyclohexadienyl dehydratase is insensitive to allosteric control. In the periplasmic system a mixture of flow routes (chorismate → prephenate → phenylpyruvate → phenylalanine or chorismate → prephenate → arogenate → phenylalanine) exist because of the ability of cyclohexadienyl dehydratase to accept either prephenate or arogenate and because of the ability of periplasmic aminotransferase (whose detailed characterization is not within the scope of this paper) to transaminate either phenylpyruvate or prephenate.
The spatial location of the overflow pathway in the periplasmic compartment was first indicated by the demonstration that cyclohexadienyl dehydratase had a cleavable amino terminus and was translocated to the periplasm. Indirect evidence favoring a periplasmic location for chorismate mutase as well in P. aeruginosa was the demonstration of a periplasmic chorismate mutase in E. herbicola [12]. The possible periplasmic location of an aromatic aminotransferase has been anticipated, but not directly examined until now. This paper demonstrates the identity and presence of all three enzymes constituting the overflow pathway in a single organism.
On the basis of broad comparative enzymological surveys [18, 29], a similar intact overflow pathway can be anticipated to be present in Serratia and Erwinia. The application of queries to the finished and unfinished genomes at the National Center for Biotechnological Information [30] returned hits for periplasmic cyclohexadienyl dehydratase and periplasmic chorismate mutase in Yersinia pestis. Although no aminotransferase gene has yet been found in Y. pestis that has a predicted signal peptide, one is present that is predicted by P-Sort to be an inner-membrane protein.
Periplasmic pathway remnants?
Before the recognition of their periplasmic location, it was known [31] that some organisms might have *AroQ but not *PheC (Salmonella) or *PheC but not *AroQ (Klebsiella). It was pointed out that appropriate mutants are better producers of prephenate in Salmonella than in Escherichia because of the presence of *AroQ in the former but not in the latter. Detailed comparative enzymological work in enteric bacteria [18, 29, 32] indicates that many additional *aroQ genes will be identified. Within Enterocluster-1, Enterobacter cloacae resembles S. typhimurium in lacking *pheC but having *aroQ, whereas Enterobacter aerogenes resembles K. pneumoniae in its possession of *pheC but its absence of *aroQ. Other Enterocluster-1 members (Escherichia, Shigella, Enterobacter agglomerans (ATCC 29915), and Citrobacter,) lack both periplasmic enzymes. On the other hand, all members of Enterocluster-2 possess both *aroQ and *pheC, except for Erwinia carotovora which lacks *pheC. These include Serratia marcescens, Serratia rubidae, Enterobacter agglomerans (ATCC 27155), Erwinia herbicola, E. milletiae, E. ananas, E. tracheiphila, E. chrysanthemi and E. amylovora. Members of Enterocluster-3 (species of Cedecea, Kluyvera, Edwardsiella, Hafnia, Yersinia, Proteus, Providencia and Morganella) lack both *aroQ and *pheC.
All four species of Mycobacterium in the finished genomes resemble Salmonella in having *aroQ but lacking *pheC. The physiological function of a partial pathway is unknown; perhaps *AroQ interfaces with a different pathway. In fact, it must be conceded that even the function of the intact periplasmic pathway to phenylalanine is unknown. An important clue presumably is the spatial location at the pole(s) of the cell. The polar, periplasmic location of a number of protein systems is receiving increasing attention [33].
Aromatic aminotransferases
Most aminotransferases that are capable of functioning as aromatic aminotransferases are members of family I within the aminotransferase protein superfamily [3]. These can usually be assigned to one of three subfamilies, Subfamily Iα partitions into narrow-specificity aspartate aminotransferases and broad-specificity aspartate aminotransferases that can accommodate the aromatic ring. Likewise, subfamily Iβ partitions into narrow-specificity imidazole acetol phosphate (IAP) aminotransferases and broad-specificity IAP aminotransferases that utilize aromatic amino/keto acids. Subfamily Iγ partitions into subgroups having a variety of substrate specificities, with aspartate aminotransferases being the most prominent subcluster. Among these are broad-specificity aspartate aminotransferases with ability to function with aromatic amino acids.
The periplasmic aminotransferase of P. aeruginosa belongs to the Iβ subfamily and exhibits catalytic activity as IAP aminotransferase in vitro assayed as described by Gu et al. [34]. It exhibits signature motifs corresponding to the regions of the active-site lysine and the interdomain interface [3]. Like its close relative, HisH of Bacillus subtilis, it has the potential to function in both histidine and aromatic amino-acid biosynthesis. Of course, the spatial location of *AatIβ in the periplasm would seem to rule out in vivo function in histidine biosynthesis. This expectation is accommodated by the observation that P. aeruginosa possesses a paralog encoded by gene PA4447 [19] that must function in vivo as IAP aminotransferase as it is closely linked with other genes of histidine biosynthesis. Yet another paralog is encoded by gene PA3165 and carries out aromatic amino-acid biosynthesis in the cytoplasm. Thus, three subfamily Iβ paralogs of aminotransferase are expressed in P. aeruginosa: one for aromatic biosynthesis in the cytoplasm, one for histidine biosynthesis in the cytoplasm, and one for phenylalanine synthesis in the periplasm. Previous enzymological work had demonstrated the existence of five aminotransferases in P. aeruginosa that were capable of functioning as aromatic aminotransferases in vitro [35].
Materials and methods
Bacterial strains, plasmids, and media
All bacterial strains and plasmids used in this study are listed in Table 6. S. typhimurium tyrA19 was grown on Luria-Bertani (LB) enriched medium or M-9 minimal media [36] plus growth-limiting L-tyrosine (0.5 μg/ml). E. coli DH5α and E. coli BL21 were grown on LB medium plus kanamycin when transformed with plasmid. Klebsiella pneumoniae 62–1 was grown according to the method of Gibson [37] for accumulation and purification of chorismate as the free acid form (about 91% pure). Prephenate and L-arogenate were obtained as specified by Bonner et al. [31]. Genomic DNA prepared from S. typhimurium was a gift from Thomas Bobik, University of Florida. Where indicated, ampicillin was added to media at 100 μg/ml, kanamycin at 35 μg/ml, and thiamine at 17 μg/ml. Medium was solidified with 1.5% (w/v) agar.
Table 6.
Bacterial strains and plasmids
| Strain or plasmid | Genotype or description | Source or reference |
| Escherichia coli | ||
| BL21(DE3) | F-ompT hsdSB (rB- mB-) gal dcm; with DE3, a λ prophage carrying the T7 RNA polymerase gene | Novagen |
| BL21(pJB-1) | BL21(DE3) transformed with pJB-1 | This study |
| DH5α | F-φ 80dlacZΔM15 Δ (lacZYA-argF) U169 deoR recA1 endA1 up44gλ-hsdR17 (rK-, mk+) thi-1 gyrA96 relA1 | Gibco-BRL |
| Salmonella typhimurium | ||
| CAB101 | tyrA19 | [48] |
| ATCC 15277 | Prototroph | ATCC |
| Klebsiella pneumoniae | ||
| 62–1 (ATCC 25306) | tyrA pheA trpD | ATCC |
| Pseudomonas aeruginosa | ||
| PAT1051 | aroQpheA (leaky phenylalanine auxotroph) | [49] |
| PAO1 | Wild-type prototroph | B.W. Holloway |
| Plasmids | ||
| pET-24b(+) | T7 lac promoter, lacI+ Kmr HisTag sequence, T7Tag (11aa) sequence | Novagen |
| pJB-1 | pET-24b(+) containing S. typhimurium*aroQ insert | This study |
Sequences from the public databases are identified with their gene identification (GI) numbers in Table 7.
Table 7.
Key to sequence identifiers
| Species name | Acronym | Gene name | GI number |
| Actinobacillus actinomycetemcomitans | Aac | AroQ•PheA | N/A |
| Actinobacillus actinomycetemcomitans | Aac | AroQ•TyrA | N/A |
| Aquifex aeolicus | Aae | AroQ•PheA | 8134616 |
| Aeropyrum penix | Ape | AroQ | N/A |
| Archaeoglobus fulgidus | Afu | TyrA•AroQ•PheA | 11497843 |
| Bacillus subtilis | Bsu | AroQ•AroA | 728897 |
| Bordetella pertussis | Bpe | AroQ•PheA | N/A |
| Campylobacterium jejeuni | Cje | AroQ•PheA | 11270735 |
| Chlorobium tepidum | Cte | AroQ | N/A |
| Clostridium acetobutylicum | Cac | AroQ•AroD | N/A |
| Clostridium difficile | Cdi | AroQ•PheA | N/A |
| Corynebacterium diphtheriae | Cdip | AroQ | N/A |
| Deinoccoccus radiodurans | Dra | AroQ•AroA | 7473313 |
| Enterococcus facaelis | Efa | AroQ | N/A |
| Erwinia herbicola | Ehe | AroQ•PheA | 266771 |
| Erwinia herbicola | Ehe | *AroQ | 1168941 |
| Escherichia coli | Eco | AroQ•PheA | 130050 |
| Escherichia coli | Eco | AroQ•TyrA | 136592 |
| Haemophilus influenzae | Hin | AroQ•PheA | 1172476 |
| Haemophilus influenzae | Hin | AroQ•TyrA | 1174832 |
| Helicobacter pylori | Hpy | AroQ | 7464214 |
| Klebsiella pneumoniae | Kpn | AroQ•PheA | N/A |
| Klebsiella pneumoniae | Kpn | AroQ•TyrA | N/A |
| Meloidogyne javanica | Mjav | *AroQ | 5353514 |
| Methanobacterium thermoautotrophicum | Mth | AroQ | 2621894 |
| Methanococcus jannaschii | Mjan | AroQ | 2495875 |
| Mycobacterium avium | Mav | AroQ | N/A |
| Mycobacterium avium | Mav | *AroQ | N/A |
| Mycobacterium bovis | Mbo | AroQ | N/A |
| Mycobacterium bovis | Mbo | *AroQ | N/A |
| Mycobacterium tuberculosis | Mtu | AroQ | 1524214 |
| Mycobacterium tuberculosis | Mtu | *AroQ | 7476951 |
| Neisseria gonorrhoeae | Ngo | AroQ•PheA | 8134631 |
| Neisseria meningitidis | Nme | AroQ•PheA | 11270737 |
| Pasteurella multocidans | Pmu | AroQ•PheA | 12720367 |
| Pasteurella multocidans | Pmu | AroQ•TyrA | 12720942 |
| Porphyromonas gingivalis | Pgi | AroA•AroQ | N/A |
| Pseudomonas aeruginosa | Pae | AroQ•PheA | 11347571 |
| Pseudomonas aeruginosa | Pae | PchB | 2498748 |
| Pseudomonas aeruginosa | Pae | *AroQ | 11350384 |
| Pseudomonas fluorescens | Pfl | PchB | 1771427 |
| Pseudomonas stutzeri | Pst | AroQ•PheA | 5712091 |
| Pyrococcus furiosus | Pfu | AroQ | N/A |
| Salmonella typhimurium | Sty | AroQ•PheA | N/A |
| Salmonella typhimurium | Sty | AroQ•TyrA | N/A |
| Salmonella typhimurium | Sty | *AroQ | N/A |
| Shewanella putrefaciens | Spu | AroQ•PheA•AroA | N/A |
| Shewanella putrefaciens | Spu | AroQ•TyrA | N/A |
| Sinorhizobium meliloti | Sme | AroQ | N/A |
| Sinorhizobium meliloti | Sme | PchB | N/A |
| Staphylococcus aureus | Sau | AroQ•AroA | 13701532 |
| Streptomyces coelicolor | Sco | PchB | 5689912 |
| Streptomyces coelicolor | Sco | AroQ | 7320903 |
| Streptococcus mutans | Smu | AroQ | N/A |
| Streptococcus pneumoniae | Spn | AroQ | N/A |
| Streptococcus pyogenes | Spy | AroQ | 13621905 |
| Streptomyces pristinaespiralis | Spr | PapB | 1575338 |
| Thermotoga maritima | Tma | AroQ•PheA | 7437419 |
| Thiobacillus ferrioxidans | Tfe | AroQ•PheA | N/A |
| Vibrio cholerae | Vch | AroQ•PheA | 11270725 |
| Vibrio cholerae | Vch | AroQ•TyrA | 11270723 |
| Vibrio vulnificans | Vvu | PchB | 1657699 |
| Xanthomonas campestris | Xca | AroQ•PheA | 3153199 |
| Yersinia pestis | Ype | AroQ•PheA | N/A |
| Yersinia pestis | Ype | AroQ•TyrA | N/A |
| Yersinia pestis | Ype | *AroQ | N/A |
aN/A refers to sequences obtained from contigs of unfinished genomes at NCBI or TIGR. FASTA files are available from the authors.
Extract preparation
E. coli transformants or P. aeruginosa cells were grown at 37°C with vigorous shaking in LB or minimal medium supplemented with appropriate antibiotics. Cells were harvested in the late-exponential phase of growth by centrifugation and resuspended in 50 mM potassium phosphate buffer (pH 7.0), containing 1 mM dithiothreitol (DTT). The cells were disrupted by sonication using an Ultratip Labsonic System (Lab-Line Instruments), and then centrifuged at 150,000 × g for 65 min at 4°C. The supernatant fraction was collected and passed through a 1.5 × 5.5 cm DG-10 Sephadex column (Bio-Rad). The resulting desalted preparation is designated as crude extract.
Pellets from BL21(DE3), the BL21(pJB-1) clone, and S. typhimurium were suspended into 25 mM potassium phosphate buffer at pH 7.0 containing 1.0 mM DTT. Extracts were sonicated using three pulses to maintain cooling. The extract was ultracentrifuged (Beckman) at 150,000 × g and 4°C. Crude extracts to be used for enzyme assays were desalted on DG-10 columns (Bio-Rad).
Enzyme assays
For routine assays with unpurified protein preparations, chorismate mutase and prephenate dehydratase were assayed spectrophotometrically by the method of Cotton and Gibson [38] as modified by Ahmad and Jensen [18], using 1 mM chorismate and 1 mM prephenate, respectively. Arogenate dehydratase was assayed by measuring the formation of L-phenylalanine using high-performance liquid chromatography (HPLC) [17]. Aromatic aminotransferase was assayed as described before [34], and phenylalanine and α-ketoglutarate were used as the substrates, unless indicated otherwise. Shikimate dehydrogenase was assayed by monitoring the reduction of NADP+ at 340 nm [39]. Alkaline phosphatase was assayed as detailed by Brickman and Beckwith [40].
For kinetic determinations made with purified preparations of chorismate mutase, initial rates were followed as disappearance of chorismate. This was monitored spectrophotometrically at 274 nm using an extinction coefficient of 2,630 M/cm. Steady-state parameters of Km and kcat were calculated from initial rates using a series of substrate concentrations over a 15-fold range below and above Km. Protein concentrations were estimated by the Bradford assay [41] with bovine serum albumin as the protein standard.
Separation of periplasmic and cytoplasmic fractions of P. aeruginosa
A modified magnesium shock procedure [42] was used to fractionate cytoplasmic and periplasmic enzymes in P. aeruginosa. Cultures were grown to late-exponential phase in 300 ml of M-9 salts/glucose medium. The culture was divided into two 150-ml parts and harvested by centrifugation at 6,000 × g at 4°C for 20 min. One part was resuspended in 5 ml of 50 mM potassium phosphate buffer (pH 7.0), and broken by sonication to make crude extract. The other part was resuspended in 20 ml of concentrated magnesium solution (0.2 M MgCl2, 0.01 M Tris-HCl pH 8.4), incubated on ice for 20 min, then centrifuged at 6,000 × g at 4°C for 20 min. The resulting supernatant was dialyzed against 50 mM (pH 7.0) potassium phosphate buffer overnight (using at least ten times the volume of the buffer, with one change of the buffer after about 5–6 h), concentrated on a YM-10 membrane apparatus to 4 ml, and denoted as the periplasmic fraction. The remaining pellet was resuspended in 3 ml of 50 mM phosphate buffer (pH 7.0) and sonicated to yield the cytoplasmic fraction.
*PheC [16] activity (assayed as prephenate dehydratase) was the marker protein for the periplasmic fraction [13], and shikimate dehydrogenase activity was the marker protein for the cytoplasmic fraction.
Purification of *AroQ and a periplasmic aromatic aminotransferase from P. aeruginosa
Twenty-liter cultures of P. aeruginosa strain PAO1 (1 l per 2.8–1 flask) were grown to late-exponential phase in M-9 salts/glucose medium, and the periplasmic fraction was separated from the rest of the cells as described above, but with a scaled-up protocol. All the buffers contained 1 mM DTT.
A 2.5 × 16 cm Bio-Gel HTP column previously equilibrated with 5 mM phosphate buffer (pH 7.0) containing 1 mM DTT was loaded with a 240-mg amount of protein, washed with 500 ml of starting buffer, and bound proteins were eluted with 800 ml of phosphate buffer at pH 7.0 (potassium phosphate gradient from 5–400 mM). Fractions of 3.5 ml were collected, and chorismate mutase and aromatic aminotransferase activities were assayed.
Fractions with high chorismate mutase activity were pooled and concentrated by means of an Amicon YM-10 membrane. The concentrated preparation was dialyzed overnight against 50 mM phosphate buffer (pH 7.0) containing 1 mM DTT. An FPLC-connected Mono Q HR 5/5 anion-exchange column (Pharmacia Biotech) equilibrated with 50 mM phosphate buffer (pH 7.0) containing 1 mM DTT was loaded with the concentrated *AroQ fraction. The column was washed with 10 ml of the starting buffer, and bound proteins were gradient-eluted with 20 ml of phosphate buffer (KCl gradient between 0 to 1 M). Fractions of 0.5 ml were collected, and those showing high chorismate mutase activity were pooled and concentrated by means of an Amicon YM-10 membrane. The concentrated preparation was dialyzed against 50 mM Tris-HCl buffer (pH 8.4) containing 1 mM DTT overnight, loaded onto the same Mono Q column that had been equilibrated with the same Tris-HCl buffer. The same elution regimen was used, except that the KCl gradient was from 0 to 350 mM. Fractions showing high chorismate mutase activity were pooled and dialyzed overnight against the above phosphate buffer containing 1 mM DTT and 150 mM NaCl. The fractions were concentrated by an Ultrafree-15 centrifugal filter device (Millipore Corporation), and loaded onto an FPLC-connected Superdex 75 HR 10/30 (Pharmacia Biotech) column which had been equilibrated with 50 mM phosphate buffer (pH 7.0) containing 1 mM DTT and 150 mM NaCl. The same buffer was used for protein elution and fractions of 0.5 ml were collected. The fractions showing high chorismate mutase activity were pooled and concentrated by centrifugal filtration.
Two peaks with aromatic aminotransferase activity eluted from the hydroxylapatite column mentioned above. When the major fractions with highest levels of activity were pooled and subjected to Mono Q chromatography as described above, two peaks of activity were again resolved, one in the wash fraction (minor) and one in the gradient fraction (major). This heterogeneity might be explained by the apparent existence of a second, minor, cleavage site for the signal peptide (see Results). The major peak of aromatic aminotransferase was pooled and subjected to Superdex gel filtration as described above. The fractions showing high aromatic aminotransferase activity were pooled and concentrated by centrifugal filtration.
Molecular mass determination
The molecular mass of the native enzyme was determined by gel filtration using an FPLC-connected Superdex 75 HR 10/30 column (Pharmacia Biotech). The column was operated as described above using a flow rate of 0.5 ml/min. Calibration standards were cytochrome c (Mr 12,400), carbonic anhydrase (Mr 31,000), bovine serum albumin (Mr 66,000), and alcohol dehydrogenase (Mr 150,000). The void volume was determined by blue dextran (Mr 2,000,000). SDS-PAGE was carried out to determine the subunit molecular mass of the enzyme. Lysozyme (Mr 14,400), soybean trypsin inhibitor (Mr 21,500), carbonic anhydrase (Mr 31,000), ovalbumin (Mr 45,000), bovine serum albumin (Mr 66,000) and phosphorylase b (Mr 97,400) were used as molecular mass standards.
Biochemicals and chemicals
Ampicillin, kanamycin, α-ketoglutarate, shikimate, and amino acids were purchased from Sigma Chemical Company, DTT was from Research Organics, DEAE-cellulose was from Whatman, hydroxylapatite was from Bio-Rad. Molecular mass standards for gel filtration and for SDS-PAGE were from Sigma and Bio-Rad, respectively. Growth medium components and agar were from Difco. Chorismate, prephenate, and L-arogenate are routinely prepared in this laboratory as the accumulated products of mutant microbial strains.
Cloning of the Salmonella typhimurium *aroQ gene and expression in E. coli
Standard molecular genetics techniques were used [43]. The *aroQ gene was amplified by PCR using genomic DNA from S. typhimurium as template. The forward primer (5'-GTTATTCATATGATTCGTCATATCGCCATT-3') was designed to introduce an NdeI restriction site (CATATG) overlapping the start codon. The reverse primer (5'-GATAAACTCGAGT-TAACGCGCGTGTCGCACCA-3') was designed to introduce an XhoI site (CTCGAG) downstream of the stop codon. The PCR products were gel-purified using low melting temperature agarose and subjected to restriction enzyme digestion with NdeI and XhoI. The same two restriction enzymes were used to digest plasmid pET-24b+ DNA. The PCR fragments were ligated to the plasmid DNA and the ligation products were used to transform strain DH5α. Plasmid DNA from several kanamycin resistant colonies was used for DNA sequence analysis. A clone designated pJB-1 with the correct nucleotide sequence was used to transform strain BL21(DE3) for analysis of *aroQ expression. A well-isolated single colony was inoculated into 10 ml of LB/kanamycin medium and grown overnight at 30°C and 250 rpm. This culture was used to inoculate 100 ml of LB/kanamycin medium the following morning. Iso-propyl-thiogalactoside (IPTG) was added when cultures reached a density of about 0.6 A600, the cultures were harvested by centrifugation about 2.5 h later, and the pellets were used immediately to prepare extracts for enzyme assay.
GenBank accession numbers for the S. typhimurium *aroQ and the P. aeruginosa *aroQ and *aat sequences are AY039113, AF389865 and AF389866, respectively.
Analysis by SDS-PAGE and western blot
SDS-PAGE was performed with a Mini-PROTEIN II cell (Bio-Rad) using the method of Laemmli [44]. Samples from pellets of centrifuged cells were used to make crude extracts or were treated for whole-cell lysis. Crude extracts were usually diluted 1:1 (v/v) with solubilization buffer and heated to 100°C for 10 min. Whole-cell lysis was performed by resuspending the pellet in 2 × SDS sample buffer, vortexing vigorously to shear the chromosomal DNA, and heating to 70°C for about 5 min. Samples from 5 to 10 μl were loaded onto SDS-12% polyacrylamide gels. MultiMark (Novex) multicolored standards were used as protein markers in order to estimate the efficiency of protein transfer and to determine the molecular mass of proteins of interest.
A Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad), which transfers in a horizontal apparatus with a platinum-coated titanium plate as anode and a stainless steel plate as cathode, was used to transfer the protein from the mini-gels to nitrocellulose membranes sandwiched between Extra Thick Blot Absorbent Filter Paper (Bio-Rad) in about 15 to 30 min.
Antigens were detected with a chemiluminescence detection system (femtoLUCENT, GENO TECH). Membranes were blocked in 5% BLOT-QuickBlocker PBST buffer (plus 0.1% Tween-20), washed three times with PBST buffer (plus 0.1% Tween-20), incubated in primary antibody diluted in femto/TBST buffer in a tray for 90 min at room temperature (or overnight at 4°C), washed five times (as above), incubated in secondary antibody-HRP (horseradish peroxidase)-conjugate (goat anti-rabbit) as above, washed four times with femto/TBST buffer followed by transfer to detection buffer and detection reagent. The blotted membranes were placed protein sides up into the film cassette with the film (Kodak X-ray, full speed blue) on top and exposed for 10 to 20 sec.
Acknowledgments
Acknowledgements
This work was supported in part by NIH grant RCMI RR08168.
References
- Bentley R. The shikimate pathway: a metabolic tree with many branches. Crit Rev Biochem Mol Biol. 1990;25:307–384. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Walker DR, Koonin EV. Analogous enzymes: independent inventions in enzyme evolution. Genome Res. 1998;8:779–790. doi: 10.1101/gr.8.8.779. [DOI] [PubMed] [Google Scholar]
- Jensen RA, Gu W. Evolutionary recruitment of biochemically specialized subdivisions of Family-I within the protein superfamily of aminotransferases. J Bacteriol. 1996;178:2161–2171. doi: 10.1128/jb.178.8.2161-2171.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir LO, Nikolakakis A, Bonner CA, Jensen RA. Evidence for enzymatic formation of isoprephenate from isochorismate. Bioorg Med Chem Lett. 1993;7:1441–1446. [Google Scholar]
- Blanc V, Gil P, Bamas-Jacques N, Lorenzon S, Zagorec M, Schleuniger J. Identification and analysis of genes from Streptomyces pristinaespiralis encoding enzymes involved in the biosynthesis of the 4-dimethylamino-L-phenylalanine precursor of pristinamycin.I. Mol Microbiol. 1997;23:191–202. doi: 10.1046/j.1365-2958.1997.2031574.x. [DOI] [PubMed] [Google Scholar]
- Lee AY, Karplus PA, Ganem B, Clardy J. Atomic structure of the buried catalytic pocket of Escherichia coli chorismate mutase. J Am Chem Soc. 1995;117:3627–3628. [Google Scholar]
- Chook YM, Ke H, Lipscomb WN. Crystal structure of the monofunctional chorismate mutase from Bacillus subtilis and its complex with a transition state analog. Proc Natl Acad Sci USA. 1993;90:8600–8603. doi: 10.1073/pnas.90.18.8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacBeath G, Kast P, Hilvert D. A small, thermostable, and monofunctional chorismate mutase from the archeon Methanococcus jannaschii. Biochemistry. 1998;37:10062–10073. doi: 10.1021/bi980449t. [DOI] [PubMed] [Google Scholar]
- Sträter N, Hakansson K, Schnappauf G, Braus G, Lipsocomb WN. Crystal structure of the T state of allosteric yeast chorismate mutase and comparison with the R state. Proc Natl Acad Sci USA. 1996;93:3330–3334. doi: 10.1073/pnas.93.8.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goers SK, Jensen RA. The differential allosteric regulation of two chorismate-mutase isoenzymes of Nicotiana silvestris. Planta. 1984;162:117–124. doi: 10.1007/BF00410207. [DOI] [PubMed] [Google Scholar]
- Jensen R, Nester EW. The regulatory significance of intermediary metabolites: control of aromatic acid biosynthesis by feedback inhibition in Bacillus subtilis. J Mol Biol. 1965;12:468–481. doi: 10.1016/s0022-2836(65)80270-4. [DOI] [PubMed] [Google Scholar]
- Xia T, Song J, Zhao G, Aldrich H, Jensen RA. The aroQ encoded monofunctional chorismate mutase (CM-F) protein is a periplasmic enzyme in Erwinia herbicola. J Bacteriol. 1993;175:4729–4737. doi: 10.1128/jb.175.15.4729-4737.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Xia Aldrich TH, Jensen RA. Cyclohexadienyl dehydratase from Pseudomonas aeruginosa is a periplasmic protein. J Gen Microbiol. 1993;139:807–813. doi: 10.1099/00221287-139-4-807. [DOI] [PubMed] [Google Scholar]
- Gosset G, Bonner CA, Jensen RA. Microbial origin of "plant-type" 2-keto-3-D-arabino-heptulosonate 7-phosphate synthases, exemplified by the chorismate/tryptophan-regulated enzyme from Xanthomonas campestris. J Bacteriol. 2001;183:4061–4070. doi: 10.1128/JB.183.13.4061-4070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty M, Vonstein V, Overbeek R, Osterman A. Archaeal shikimate kinase, a new member of the GHMP-kinase family. J Bacteriol. 2001;183:292–300. doi: 10.1128/JB.183.1.292-300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Xia T, Fischer RS, Jensen RA. Cyclohexadienyl dehydratase from Pseudomonas aeruginosa: molecular cloning of the gene and characterization of the gene product. J Biol Chem. 1992;267:2487–2493. [PubMed] [Google Scholar]
- Fischer R, Jensen RA. Arogenate dehydratase. Methods Enzymol. 1987;142:503–507. doi: 10.1016/s0076-6879(87)42061-2. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Jensen RA. Phylogenetic distribution of components of the overflow pathway to L-phenylalanine within the enteric lineage of bacteria. Curr Microbiol. 1988;16:295–302. [Google Scholar]
- Pseudomonas Genome Project http://www.pseudomonas.com
- Neu HC, Heppel LA. The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts. J Biol Chem. 1965;40:3685–3692. [PubMed] [Google Scholar]
- Lambert KN, Allen KD, Sussex IM. Cloning and characterization of an esophageal-gland-specific chorismate mutase from the phytoparasitic nematode Meloidogyne javanica. Mol Plant Microbe Interact. 1999;12:328–336. doi: 10.1094/MPMI.1999.12.4.328. [DOI] [PubMed] [Google Scholar]
- Xie G, Brettin TS, Bonner CA, Jensen RA. Mixed-function supraoperons that exhibit overall conservation, albeit shuffled gene organization, across wide intergenomic distances within eubacteria. Microb Comp Genomics. 1999;4:5–28. doi: 10.1089/omi.1.1999.4.5. [DOI] [PubMed] [Google Scholar]
- Serino L, Reimann C, Baur H, Beyeler M, Visca P, Haas D. Structural genes for salicylate biosynthesis from chorismate in Pseudomonas aeruginosa. Mol Gen Genet. 1995;249:217–228. doi: 10.1007/BF00290369. [DOI] [PubMed] [Google Scholar]
- Subramaniam PS, Xie G, Xia T, Jensen RA. Substrate ambiguity of 3-deoxy-D -manno-octulosonate 8-phosphate synthase from Neisseria gonorrhoeae in the context of its membership in a protein family containing a subset of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthases. J Bacteriol. 1998;180:119–127. doi: 10.1128/jb.180.1.119-127.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin CM, Rayback TW, Skinner J. Role of catechol siderophore synthesis in Vibrio vulnificus virulence. Infect Immun. 1996;64:2834–2838. doi: 10.1128/iai.64.7.2834-2838.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff EE, Stoebner JA, Reed KE, Payne SM. Cloning of a Vibrio cholerae vibriobactin gene cluster: Identification of genes required for early steps in siderophore biosynthesis. J Bacteriol. 1997;179:7055–7062. doi: 10.1128/jb.179.22.7055-7062.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, Stenmark-Cox S, Jensen RA. Enzymological basis of reluctant auxotrophy for phenylalanine and tyrosine in Pseudomonas aeruginosa. J Biol Chem. 1978;253:2972–2978. [PubMed] [Google Scholar]
- Fiske MJ, Whitaker RJ, Jensen RA. Hidden overflow pathway to L-phenylalanine in Pseudomonas aeruginosa. J Bacteriol. 1983;154:623–631. doi: 10.1128/jb.154.2.623-631.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S, Weisburg WG, Jensen RA. Evolution of aromatic amino acid biosynthesis and application to the fine-tuned phylogenetic positioning of enteric bacteria. J Bacteriol. 1990;172:1051–1061. doi: 10.1128/jb.172.2.1051-1061.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/
- Bonner CA, Fischer RS, Ahmad S, Jensen RA. Remnants of an ancient pathway to L-phenylalanine and L-tyrosine in enteric bacteria: evolutionary implications and biotechnological impact. Appl Environ Microbiol. 1990;56:3741–3747. doi: 10.1128/aem.56.12.3741-3747.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Jensen RA. Monofunctional chorismate mutase from Serratia rubidaea: a paradigm system for the three-isozyme gene family of enteric bacteria. Arch Biochem Biophys. 1992;294:147–153. doi: 10.1016/0003-9861(92)90149-q. [DOI] [PubMed] [Google Scholar]
- Maddock JR, Shapiro L. Polar location of the chemoreceptor complex in the Escherichia coli cell. Science. 1993;259:1717–1723. doi: 10.1126/science.8456299. [DOI] [PubMed] [Google Scholar]
- Gu W, Zhao G, Eddy C, Jensen RA. Imidazole acetol phosphate aminotransferase in Zymomonas mobilis: molecular-genetic, biochemical and evolutionary analyses. J Bacteriol. 1995;177:1576–1584. doi: 10.1128/jb.177.6.1576-1584.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker RJ, Gaines CG, Jensen RA. A multi-specific quintet of aromatic aminotransferases that overlap different biochemical pathways in Pseudomonas aeruginosa. J Biol Chem. 1982;257:13550–13556. [PubMed] [Google Scholar]
- Silhavy TJ, Berman ML, Enquist LW, (Eds) Experiments with Gene Fusions Cold Spring Harbor, NY: Cold Spring Harbor Laboratory, 1984.
- Gibson F. Chorismic acid: purification and some chemical and physical studies. Biochem J. 1964;90:256–261. doi: 10.1042/bj0900256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotton RGH, Gibson F. The biosynthesis of phenylalanine and tyrosine: enzymes converting chorismic acid into prephenic acid and their relationship to prephenate dehydratase and prephenate dehydrogenase. Biochim Biophys Acta. 1965;100:75–88. doi: 10.1016/0304-4165(65)90429-0. [DOI] [PubMed] [Google Scholar]
- Chaudhuri S, Anton IA, Coggins JR. Shikimate dehydrogenase from Escherichia coli. Methods Enzymol. 1987;142:315–320. doi: 10.1016/s0076-6879(87)42042-9. [DOI] [PubMed] [Google Scholar]
- Brickman E, Beckwith J. Analysis of the regulation of Escherichia coli alkaline phosphatase synthesis using deletions and transducing phages. J Mol Biol. 1975;96:307–316. doi: 10.1016/0022-2836(75)90350-2. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Kessler K, Safrin M. The propeptide Pseudomonas aeruginosa elastase acts as an elastase inhibitor. J Biol Chem. 1994;269:22726–22731. [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual 2nd edn Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1989.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Jensen RA. The evolutionary history of two bifunctional proteins that emerged in the purple bacteria. Trends Biochem Sci. 1986;11:108–112. [Google Scholar]
- Gu W, Williams D, Aldrich H, Xie G, Gabriel DW, Jensen RA. The AroQ and PheA domains of the bifunctional P-protein from Xanthomonas campestris in a context of genomic comparison. Microb Comp Genomics. 1997;2:141–158. doi: 10.1089/omi.1.1997.2.141. [DOI] [PubMed] [Google Scholar]
- ExPASY Molecular Biology Server http://www.expasy.ch
- Nishioka M, Demerec M, Eisenstark A. Genetic analysis of aromatic mutants of Salmonella typhimurium. Genetics. 1967;56:341–351. doi: 10.1093/genetics/56.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry A, Bhatnagar RK, Jensen RA. Enzymic basis for leakiness of auxotrophs for phenylalanine in Pseudomonas aeruginosa. J Gen Microbiol. 1987;133:3257–3263. doi: 10.1099/00221287-133-11-3257. [DOI] [PubMed] [Google Scholar]
- Walker GE, Dunbar B, Hunter IS, Nimmo HG, Coggins JR. Evidence for a novel class of microbial 3-deoxy-D -arabino-heptulosonate-7-phosphate synthase in Streptomyces coelicolor A3(2), Streptomyces rimosus and Neurospora crassa. Microbiology. 1996;142:1973–1982. doi: 10.1099/13500872-142-8-1973. [DOI] [PubMed] [Google Scholar]
- Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]