

ABSTRACT
Chronic hepatitis B virus (HBV) infection is a global public health problem. Although the currently approved medications can reliably reduce the viral load and prevent the progression of liver diseases, they fail to cure the viral infection. In an effort toward discovery of novel antiviral agents against HBV, a group of benzamide (BA) derivatives that significantly reduced the amount of cytoplasmic HBV DNA were discovered. The initial lead optimization efforts identified two BA derivatives with improved antiviral activity for further mechanistic studies. Interestingly, similar to our previously reported sulfamoylbenzamides (SBAs), the BAs promote the formation of empty capsids through specific interaction with HBV core protein but not other viral and host cellular components. Genetic evidence suggested that both SBAs and BAs inhibited HBV nucleocapsid assembly by binding to the heteroaryldihydropyrimidine (HAP) pocket between core protein dimer-dimer interfaces. However, unlike SBAs, BA compounds uniquely induced the formation of empty capsids that migrated more slowly in native agarose gel electrophoresis from A36V mutant than from the wild-type core protein. Moreover, we showed that the assembly of chimeric capsids from wild-type and drug-resistant core proteins was susceptible to multiple capsid assembly modulators. Hence, HBV core protein is a dominant antiviral target that may suppress the selection of drug-resistant viruses during core protein-targeting antiviral therapy. Our studies thus indicate that BAs are a chemically and mechanistically unique type of HBV capsid assembly modulators and warranted for further development as antiviral agents against HBV.
IMPORTANCE HBV core protein plays essential roles in many steps of the viral replication cycle. In addition to packaging viral pregenomic RNA (pgRNA) and DNA polymerase complex into nucleocapsids for reverse transcriptional DNA replication to take place, the core protein dimers, existing in several different quaternary structures in infected hepatocytes, participate in and regulate HBV virion assembly, capsid uncoating, and covalently closed circular DNA (cccDNA) formation. It is anticipated that small molecular core protein assembly modulators may disrupt one or multiple steps of HBV replication, depending on their interaction with the distinct quaternary structures of core protein. The discovery of novel core protein-targeting antivirals, such as benzamide derivatives reported here, and investigation of their antiviral mechanism may lead to the identification of antiviral therapeutics for the cure of chronic hepatitis B.
KEYWORDS: antiviral agents, capsid assembly, hepatitis B virus
INTRODUCTION
Hepatitis B virus (HBV) is a small DNA virus that chronically infects 240 million people worldwide and causes 686,000 deaths annually due to various severe liver diseases, including cirrhosis, hepatocellular carcinoma (HCC), and liver failure (1, 2). The current standard care of chronic hepatitis B, including two formulations of alpha-interferon (IFN-α) and six nucleos(t)ide analogues (NUCs), can efficiently reduce viral load and prevent liver disease progression (3, 4) but fails to induce a durable off-drug control of HBV replication in the vast majority of treated patients (5). This is mainly due to their failure to completely suppress viral replication, eliminate the covalently closed circular DNA (cccDNA), the nuclear form of the HBV genome and transcriptional template of viral mRNA, and restore the dysfunctional host antiviral immune response against the virus (6, 7). Accordingly, the discovery and development of novel therapeutics to completely stop HBV replication and eradicate or functionally inactivate cccDNA, as well as activate the host antiviral immune response against HBV, are the primary goals toward finding a cure for chronic hepatitis B (8–10).
HBV is the prototype member of the Hepadnaviridae family and contains a relaxed circular DNA (rcDNA) genome, partially double stranded and 3.2 kb in length (11). In spite of being a DNA virus, HBV replicates its genome via the protein-primed reverse transcription of an RNA pregenome in a cytoplasmic nucleocapsid (12). HBV core protein is a small polypeptide of 183 amino acid residues but exists in infected hepatocytes as several distinct quaternary structures and plays multiple roles in the viral replication cycle (13, 14). Specifically, in addition to assembly into capsids with 120 core protein dimers, the core protein dimers assemble the pregenomic RNA (pgRNA)-viral DNA polymerase complex to form nucleocapsids, where HBV DNA synthesis takes place. It is important to note that the nucleocapsid is not an inert container for viral pgRNA and DNA. Rather, it is structurally metastable, functionally participates in the process of reverse transcriptional replication of viral DNA, and plays critical roles in the regulation of virion assembly and egress (15, 16) as well as rcDNA nuclear import, nucleocapsid uncoating, and cccDNA formation via de novo infection and intracellular amplification pathways (17–19). It has also been speculated that HBV core proteins may associate with the cccDNA minichromosome, in an as-yet-undefined structural nature, to modulate its structure and transcription activity (20–22). Intriguingly, the core protein had also been shown to recruit cytokine-induced DNA cytosine deaminase APOBEC3A or APOBEC3B to cccDNA minichromosome, which results in cytosine deamination and decay of cccDNA (23). This remarkable finding implies that core protein can also be “hijacked” by the host immune system for destabilization and elimination of cccDNA (24).
In the effort toward discovery and development of novel antiviral agents against HBV, five chemotypes of small molecules have, thus far, been reported to alter the kinetics and pathway of core protein assembly (10). While heteroaryldihydropyrimidines (HAPs), such as Bay 41-4109 and GLS4, misdirect capsid assembly to form noncapsid polymers of core proteins (25–28), all other reported chemotypes of core protein assembly modulators induce the formation of various sizes of capsids devoid of viral pgRNA and DNA polymerase (29–32). Structural biology studies suggest that HAPs, phenylpropenamides (PPAs), and sulfamoylbenzamides (SBAs) all bind to a hydrophobic pocket formed at the dimer-dimer interface near the C termini of core protein subunits, with contributions from two neighboring core protein dimers (33). Binding of these molecules in the HAP pocket induces large-scale allosteric conformational changes in core protein subunits and results in quaternary and/or tertiary structure changes of capsids (34, 35). Thus far, several core protein assembly modulators have been shown to inhibit HBV replication in mouse models in vivo and are currently under preclinical or clinical development (28, 36).
We report here the discovery and mechanistic analysis of a novel chemotype of core protein assembly modulators benzamide derivatives (BAs). Our results indicate that similar to SBAs, BAs promote the formation of empty capsids through specific interaction with HBV core protein by binding to the HAP pocket. We also provide evidence suggesting that the C-terminal arginine-rich domain of core protein does not play a role in SBA- and BA-induced alteration of HBV capsid assembly. However, unlike SBAs, BA compounds uniquely induce the formation of empty capsids that migrate significantly slower in native agarose gel electrophoresis in cells replicating a genotype C HBV containing core protein with A36V mutation than in cells with a wild-type (WT) HBV core protein. Our studies thus indicate that BAs are a chemically and mechanistically unique type of capsid assembly modulators and warranted for further development as antiviral agents for treatment of chronic hepatitis B.
RESULTS
Discovery and optimization of benzamide derivatives that inhibit HBV replication.
Taking advantage of an immortalized murine hepatocyte (AML12)-derived stable cell line (AML12HBV10) that supports robust HBV replication in a tetracycline-inducible manner (37), we developed a high-throughput assay to screen small-molecule libraries for inhibitors of HBV replication (29). The primary screening of 26,900 compounds revealed two groups of structurally distinct compounds, 36 sulfamoylbenzamide derivatives (SBAs) (29) and 4 benzamide derivatives (BAs), that selectively inhibited HBV replication. The chemical structures of two representative BAs that inhibit HBV replication with 50% effective concentrations (EC50s) of 2.3 μM (compound 1) and 9 μM (compound 2), without measurable cytotoxicity up to 50 μM by using the standard 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay, are presented in Fig. 1A. Our initial lead optimization efforts identified two compounds, designated BA-26019 and BA-38017 (Fig. 1A), that inhibited HBV replication with EC50s of 0.58 and 0.16 μM, respectively (Fig. 1B and C), for further mechanistic studies.
FIG 1.

Identification of benzamide derivatives (BAs) that inhibit HBV replication. (A) Chemical structures of two primary screening “hits,” two optimized lead benzamide derivatives, BA-26019 and BA-38017, as well as two reference sulfamoylbenzamide derivatives (DVR-23 and ENAN-34017) used in this study are presented. (B and C) AML12HBV10 cells were treated with the indicated concentrations of compound BA-26019 or BA-38017 for 2 days. Cytoplasmic HBV core DNAs were extracted and quantified by a qPCR assay and expressed as the percentage of the mock-treated controls. The means and standard deviations (n = 4) were plotted. The cytotoxicity was determined by an MTT assay.
BA treatment prevents formation of pgRNA-containing nucleocapsids.
To map the HBV replication step(s) inhibited by the BAs, AML12HBV10 cells were treated with the indicated concentrations of BA-26019 and BA-38017 for 48 h. As positive controls, the cells were also treated with the HBV DNA polymerase inhibitor entecavir (ETV) or capsid assembly modulators HAP derivative Bay 41-4109 (26, 38) and SBA derivative DVR-23 (29). As shown in Fig. 2, consistent with its antiviral mechanism, ETV treatment did not alter the levels of HBV pgRNA (Fig. 2A), core protein (Fig. 2B), encapsidated pgRNA (Fig. 2C), and capsids (Fig. 2D) but significantly reduced the amount of viral DNA (Fig. 2E). Also in agreement with previous observations and their antiviral mechanisms, the particle gel assay revealed that Bay 41-4109 treatment abolished capsid formation (Fig. 2D) and thus pgRNA encapsidation and DNA synthesis (Fig. 2C and E). In contrast, DVR-23 treatment did not alter the amount of capsids (Fig. 2D) but reduced the amounts of encapsidated pgRNA (Fig. 2C) and the capsid-associated HBV DNA (Fig. 2E). Similar to DVR-23, both BA-26017 and BA-38019 did not significantly alter the amounts of viral pgRNA (Fig. 2A), core protein (Fig. 2B), and total HBV capsids (Fig. 2D) but reduced encapsidated pgRNA and capsid-associated HBV DNA in a dose-dependent manner (Fig. 2C and E). The results thus imply that, similar to SBAs, the BAs did not inhibit the formation of capsids per se but did dose-dependently inhibit the formation of pgRNA-containing nucleocapsids, which precludes viral DNA synthesis.
FIG 2.
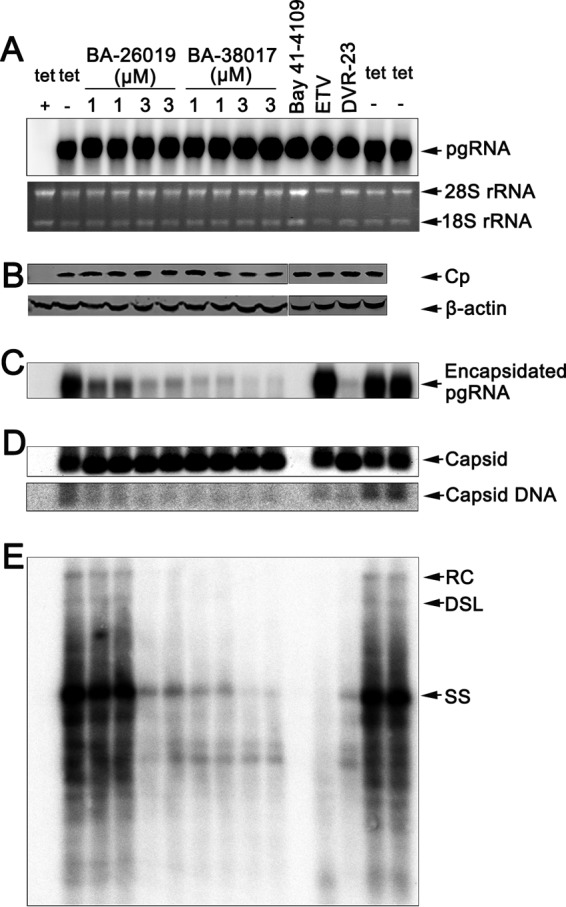
Antiviral mechanism of BA derivatives against HBV. AML12HBV10 cells were cultured in the presence of tetracycline (tet +) or in the absence of tetracycline and mock treated (tet −) or treated with the indicated concentrations of the compound BA-26019 (1 and 3 μM), BA-38017 (1 and 3 μM), ETV (1 μM), DVR-23 (2 μM), or Bay 41-4109 (2 μM) for 2 days. (A) Intracellular viral RNA was measured by Northern blotting hybridization. 28S and 18S rRNA were used as loading controls. (B) HBV core protein (Cp) expression was detected by Western blotting with a rabbit polyclonal antibody (Dako). β-Actin served as the loading control. The results were derived from two separate gels, as indicated. (C) Encapsidated pgRNA was determined by Northern blotting. (D) The total amounts of capsids were determined by a particle gel assay in a 1.0% agarose gel electrophoresis. Capsid-associated HBV DNA was detected by hybridization upon alkaline treatment of nucleocapsids on the membrane following the particle gel assay. (E) HBV DNA replication intermediates were extracted and determined by Southern blotting. A 32P-labeled full-length minus-strand-specific riboprobe was used for Southern blot analysis. RC, relaxed circular DNA; DSL, double-stranded linear DNA; SS, single-strand, negative-polarity DNA.
BAs alter capsid assembly and pgRNA encapsidation via specific interaction with HBV core protein.
HBV nucleocapsid assembly depends on the orchestrated interaction of multiple viral and host cellular components. The process begins with the binding of viral DNA polymerase, assisted by the host cellular heat shock protein 90 (HSP90) chaperone complex, to the stem-loop structure (ε) located at 5′-terminal region of pgRNA (39, 40). HBV core protein dimers are then recruited to assemble the protein-pgRNA complex into a nucleocapsid (41). In order to investigate the role of HSP90 chaperone complex in BA inhibition of HBV replication, we first compared the effects of BAs and HSP90 inhibitor DMAG [17-(dimethylaminoethylamino)-17-demethoxygeldanamycin] on HBV pgRNA encapsidation, capsid assembly, and DNA replication in AML12HBV10 cells. As expected, both BAs and DMAG efficiently reduced the amounts of encapsidated pgRNA and capsid-associated viral DNA (Fig. 3A and C) (42). Interestingly, by increasing the agarose concentration from 1.0 to 1.5% in the particle gel assay, HBV capsids in AML12HBV10 cells can be resolved into two distinct bands (Fig. 3B). In spite of significant reduction of HBV core DNA that comigrated with the slower-migrating capsids, ETV and DMAG treatment did not alter the migration profile of HBV capsids. However, similar to DVR-23 treatment, BA-26019 and BA-38017 treatment reduced the amounts of slow-migrating capsids but increased the amounts of fast-migrating capsids. Intriguingly, the residual amounts of HBV DNA in DVR-23- and BA-treated cells comigrated with the fast-migrating capsids (Fig. 3C). To investigate whether BA treatment induces the formation of smaller capsids, cytoplasmic capsids from mock-treated and BA-38017-treated AML12HBV10 cells were purified by ultracentrifugation and analyzed by negative-staining electron microscopy (EM). The capsids from BA-38017-treated cells are similar in size and morphology to those from mock-treated cells (Fig. 3D). However, although the molecular nature of two distinct populations of capsids remains to be determined, we speculated that similar to what occurs with DVR-23, the inhibition of HBV pgRNA encapsidation by the BA derivatives is mechanistically distinct from that of the HSP90 inhibitor and most likely occurs through interaction with core protein or other viral components.
FIG 3.

BA and SBA compounds alter HBV capsid migration in native agarose gel electrophoresis. AML12HBV10 cells were cultured in the presence of tetracycline (tet +) or in the absence of tetracycline and mock treated (tet −) or treated with BA-26019 (5 μM), BA-38017 (5 μM), Bay 41-4109 (2 μM), ETV (1 μM), DVR-23 (2 μM), or DMAG (0.15 μM) for 2 days. (A) Encapsidated pgRNA was extracted and detected by Northern blotting hybridization. (B) The capsids were separated in a 1.5% agarose gel electrophoresis, transferred onto a nylon membrane, and detected by a rabbit polyclonal antibody against core protein (Dako). (C) Capsid-associated HBV DNA was detected by hybridization with a 32P-labeled full-length HBV minus-strand-specific riboprobe upon alkaline treatment of the membrane following the particle gel assay. (D) Two representative electron micrographs of capsids purified from AML12HBV10 cells mock treated (upper panel) or treated with 5 μM BA-38017 (lower panel) for 2 days. Bar, 100 nm.
In order to identify the viral target of BA derivatives, we examined the effects of BAs on HBV capsids and capsid-associated DNA in HepG2 cells transfected with plasmids supporting HBV replication (pCMV-HBV or pHBV1.3), a pCMV-HBV-derived plasmid deficient in polymerase protein expression (pCMV-HBVΔpol), or a pHBV1.3-derived plasmid expressing a Y63F mutant DNA polymerase that is deficient in priming negative-strand DNA synthesis (pHBV1.3polY63F). Particle gel assays revealed that independent of the expression and functionality of viral DNA polymerase, Bay 41-4109 drastically reduced viral capsids. DVR-23 as well as the two BA derivatives induced the accumulation of the fast-migrating capsids (Fig. 4). The results thus suggest that like DVR-23, the BAs most likely target core protein, but not DNA polymerase, to inhibit pgRNA encapsidation.
FIG 4.
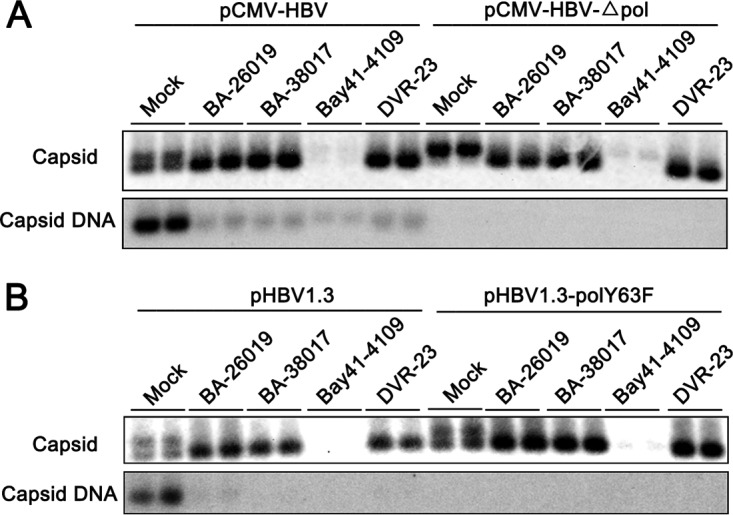
BA derivatives alter the capsid assembly in a viral DNA polymerase-independent manner. HepG2 cells were transiently transfected with plasmid pCMV-HBV or pCMV-HBV-Δpol (A) or with pHBV1.3 or pHBV1.3polY63F (B). Six hours posttransfection, the cells were mock treated or treated with 5 μM BA-26019, 5 μM BA-38017, 2 μM Bay 41-4109, or 2 μM DVR-23 for 48 h. The capsids were separated by 1.5% agarose gel electrophoresis, transferred onto a nylon membrane, and detected by a rabbit polyclonal antibody against HBV core protein (Dako). Capsid-associated HBV DNA was detected by hybridization with a 32P-labeled full-length HBV minus-strand-specific riboprobe upon alkaline treatment of the membrane following the particle gel separation.
Molecular docking studies suggest that BA and SBA derivatives bind to the HAP pocket.
The assembly of capsids from core protein dimers is driven by hydrophobic interactions at the dimer-dimer interface. Structural biology studies have identified a hydrophobic pocket, designated the HAP pocket, at the dimer-dimer interfaces near the C termini of core protein subunits (35). Binding of HAPs or PPAs in this pocket strengthens protein-protein interactions by filling that gap, which induces large-scale allosteric conformational changes in core protein subunits and results in quaternary and/or tertiary structure changes of capsids (34, 35). Residue V124 of core protein forms part of the wall of the HAP pocket. Replacing V124 with amino acid residues with a hydrophobic side chain of different size partially fills the HAP pocket and alters capsid assembly and interaction with capsid assembly modulators (43, 44). Specifically, the V124A mutation (Fig. 5A, residue 124 in sticks), in which a smaller amino acid is substituted, causes the pocket to open, unlike what is seen in the wild type, while the V124F and V124W mutations insert more steric bulk into the binding site and begin to close the pocket. In this study, we examined the effect of the core protein mutations on ligand binding using computational docking. The docking grid, which specifies the surface of the core protein to be sampled, was centered on residue W102, and the box dimensions were large enough to span the dimer-dimer interface. All the docked poses found the HAP binding site (Fig. 5B, C, and D), and all have a hydrogen bond with W102, a key interaction that is present in cocrystal structures with capsid assembly modulators (33–35). BA-38017 and ENAN-34017 were able to find binding poses on wild-type and all of the mutant core proteins, but for Bay 41-4109, only binding poses on wild-type and V124A were reported, with no poses being reported for the V124F and V124W mutations, suggesting that these mutations could prevent binding of Bay 41-4109. When examining the local binding energies' docking scores, the scores become less favorable when the mutations are introduced. Overall, the V124W mutation had the least-favorable scores, followed by V124F and then V124A (Fig. 5E).
FIG 5.
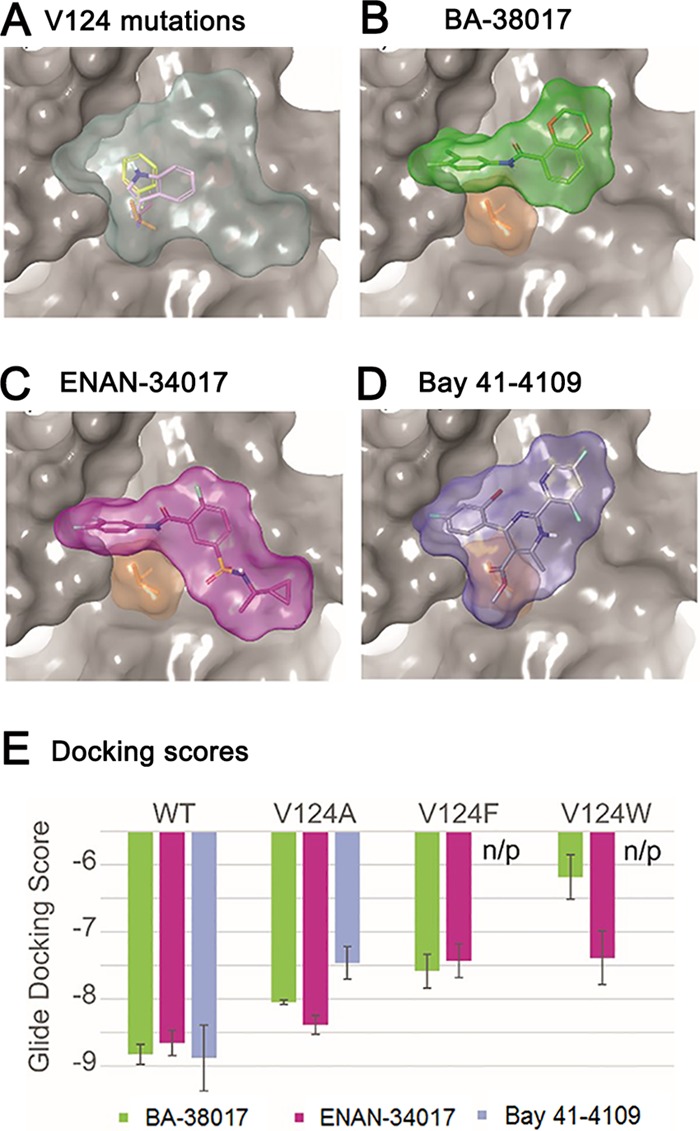
Structural simulation and docking analysis. (A) Binding site (light blue area) between the core protein dimer-dimer interface, with A, F, and W mutations of V124 shown as sticks. All the compounds bound at the HAP pocket. (B) Binding pose of 38017, green compound; (C) binding pose of 34017, magenta compound; (D) binding pose of Bay 41-4109, gray compound. V124 is shown in tan for each pose, and part of chain C was hidden to visualize the binding site. (E) Docking energies for each of the compounds with each mutant. n/p, no pose reported during docking.
Genetic evidence suggesting that BA and SBA derivatives bind to the HAP pocket.
In order to experimentally confirm that SBAs and BAs do bind the HAP pocket to disrupt capsid assembly and pgRNA encapsidation, we examined the effects of those compounds, with ETV and Bay 41-4109 as controls, on the capsid assembly and viral DNA replication in HepG2 cells transfected with pHBV1.3-derived plasmids with core protein V124A, V124L, V124F, or V124W mutation. In addition, a naturally occurring core protein mutation, F97L, which is not involved in the contact between core protein dimers but enhances both the rate and extent of capsid assembly relative to those for the wild-type core protein, was also included as a control (45). As shown in Fig. 6A, except for V124L mutant core protein that accumulated in the transfected cells at a reduced level, all other mutant core proteins accumulated in the transfected HepG2 cells to a level similar to that of cells transfected with parental pHBV1.3. However, particle gel assays showed that like wild-type core protein, F97L core protein assembled two distinct populations of capsids, but V124A, V124F, and V124W core proteins assembled predominantly slower- and fast-migrating capsids, respectively (Fig. 6B). Analyses of core DNA by quantitative PCR (qPCR) (Fig. 6C) and Southern blot (Fig. 6D) assays consistently demonstrated that compared to that in the cells transfected with wild-type HBV genome, the levels of HBV DNA replication intermediates in cells transfected with each of the genomes expressing mutant core proteins were significantly reduced. The reduced amount of core protein and undetectable levels of capsids and viral DNA in pHBV1.3coreV124L-transfected cells strongly imply that the V124L mutant core protein is incompetent to assemble stable capsids in HepG2 cells, and it was thus eliminated from further studies.
FIG 6.

Replacement of core protein valine 124 with amino acids with different sizes of hydrophobic side chains confers resistance to BA and SBA compound-induced alteration of capsid assembly. HepG2 cells were transfected with plasmid pHBV1.3 or pHBV1.3-derived plasmids expressing core protein with F97L, V124A, V124L, V124F, or V124W mutation. The cells were harvested at 48 h posttransfection. (A) HBV core protein (Cp) expression was detected by Western blotting with a rabbit polyclonal antibody (Dako). (B) Capsids and capsid-associated HBV DNA were detected by a particle gel assay. (C) Capsid-associated HBV DNAs in the transfected cells were quantified by a qPCR assay, and the results were plotted as the percentage of the amounts in pHBV1.3-transfected cells (n = 4). (D) HBV DNA replication intermediates were detected by Southern blotting. (E) HepG2 cells were transfected with plasmid pHBV1.3 or pHBV1.3-derived plasmids expressing core protein with F97L, V124A, V124L, V124F, or V124W. Six hours posttransfection, the cells were mock treated or treated with 5 μM BA-38017, 10 μM 3TC, 2 μM Bay 41-4109, or 5 μM ENAN-34017 for 48 h. Cytoplasmic capsids were determined by a 1.5% agarose particle gel assay. (F) Capsid-associated HBV DNAs in the transfected cells were quantified by a qPCR assay, and the results were plotted as the percentage of the amounts in pHBV1.3-transfected cells (n = 4). (G) HepG2 cells were cotransfected with pHBV1.3 and pHBV1.3coreV124F or pHBV1.3coreV124W in a molar ratio of 1:1. Six hours posttransfection, the cells were mock treated or treated with BA-38017 (5 μM), 3TC (10 μM), Bay 41-4109 (2 μM), or ENAN-34017 (5 μM) for 72 h. Cytoplasmic capsids were analyzed by a particle gel assay. Capsid-associated HBV DNA was quantified by hybridization with a 32P-labeled full-length HBV minus-strand-specific riboprobe.
Next, we tested the sensitivity of the mutant core proteins to the different chemotypes of capsid assembly modulators on assembly of capsids in human hepatoma cells. As shown in Fig. 6E and Table 1, while Bay 41-4109 efficiently abolished the accumulation of capsids and inhibited viral DNA replication in the cells transfected with pHBV1.3, pHBV1.3coreF97L, or pHBV1.3coreV124A, the HAP compound failed to alter the migration of capsids and did not inhibit viral DNA replication in the cells transfected with pHBV1.3coreV124W. In contrast, although Bay 41-4109 treatment efficiently induced the assembly of slower-migrating capsids in cells transfected with pHBV1.3coreV124F, the compound failed to reduce the viral DNA replication in these cells. Overall, these results are in agreement with the prediction of molecular docking results (Fig. 5E). Interestingly, while treatment of BA-38017 and ENAN-34017 induced the assembly of fast-migrating capsids in the cells transfected with pHBV1.3 or pHBV1.3coreF97L, the mutant core protein V124A was partially resistant to BA-38017-induced change of capsid assembly (Fig. 6E). Moreover, as shown in Table 1, HBV DNA replication in cells transfected with pHBV1.3coreV124A was 3.8- and 5-fold more resistant to BA-38017 and ENAN-34017, respectively, than that in cells transfected with wild-type pHBV1.3. In addition, while BA-38017 and ENAN-34017 did not alter the electrophoresis migration profile of capsids assembled from V124F and V124W core protein, HBV DNA replication in cells transfected with a V124F core protein-expressing replicon is sensitive to BA-38017 and ENAN-34017. In contrast, an HBV replicon expressing V124W core protein was 13- and 100-fold more resistant to BA-38017 and ENAN-34017, respectively, than wild-type HBV (Table 1). Those experimental data are generally in agreement with the predictions of molecular docking studies and thus strongly suggest that BA-38017 and ENAN-34017 modulate HBV capsid assembly by binding to the HAP pocket.
TABLE 1.
Antiviral activity of representative compounds against wild-type and mutant HBV
| Compound | Mean EC50 ± SD (μM) against HBV with indicated mutation(s)a |
||||||
|---|---|---|---|---|---|---|---|
| None (WT) | F97L | V124A | V124F | V124W | WT+V124F | VT+V124W | |
| Bay 41-4109 | 0.09 ± 0.01 | 0.13 ± 0.01 | 0.08 ± 0.02 | >2.00 | >2.00 | 0.09 ± 0.02 | 0.91 ± 0.09 |
| BA-38017 | 0.24 ± 0.01 | 0.67 ± 0.03 | 0.92 ± 0.13 | 0.23 ± 0.02 | 3.10 ± 1.07 | 0.47 ± 0.02 | 0.66 ± 0.10 |
| ENAN-34017 | 0.06 ± 0.01 | 0.05 ± 0.01 | 0.30 ± 0.03 | 0.03 ± 0.01 | 6.66 ± 0.80 | 0.04 ± 0.01 | 0.29 ± 0.08 |
From quadruplicate experiments.
BAs and SBAs efficiently modulate the assembly of chimeric HBV capsids.
It has been demonstrated that wild-type core protein can coassemble with mutant V124 core proteins to form chimeric capsids and partially rescue viral DNA replication (43) (Fig. 6F). However, it is possible that capsid assembly modulators can bind to wild-type core protein and efficiently disrupt the assembly of replication-competent chimeric nucleocapsids. To test this hypothesis, HepG2 cells were cotransfected with pHBV1.3 and pHBV1.3coreV124F or pHBV1.3coreV124W at a molar ratio of 1:1. The cells were mock treated or treated with the indicated compounds. As shown in Fig. 6G, two populations of capsids were detected in the cotransfected cells. Intriguingly, while Bay 41-4109 abolished the accumulation of the wild-type and V124F core protein chimeric capsids, the compound induced the formation of slow-migrating capsids in pHBV1.3- and pHBV1.3coreV124W-cotransfected cells. Consistent with the particle gel assay results, viral DNA replication in cells cotransfected with HBV replicons expressing wild-type and V124F core proteins was completely sensitive to Bay 41-4109. However, although the viral DNA replication in cells cotransfected with HBV replicons expressing wild-type and V124W core proteins became sensitive to Bay 41-4109, compared to wild-type capsids, it was still approximately 10-fold more resistant to Bay 41-4109 (Table 1). Interestingly, similar to what was seen in pHBV1.3-transfected cells, BA-38017 and ENAN-34017 induced the assembly of fast-migrating capsids in cells cotransfected with pHBV1.3 and pHBV1.3coreV124F or pHBV1.3coreV124W (Fig. 6G). As expected, BA-38017 and ENAN-34017 reduced the amount of viral DNA at a similar efficiency in cells transfected with pHBV1.3 or pHBV1.3coreV124F alone or in combination. However, compared to that in cells transfected with pHBV1.3coreV124W, viral DNA replication in cells cotransfected with pHBV1.3 and pHBV1.3coreV124W was approximately 5- and 30-fold more sensitive to BA-38017 and ENAN-34017, respectively (Table 1). Our results thus indicate that HBV core protein is a dominant target that can override the drug resistance of mutant core proteins, in a variable degree, in the context of chimeric capsid assembly.
The core protein C-terminal arginine-rich domain does not play a role in BA and SBA modulation of capsid assembly.
The amino acid residues 150 to 183 constitute the C-terminal domain (CTD) of HBV core protein. This arginine-rich domain contains 3 major and 4 minor phosphorylation sites and plays an important role in the regulation of pgRNA encapsidation, DNA replication, nucleocapsid uncoating, and nuclear transport of rcDNA (46–50). Because a recent report showed that phosphorylation of the three major serines in the CTD stabilized capsids (51), we tested whether the phosphorylation status of CTD modified the effects of capsid assembly modulators. To this end, plasmid encoding wild-type or mutant core protein (in which, at the three major or all seven potential phosphorylation sites, substitutions of either alanines to mimic unphosphorylated protein or glutamates to mimic phosphorylated protein were performed) was transfected into AML12 cells. As shown in Fig. 7A, expression of all the mutant core proteins resulted in the assembly of capsids with a higher speed of migration in native agarose gel electrophoresis. However, similar to the capsids assembled from wild-type core protein, Bay 41-4109 treatment completely abolished the accumulation of capsids, and BA-38017 and DVR-23 treatment efficiently induced the formation of faster-migrating capsids in cells expressing all the mutant core proteins (Fig. 7B). The result thus implies that the CTD phosphorylation status does not affect core protein interaction with the three tested capsid assembly modulators.
FIG 7.
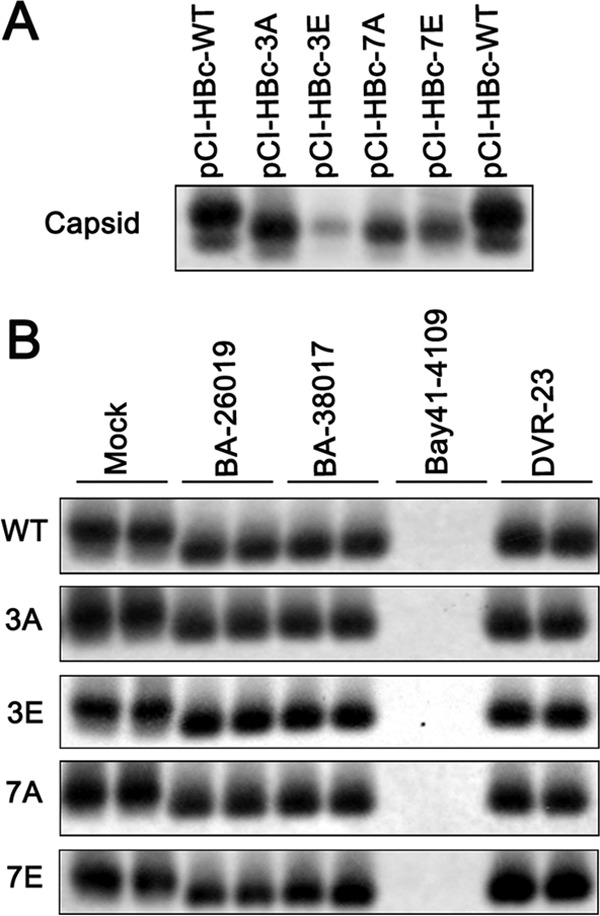
The phosphorylation status of the CTD does not affect BA and SBA modulation of capsid assembly. (A) AML12 cells were transfected with plasmid pCI-HBc, pCI-HBc-3A, pCI-HBc-3E, pCI-HBc-7A, or pCI-HBc-7E. The cells were harvested at 48 h posttransfection. The cytoplasmic capsids were analyzed by a particle gel assay. (B) AML12 cells were transfected with the indicated core protein-expressing plasmid. Six hours posttransfection, the cells were left untreated or treated with 5 μM BA-26019, 5 μM BA-38017, 2 μM Bay 41-4109, or 2 μM DVR-23 for 48 h. The cytoplasmic capsids were analyzed by a particle gel assay.
To further investigate the role of arginine-rich CTD in SBA and BA modulation of capsid assembly, a panel of pTRE2-derived plasmids expressing full-length or a series of C-terminally truncated core proteins were constructed (Fig. 8A). Their proper expression and the assembly of capsids in transfected AML12 cells were demonstrated by Western blot and particle gel assays (Fig. 8B). Consistent with a previous report, although Cp144 was able to assemble into capsids, Cp149 failed to accumulate in AML12 cells, presumably due to assembly of unstable capsids (46). Interestingly, like wild-type core protein, most of the truncated core proteins assembled into a major species of slower-migrating and minor species of faster-migrating capsids. However, the capsids assembled from all truncated core proteins, except Cp177 and Cp144, migrated slightly slower than those assembled from wild-type core protein (Fig. 8B, lower panel). As shown in Fig. 8C, while Bay 41-4109 treatment completely abolished the accumulation of capsids, BA-38017 and DVR-23 treatment efficiently induced the shift of slow-migrating capsid species toward fast-migrating species in cells transfected with plasmids expressing either full-length or each of the C-terminally truncated core proteins.
FIG 8.

The arginine-rich CTD of HBV core protein does not play a role in BA and SBA modulation of capsid assembly. (A) Sequences of wild-type or C-terminally truncated HBV core proteins expressed by plasmids. (B) AML12 cells were transfected with vector plasmid or a plasmid expressing either wild-type full-length core protein or the indicated C-terminally truncated core protein. The cells were harvested at 48 h posttransfection. Core protein (Cp) expression was detected by Western blotting (top), and β-actin served as the loading control (middle). The cytoplasmic capsids were analyzed by a particle gel assay (bottom). (C) AML12 cells were transfected with a plasmid expressing either wild-type core protein or the indicated C-terminally truncated core protein. Six hours posttransfection, the cells were left untreated (NT) or treated with 5 μM 26019, 5 μM BA-38017, 2 μM Bay 41-4109, or 2 μM DVR-23 for 48 h. The cytoplasmic capsids were analyzed by a particle gel assay.
Taken together, the results presented in Fig. 7 and 8 clearly demonstrated that although the CTD of core protein does not play a direct role in capsid assembly (14), posttranslational modification and mutation of CTD may change its exposure on the surface of capsids and/or alter the net charge of capsids, which consequentially alters the migration of capsids in native agarose gel electrophoresis. However, neither phosphorylation nor arginine-rich motifs of the CTD play a role in HAP, SBA, and BA modulation of capsid assembly.
BAs efficiently inhibit the replication of Nuc-resistant strains of HBV.
Long-term treatment of chronic hepatitis B with NUCs can lead to the emergence of drug-resistant HBV variants with specific mutations in the polymerase gene and result in treatment failure. Based on their distinct antiviral mechanism, all five previously reported chemotypes of capsid assembly modulators had been demonstrated to effectively inhibit the replication of NUC-resistant HBV (29, 52). Not surprisingly, as shown in Table 2, BA-38017 inhibited the replication of wild-type HBV and mutant HBV bearing specific mutations in the polymerase gene conferring resistance to lamivudine, adefovir, and/or entecavir with similar efficacy.
TABLE 2.
Antiviral activity of representative compounds against drug-resistant HBV mutants
| Compound | Mean EC50 ± SD (μM)a |
||||
|---|---|---|---|---|---|
| NUC sensitive, WT | ADV resistant |
3TC resistant, rtM204I | 3TC/ETV resistant, rtL180M, rtM204V, rtT184G, rtS202I | ||
| rtA181V | rtN236T | ||||
| 3TC | 0.029 ± 0.0007 | 0.110 ± 0.007 | 0.064 ± 0.09 | >10.000 | >10.000 |
| ETV | 0.001 ± 0.0001 | 0.002 ± 0.001 | 0.0002 ± 0.0001 | 0.055 ± 0.0007 | 1.100 ± 0.140 |
| BA-38017 | 1.04 ± 0.01 | 1.51 ± 0.18 | 1.91 ± 0.07 | 0.75 ± 0.07 | 1.59 ± 0.07 |
From duplicate experiments.
Furthermore, we also tested the effects of BA-38017 and ENAN-34017 on three genotype B and one genotype C HBV clones derived from Chinese patients that are sensitive or resistant to lamivudine or adefovir (ADV). Their GenBank accession numbers and drug resistance profiles are presented in Table 3. As expected, while pHY634 demonstrated a high resistance to lamivudine and modest resistance to ETV, pHY6923 and pHY6945 were modestly resistant to adefovir. Interestingly, particle gel assays revealed that all four HBV clones supported the accumulation of predominantly slow-migrating capsids in transfected HepG2 cells and that treatment with any of the three NUCs did not alter the electrophoresis migration profile of capsids in the cells (Fig. 9A). However, treatment with either BA-38017 or ENAN-34017 efficiently shifted capsids from slow- to fast-migrating species in cells transfected with pHY536207, pHY634, or pHY692. However, while ENAN-34017 treatment of pHY6945-transfected cells failed to induce the accumulation of faster-migrating capsids, BA-38017 treatment induced the accumulation of novel capsid species that migrated slower than that in untreated cells. Moreover, the total amount of capsids was reduced in pHY6945-transfected cells treated with ENAN-34017 but not in those treated with BA-38017 (Fig. 9A). Consistent with the observations made using the particle gel assay, examination of the SBA and BA compounds on intracellular HBV core DNA revealed that only pHY6945 is modestly resistant to BA-38017 (3-fold) and ENAN-34017 (6-fold); all other strains of HBV can be efficiently inhibited by the two compounds (Table 3).
TABLE 3.
Antiviral activities of representative compounds against drug-resistant HBV mutants
| Plasmid (GenBank accession no.) | Genotype | Mean EC50 ± SD (μM)a |
||||
|---|---|---|---|---|---|---|
| 3TC | ETV | ADV | BA-38017 | ENAN-34017 | ||
| pHY536207 (AY220698.1) | B | 0.040 ± 0.004 | 0.0002 ± 0.00003 | 0.017 ± 0.004 | 0.470 ± 0.012 | 0.029 ± 0.003 |
| pHY634 (AY220697.1) | B | >3.000 | 0.007 ± 0.004 | 0.028 ± 0.011 | 0.390 ± 0.047 | 0.021 ± 0.007 |
| pHY6923 (FJ518812.1) | B | 0.039 ± 0.005 | 0.0003 ± 0.00003 | 0.065 ± 0.009 | 0.420 ± 0.051 | 0.046 ± 0.007 |
| pHY6945 (FJ518813.1) | C | 0.032 ± 0.005 | <0.0002 | 0.057 ± 0.009 | 1.500 ± 0.565 | 0.180 ± 0.038 |
From quadruplicate experiments.
FIG 9.

Effects of BA and SBA compounds on capsid assembly and DNA replication of clinically isolated strains of HBV. (A) HepG2 cells were transfected with the indicated plasmid containing a replication-competent HBV genome derived from Chinese patients. Six hours posttransfection, the cells were mock treated or treated with BA-38017 (5 μM), ETV (0.125 μM), 3TC (1 μM), ADV (0.2 μM), or ENAN-34017 (5 μM) for 72 h. Cytoplasmic capsids were analyzed by a particle gel assay. Capsid-associated HBV DNA was quantified by hybridization with a 32P-labeled full-length HBV minus-strand-specific riboprobe upon alkaline treatment of the membrane following the particle gel assay. (B) Amino acid sequence alignment with highlights of mutations. (C) HepG2 cells were transfected with plasmid pTRE2-HBc or pTRE2-HBcA36V. The transfected cells were treated with BA-38017 (5 μM), Bay 41-4109 (2 μM), 3TC (1 μM), or ENAN-34017 (5 μM) for 72 h. Cytoplasmic capsids were analyzed by a particle gel assay.
Interestingly, as shown in Fig. 9B, the core protein encoded by pHY634 has 7 amino acid substitutions that are distant from the dimer-dimer interface, whereas the core protein of pHY6945 bears an A36V substitution that is approximately 10 Å from the HAP pocket. However, molecule docking analysis revealed no differences in binding pose or docking scores of BA-38017 or ENAN-34017 to wild-type and A36V mutant core proteins. This may be due to the long-range changes introduced by this mutation not being sampled by the current simulations. To experimentally test if this point mutation confers resistance to the SBA and BA compounds, HepG2 cells were transfected with pTRE2-derived plasmid expressing either wild-type or A36V mutant core protein and treated with the indicated compounds for 48 h. In agreement with the results presented above, particle gel assays showed that while both BA-38017 and ENAN-34017 induced the accumulation of fast-migrating capsids in cells expressing wild-type core protein, ENAN-34017 treatment did not alter A36V capsid migration but apparently reduced the amount of capsids. In contrast, BA-38017 treatment induced the accumulation of slower-migrating A36V capsids (Fig. 9C).
In summary, the results presented in this section further support the notion that the antiviral activity of SBA and BA compounds depends solely on core protein and not polymerase. Moreover, although both SBAs and BAs modulate capsid assembly by binding to the HAP pocket, the two compounds distinctly interact with core protein and induce different structural alterations of capsids (53).
DISCUSSION
Traditionally, virus-encoded enzymes have been the primary antiviral targets (54–56). However, nonenzymatic viral structural and nonstructural proteins have now been demonstrated to be viable targets of highly selective and potent antiviral agents, as highlighted by the FDA approval of HCV NS5A inhibitors for treatment of chronic hepatitis C (57) as well as the development of antiviral agents against distinct nonstructural proteins of many other RNA viruses (58). The genomes of all the viruses are wrapped with capsid protein(s) to form nucleocapsids. Unlike viral enzymes, which usually have their host cellular homologues, host cells do not encode any proteins that are structurally and functionally similar to viral capsid proteins. Therefore, viral capsid proteins are attractive and highly selective antiviral targets. Indeed, disruption of, or interference with, nucleocapsid assembly and/or disassembly (uncoating) with small-molecular capsid protein allosteric modulators represents a new frontier in the development of novel antiviral agents against many medically important viruses, such as human immunodeficiency virus (HIV) (59), HBV (14, 60), and dengue virus (61).
The results reported here extend our efforts in the discovery and development of HBV core protein-targeting antiviral agents and demonstrate a novel chemotype of small molecular compounds, benzamide derivatives, that specifically disrupt HBV core protein assembly through binding to the hydrophobic HAP pocket between core protein dimer-dimer interfaces. Similar to SBAs and PPAs, the BAs induce the formation of capsids that are devoid of viral RNA and migrate faster in native agarose gel electrophoresis than do wild-type capsids. However, it is not yet known how those capsid assembly modulators reduce pgRNA encapsidation and favor the empty-capsid assembly. One possibility is that those compounds interact with core protein dimers to accelerate the assembly of empty capsids, which results in the depletion of core protein dimers for encapsidation of the pgRNA-viral DNA polymerase complex. Alternatively, encapsidation of the pgRNA-viral DNA polymerase complex requires distinct interactions between core protein subunits and binding of core protein assembly modulators alters the geometry of interdimer contacts and consequentially inhibits nucleocapsid assembly. Interestingly, although the phosphorylation and the arginine-rich motifs in the CTD of core protein have been demonstrated to play important roles in pgRNA encapsidation via unknown mechanisms, our studies strongly suggest that the CTD does not play a significant role in HAP, BA, or SBA modulation of capsid assembly.
It is quite interesting that binding of different core protein assembly modulators into the HAP pocket induces the formation of different assembled products, ranging from noncapsid polymers to empty capsids of different sizes, as revealed by electron microscopic (EM) observation. Moreover, determination of the crystal structure of the HBV capsids in complex with three different HAPs demonstrated that each HAP compound induces distinct allosteric responses of core protein and results in different quaternary and tertiary structural changes of core protein subunits in assembled capsids (53). In this study, we showed that a 1.5% native agarose gel electrophoresis-based particle gel assay is a very sensitive and convenient method to reveal the alterations of assembled capsids, due to either core protein mutation or treatment of capsid assembly modulators. In principle, alteration of electrophoresis mobility in the native agarose gel may indicate the change of capsids in size, shape, and/or net charge on surface. However, the correlation between the capsid electrophoresis mobility shifts and structural/biophysical changes of capsids remains to be established. Particularly, although previous cryo-EM studies showed that full-length and C-terminally truncated (Cp154, Cp164, and Cp167) core proteins self-assembled into structurally very similar capsids (62), the particle gel assay clearly demonstrated that the C-terminally truncated cores migrated slower than did wild-type cores (Fig. 8). Moreover, despite the fact that EM analysis did not reveal differences in size between the capsids from untreated and BA-38017-treated cells (Fig. 3D), induction of faster-migrating-capsid formation from wild-type and most of the tested mutant core proteins is a common feature of SBA and BA compounds. Similarly, filling the HAP pocket with larger amino acid side chains also induces the formation of faster-migrating capsids (Fig. 6). Interestingly, as shown in Fig. 3, the residual HBV DNAs in SBA- and BA-treated cells are also associated with the faster-migrating capsids. Hence, formation of faster-migrating capsids should be one of the reliable diagnostic parameters for compounds that bind the HAP pocket and allosterically alter capsid assembly. In contrast, failure to induce the formation of faster-migrating capsids is an indicator of resistance of a core protein to a capsid assembly modulator. However, the formation of slower-migrating A36V capsids in BA compound-treated cells is an exception. In addition, Bay 41-4109 treatment also induced the assembly of slower-migrating V124F capsids. Those observations imply that the structures of core protein assembly products are determined by distinct interactions between the specific core assembly modulators and core proteins. Nevertheless, further investigation with additional mutant HBV core proteins and capsid assembly modulators, in combination with structural biological and biophysical analyses of the assembled capsids, is required to establish the structural and biophysical basis for the electrophoresis mobility changes of capsids.
The emergence of drug-resistant viruses frequently results in treatment failure with direct-acting antiviral agents. This is, particularly, a tremendous problem for chronic viral infections requiring a long-term antiviral therapy. The common practice to reduce the selection of drug resistance is combination therapy with antivirals targeting multiple viral functions (63, 64). However, although drug-resistant viruses usually exist in inocula and pretreated individuals at a very low frequency and can be selected during antiviral therapies, a drug-resistant mutation first occurs in an infected cell in the presence of many drug-susceptible genomes. For antiviral drugs that target monomeric viral enzymes and other proteins, the presence of drug-sensitive genomes and their transcribed/translated products in infected cells does not prevent the selection of drug-resistant viruses (65). In contrast, for drugs targeting the packaging and function of oligomeric viral structures assembled in trans from single or multiple viral components, such as capsids, cccDNA minichromosomes, and ribonucleoprotein (RNP) complexes, the oligomeric structures that contain drug-resistant subunits will also contain drug-susceptible ones in infected cells. If the ratio of susceptible to resistant subunits is high enough, the assembly and function of the chimeric structures will be susceptible to the antiviral agents (65). Such genetically dominant antiviral targets have been demonstrated recently for envelope protein of vesicular stomatitis virus (66) and capsids of poliovirus (67) and dengue virus (68). In this study, we provide strong evidence suggesting that the assembly of chimeric capsids from wild-type and drug-resistant core proteins is susceptible to multiple capsid assembly modulators (Fig. 6). Hence, HBV core protein is also a dominant antiviral target. Unlike DNA polymerase inhibitors, treatment of capsid assembly modulators may suppress the selection of drug resistance. Due to the lack of cell culture systems supporting efficient multiple rounds of HBV infection and spread (22, 69), this unique therapeutic feature of capsid assembly modulators can be examined only in future clinical trials.
In conclusion, the work reported here strongly suggests that the interaction of different capsid assembly modulators with core protein dimers induces distinct allosteric changes and results in the assembly of structurally different core protein oligomers and capsids. Considering the multiple roles of core protein in the HBV life cycle, it can be anticipated that pharmacological targeting of core protein not only should disrupt pgRNA encapsidation but also may alter core protein metabolism and function in other aspects of HBV replication and virus-host interaction, such as formation and transcriptional activity of cccDNA as well as activation of the host immune response (17, 24, 70). The discovery of novel core protein-targeting molecules, such as the benzamide derivatives reported here, and investigation of their antiviral mechanism and extended biological consequences should ultimately lead to the identification of core protein-targeting antiviral agents that potently suppress HBV replication and cure chronic hepatitis B.
MATERIALS AND METHODS
Cell culture and reagents.
Human hepatoblastoma cell line HepG2 and immortalized mouse hepatocytes AML12 were maintained in Dulbecco's modified Eagle medium (DMEM)/F12 medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. AML12HBV10 is an AML12-derived stable cell line supporting replication of a stably transfected envelope protein-deficient HBV genome in a tetracycline-inducible manner (37, 71). This cell line was maintained in DMEM/F12 medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml tetracycline, and 400 μg/ml G-418. Removal of tetracycline from the culture medium will initiate HBV pgRNA transcription and DNA replication. Lamivudine (LAM) and adefovir (ADV) were purchased from Sigma. Entecavir (ETV) was provided by William S. Mason at Fox Chase Cancer Center, Philadelphia, PA. Bay 41-4109 was provided by Lai Wei at the Hepatology Institute of Beijing University, China. Sulfamoylbenzamides and benzamine derivatives used in this studies were synthesized in-house (29). 17-DMAG was purchased from InvivoGen. Rabbit anti-HBc antibody was obtained from Dako (catalog number B0586).
Plasmids.
pCMV-HBV expressing HBV pgRNA under the control of the cytomegalovirus (CMV) immediate early promoter, wild-type HBV replicon pHBV1.3mer, and pCMV-HBV-Δpol have been described previously (72–74). The pTRE2-derived plasmids containing a wild-type or mutant HBV genome in DNA polymerase gene, including pTREHBV, pTREHBV/rtA180V, pTREHBV/rtN236T, pTREHBV/rtM204I, pTREHBV/rtL180M, and pTREHBV/rtL180M/rtM204V/rt184G/rtS202I, were constructed and reported previously (71, 75). Information on the sequence and construction of the wild-type or NUC-resistant HBV genotype B and C plasmids, including pHY536207, pHY634, pHY6923, and pHY6945, has been published previously (76). The plasmids pCI-HBc-WT, pCI-HBc-3A, pCI-HBc-3E, pCI-HBc-7A, and pCI-HBc-7E were gifts of Jianming Hu at Pennsylvania State University (46).
pHBV1.3-derived plasmids expressing mutant polymerase Y63F or mutant core proteins with F97L, V124A, V124F, V124L, or V124W mutation were generated by an overlapping PCR strategy. For instance, to generate the V124A mutant core protein-expressing HBV replicon plasmid, two DNA fragments were amplified with F172 sense primer (5′-ATATATGCATGCGTGGAACCTTTTCGGCTCCTCTG-3′) and V124A antisense primer (5′-GGAGTGCGAATCCACGCTCCGAAAGACACCA-3′) or V124A sense primer (5′-TGGTGTCTTTCGGAGCGTGGATTCGCACTCC-3′) and R2116 antisense primer (5′-ATATATGAATTCCACTGCATGGCCTGAGGATGAGTGTT-3′). The PCR products were purified and mixed together in a molar ratio of 1:1. The mixture was amplified with F172 and R2116 primers. The resultant DNA fragment was digested with SphI and EcoR I for replacement of the corresponding DNA segment in pHBV1.3. The resulting plasmid was designated pHBV1.3core-V124A. For the construction of plasmids expressing full-length and C-terminally truncated core proteins, the HBV genome regions encoding the full-length and the C-terminally truncated core proteins were amplified by PCR using pHBV1.3 as a template. The PCR products were cloned into a pTRE2 vector. All plasmids were confirmed by DNA sequencing. Sequences of the primers used in the construction of those recombinant plasmids are available upon request.
Antiviral activity and cytotoxicity assays in AML12HBV10 cells.
AML12HBV10 cells were seeded into 24-well plates at a density of 1 × 105 cells per well and cultured in DMEM/F-12 medium with 1 μg/ml tetracycline. One day after seeding, the cells were mock treated or treated with a serial dilution of compounds, ranging from 10 μM to 0.16 μM, for 2 days in the absence of tetracycline. Intracellular HBV core DNA was extracted as described previously (71) and quantified by a real-time PCR assay using the LightCycler 480 SYBR green I Master PCR kit (Roche) with primers 5′-GGCTTTCGGAAAATTCCTATG-3′ (sense) and 5′-AGCCCTACGAACCACTGAAC-3′ (antisense). The PCR was carried out as follows: denaturation at 95°C for 5 min, followed by 40 cycles of amplification consisting of 95°C for 15 s and 60°C for 30 s. The antiviral efficacy of a compound was expressed as the concentration that reduced the amount of HBV DNA by 50% (EC50) in comparison with the levels of the mock-treated controls.
To determine the cytotoxicity, AML12HBV10 cells were seeded into 96-well plates at a density of 2 × 104 cells per well and cultured in DMEM/F-12 medium with 10% fetal bovine serum in the absence of tetracycline. One day after seeding, cells were left untreated or treated with a serial dilution of testing compounds, ranging from 50 μM to 0.2 μM, for 2 days. The cell viability was measured by an MTT assay by following a procedure provided by the manufacturer (Invitrogen). The cytotoxicity of a compound was expressed as the concentration of compound that reduced the viability of the cells by 50% (50% cytotoxic concentration [CC50]).
Electron microscopic analysis of capsids.
AML12HBV10 cells were mock treated or treated with 5 μM BA-38017 for 2 days. HBV capsids in the cell lysates were purified by sucrose gradient centrifugation (18) and detected by EM after negative staining with UranyLess (Electron Microscopy Sciences).
Computational analyses of the binding affinity of core protein assembly modulators.
The HBV core protein sequence used in the simulations was downloaded from the Protein Data Bank, identification number 5e0i (77). The protein structure was prepared using the Protein Preparation Wizard, a module in Schrödinger's Small Molecule Drug Discovery Suite (78). Each mutant was generated with an amino acid replacement and then subjected to energy minimization prior to docking. The minimization method used was Polak-Ribier conjugation gradient (PRCG) with a maximum number of iterations of 2,500, a convergence on a gradient, and a convergence threshold of 0.05. Ligands were prepared using the ligand preparation tool and were generated at possible states at a target pH of approximately 7.0 ± 0.5. Ligands were docked using extraprecise docking in the Glide module. The docking grid was centered on residue W102 with box dimensions of 20 Å. The energy minimization and docking protocols were run in triplicate to generate error bars.
Transient-transfection assay.
HepG2 or AML12 cells were seeded in 24-well plates and grown to approximately 70% confluence. Cells were then transfected with 0.25 μg desired plasmid(s) using 1.25 μl Lipofectamine 2000 (Invitrogen) per well. Six hours posttransfection, the culture media were replaced with fresh media or media containing the desired concentrations of compounds and cultured for an additional 72 h. Intracellular HBV core protein, encapsidated pgRNA, and core DNA were examined with the assays specified below.
HBV nucleic acid analyses.
Total cellular RNA was extracted with TRIzol reagents (Invitrogen). Encapsidated viral RNA was extracted as described previously (79). Three micrograms of total RNA or one-half of the encapsidated RNA from each well of the 12-well plates was resolved in a 1.5% agarose gel containing 2.2 M formaldehyde and transferred onto a Hybond-XL membrane in 20× SSC buffer (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). For the detection of HBV RNA, membranes were probed with an [α-32P]UTP-labeled plus-strand-specific full-length HBV riboprobe. HBV core DNA from AML12HBV10 cells or transiently transfected HepG2 cells was extracted as previously described and quantified by either a qPCR or a Southern blot hybridization assay and probed with an [α-32P]UTP-labeled minus-strand-specific full-length HBV riboprobe (29).
Western blot assay.
Cell monolayers were washed once with phosphate-buffered saline (PBS) buffer and lysed with 1× Laemmli buffer. A fraction of cell lysate was separated on 12% Nupage bis-Tris gels (Invitrogen) and electrophoretically transferred onto a nitrocellulose membrane (0.45 μm; Millipore, Billerica, MA). Membranes were probed with antibodies against HBc (Dako) or β-actin (Millipore). Bound antibody was revealed by IRDye secondary antibodies and visualized by a Li-COR Odyssey system.
Particle gel assay.
HBV capsids and associated viral DNA were analyzed by a native agarose gel electrophoresis-based assay (37).
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health, USA (AI113267), and The Commonwealth of Pennsylvania through the Baruch S. Blumberg Institute.
REFERENCES
- 1.GBD 2013 Mortality and Causes of Death Collaborators. 2015. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385:117–171. doi: 10.1016/S0140-6736(14)61682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 3.Dienstag JL. 2009. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology 49:S112–S121. doi: 10.1002/hep.22920. [DOI] [PubMed] [Google Scholar]
- 4.Perrillo R. 2009. Benefits and risks of interferon therapy for hepatitis B. Hepatology 49:S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- 5.Block TM, Gish R, Guo H, Mehta A, Cuconati A, Thomas London W, Guo JT. 2013. Chronic hepatitis B: what should be the goal for new therapies? Antiviral Res 98:27–34. doi: 10.1016/j.antiviral.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang J, Guo F, Zhao X, Guo JT. 2014. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta Pharm Sin B 4:248–257. doi: 10.1016/j.apsb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang P, Liu F, Guo F, Zhao Q, Chang J, Guo JT. 2016. Characterization of novel hepadnaviral RNA species accumulated in hepatoma cells treated with viral DNA polymerase inhibitors. Antiviral Res 131:40–48. doi: 10.1016/j.antiviral.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang TJ, Block TM, McMahon BJ, Ghany MG, Urban S, Guo JT, Locarnini S, Zoulim F, Chang KM, Lok AS. 2015. Present and future therapies of hepatitis B: from discovery to cure. Hepatology 62:1893–1908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang J, Guo JT. 2015. Treatment of chronic hepatitis B with pattern recognition receptor agonists: current status and potential for a cure. Antiviral Res 121:152–159. doi: 10.1016/j.antiviral.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang L, Zhao Q, Wu S, Cheng J, Chang J, Guo JT. 2017. The current status and future directions of hepatitis B antiviral drug discovery. Expert Opin Drug Discov 12:5–15. doi: 10.1080/17460441.2017.1255195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Block TM, Guo H, Guo JT. 2007. Molecular virology of hepatitis B virus for clinicians. Clin Liver Dis 11:685–706, vii. doi: 10.1016/j.cld.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. doi: 10.1016/0092-8674(82)90157-X. [DOI] [PubMed] [Google Scholar]
- 13.Venkatakrishnan B, Zlotnick A. 2016. The structural biology of hepatitis B virus: form and function. Annu Rev Virol 3:429–451. doi: 10.1146/annurev-virology-110615-042238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. 2015. Core protein: a pleiotropic keystone in the HBV lifecycle. Antiviral Res 121:82–93. doi: 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruss V. 2007. Hepatitis B virus morphogenesis. World J Gastroenterol 13:65–73. doi: 10.3748/wjg.v13.i1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pairan A, Bruss V. 2009. Functional surfaces of the hepatitis B virus capsid. J Virol 83:11616–11623. doi: 10.1128/JVI.01178-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo JT, Guo H. 2015. Metabolism and function of hepatitis B virus cccDNA: implications for the development of cccDNA-targeting antiviral therapeutics. Antiviral Res 122:91–100. doi: 10.1016/j.antiviral.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui X, Guo JT, Hu J. 2015. Hepatitis B virus covalently closed circular DNA formation in immortalized mouse hepatocytes associated with nucleocapsid destabilization. J Virol 89:9021–9028. doi: 10.1128/JVI.01261-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bock CT, Schwinn S, Locarnini S, Fyfe J, Manns MP, Trautwein C, Zentgraf H. 2001. Structural organization of the hepatitis B virus minichromosome. J Mol Biol 307:183–196. doi: 10.1006/jmbi.2000.4481. [DOI] [PubMed] [Google Scholar]
- 21.Schultz U, Summers J, Staeheli P, Chisari FV. 1999. Elimination of duck hepatitis B virus RNA-containing capsids in duck interferon-alpha-treated hepatocytes. J Virol 73:5459–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi Y, Gao Z, Xu G, Peng B, Liu C, Yan H, Yao Q, Sun G, Liu Y, Tang D, Song Z, He W, Sun Y, Guo JT, Li W. 2016. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog 12:e1005893. doi: 10.1371/journal.ppat.1005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T. 2014. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Block TM, Guo JT. 2016. The covalently closed circular form of hepatitis B virus genome: is there now an end in “site”? Gastroenterology 150:34–36. doi: 10.1053/j.gastro.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 25.Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. 2005. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci U S A 102:8138–8143. doi: 10.1073/pnas.0409732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stray SJ, Zlotnick A. 2006. BAY 41-4109 has multiple effects on hepatitis B virus capsid assembly. J Mol Recognit 19:542–548. doi: 10.1002/jmr.801. [DOI] [PubMed] [Google Scholar]
- 27.Bourne CR, Finn MG, Zlotnick A. 2006. Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1. J Virol 80:11055–11061. doi: 10.1128/JVI.00933-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu G, Liu B, Zhang Y, Li J, Arzumanyan A, Clayton MM, Schinazi RF, Wang Z, Goldmann S, Ren Q, Zhang F, Feitelson MA. 2013. Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly. Antimicrob Agents Chemother 57:5344–5354. doi: 10.1128/AAC.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. 2013. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol 87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.King RW, Ladner SK, Miller TJ, Zaifert K, Perni RB, Conway SC, Otto MJ. 1998. Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (-)beta-L-2′,3′-dideoxy-3′-thiacytidine. Antimicrob Agents Chemother 42:3179–3186. (Erratum, 43:726, 1999.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang YJ, Lu D, Xu YB, Xing WQ, Tong XK, Wang GF, Feng CL, He PL, Yang L, Tang W, Hu YH, Zuo JP. 2015. A novel pyridazinone derivative inhibits hepatitis B virus replication by inducing genome-free capsid formation. Antimicrob Agents Chemother 59:7061–7072. doi: 10.1128/AAC.01558-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang L, Wang YJ, Chen HJ, Shi LP, Tong XK, Zhang YM, Wang GF, Wang WL, Feng CL, He PL, Xu YB, Lu MJ, Tang W, Nan FJ, Zuo JP. 2016. Effect of a hepatitis B virus inhibitor, NZ-4, on capsid formation. Antiviral Res 125:25–33. doi: 10.1016/j.antiviral.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Z, Hu T, Zhou X, Wildum S, Garcia-Alcalde F, Xu Z, Wu D, Mao Y, Tian X, Zhou Y, Shen F, Zhang Z, Tang G, Najera I, Yang G, Shen HC, Young JA, Qin N. 2017. Heteroaryldihydropyrimidine (HAP) and sulfamoylbenzamide (SBA) inhibit hepatitis B virus replication by different molecular mechanisms. Sci Rep 7:42374. doi: 10.1038/srep42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katen SP, Tan Z, Chirapu SR, Finn MG, Zlotnick A. 2013. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure 21:1406–1416. doi: 10.1016/j.str.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klumpp K, Lam AM, Lukacs C, Vogel R, Ren S, Espiritu C, Baydo R, Atkins K, Abendroth J, Liao G, Efimov A, Hartman G, Flores OA. 2015. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc Natl Acad Sci U S A 112:15196–15201. doi: 10.1073/pnas.1513803112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiu Z, Lin X, Zhou M, Liu Y, Zhu W, Chen W, Zhang W, Guo L, Liu H, Wu G, Huang M, Jiang M, Xu Z, Zhou Z, Qin N, Ren S, Qiu H, Zhong S, Zhang Y, Zhang Y, Wu X, Shi L, Shen F, Mao Y, Zhou X, Yang W, Wu JZ, Yang G, Mayweg AV, Shen HC, Tang G. 2016. Design and synthesis of orally bioavailable 4-methyl heteroaryldihydropyrimidine based hepatitis B virus (HBV) capsid inhibitors. J Med Chem 59:7651–7666. doi: 10.1021/acs.jmedchem.6b00879. [DOI] [PubMed] [Google Scholar]
- 37.Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT. 2010. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol 84:9332–9340. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RN, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. 2003. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 299:893–896. doi: 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- 39.Tavis JE, Perri S, Ganem D. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J Virol 68:3536–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc Natl Acad Sci U S A 93:1060–1064. doi: 10.1073/pnas.93.3.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J 16:59–68. doi: 10.1093/emboj/16.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan Z, Pionek K, Unchwaniwala N, Maguire ML, Loeb DD, Zlotnick A. 2015. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J Virol 89:3275–3284. doi: 10.1128/JVI.03545-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan Z, Maguire ML, Loeb DD, Zlotnick A. 2013. Genetically altering the thermodynamics and kinetics of hepatitis B virus capsid assembly has profound effects on virus replication in cell culture. J Virol 87:3208–3216. doi: 10.1128/JVI.03014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ceres P, Stray SJ, Zlotnick A. 2004. Hepatitis B virus capsid assembly is enhanced by naturally occurring mutation F97L. J Virol 78:9538–9543. doi: 10.1128/JVI.78.17.9538-9543.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ludgate L, Liu K, Luckenbaugh L, Streck N, Eng S, Voitenleitner C, Delaney WEt, Hu J. 2016. Cell-free hepatitis B virus capsid assembly dependent on the core protein C-terminal domain and regulated by phosphorylation. J Virol 90:5830–5844. doi: 10.1128/JVI.00394-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewellyn EB, Loeb DD. 2011. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J Virol 85:1298–1309. doi: 10.1128/JVI.01957-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewellyn EB, Loeb DD. 2011. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS One 6:e17202. doi: 10.1371/journal.pone.0017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao W, Ou JH. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol 69:1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lan YT, Li J, Liao W, Ou J. 1999. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342–348. doi: 10.1006/viro.1999.9798. [DOI] [PubMed] [Google Scholar]
- 51.Selzer L, Kant R, Wang JC, Bothner B, Zlotnick A. 2015. Hepatitis B virus core protein phosphorylation sites affect capsid stability and transient exposure of the C-terminal domain. J Biol Chem 290:28584–28593. doi: 10.1074/jbc.M115.678441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Billioud G, Pichoud C, Puerstinger G, Neyts J, Zoulim F. 2011. The main hepatitis B virus (HBV) mutants resistant to nucleoside analogs are susceptible in vitro to non-nucleoside inhibitors of HBV replication. Antiviral Res 92:271–276. doi: 10.1016/j.antiviral.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 53.Venkatakrishnan B, Katen SP, Francis S, Chirapu S, Finn MG, Zlotnick A. 2016. Hepatitis B virus capsids have diverse structural responses to small-molecule ligands bound to the heteroaryldihydropyrimidine pocket. J Virol 90:3994–4004. doi: 10.1128/JVI.03058-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang J, Block TM, Guo JT. 2015. Viral resistance of MOGS-CDG patients implies a broad-spectrum strategy against acute virus infections. Antivir Ther 20:257–259. doi: 10.3851/IMP2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang J, Block TM, Guo JT. 2013. Antiviral therapies targeting host ER alpha-glucosidases: current status and future directions. Antiviral Res 99:251–260. doi: 10.1016/j.antiviral.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Clercq E, Li G. 2016. Approved antiviral drugs over the past 50 years. Clin Microbiol Rev 29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O'Boyle DR II, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. 2010. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo F, Wu S, Julander J, Ma J, Zhang X, Kulp J, Cuconati A, Block TM, Du Y, Guo JT, Chang J. 2016. A novel benzodiazepine compound inhibits yellow fever virus infection by specifically targeting NS4B protein. J Virol 90:10774–10788. doi: 10.1128/JVI.01253-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klumpp K, Crepin T. 2014. Capsid proteins of enveloped viruses as antiviral drug targets. Curr Opin Virol 5:63–71. doi: 10.1016/j.coviro.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 60.Cole AG. 2016. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr Opin Pharmacol 30:131–137. doi: 10.1016/j.coph.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 61.Byrd CM, Dai D, Grosenbach DW, Berhanu A, Jones KF, Cardwell KB, Schneider C, Wineinger KA, Page JM, Harver C, Stavale E, Tyavanagimatt S, Stone MA, Bartenschlager R, Scaturro P, Hruby DE, Jordan R. 2013. A novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob Agents Chemother 57:15–25. doi: 10.1128/AAC.01429-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu S, He J, Shih C, Li K, Dai A, Zhou ZH, Zhang J. 2010. Structural comparisons of hepatitis B core antigen particles with different C-terminal lengths. Virus Res 149:241–244. doi: 10.1016/j.virusres.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 63.Hezode C, Bronowicki JP. 2016. Ideal oral combinations to eradicate HCV: the role of ribavirin. J Hepatol 64:215–225. doi: 10.1016/j.jhep.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 64.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 65.Kirkegaard K, van Buuren NJ, Mateo R. 2016. My cousin, my enemy: quasispecies suppression of drug resistance. Curr Opin Virol 20:106–111. doi: 10.1016/j.coviro.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holland JJ, de la Torre JC, Steinhauer DA, Clarke D, Duarte E, Domingo E. 1989. Virus mutation frequencies can be greatly underestimated by monoclonal antibody neutralization of virions. J Virol 63:5030–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanner EJ, Liu HM, Oberste MS, Pallansch M, Collett MS, Kirkegaard K. 2014. Dominant drug targets suppress the emergence of antiviral resistance. Elife 3:e03830. doi: 10.7554/eLife.03830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mateo R, Nagamine CM, Kirkegaard K. 2015. Suppression of drug resistance in dengue virus. mBio 6:e01960-15. doi: 10.1128/mBio.01960-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allweiss L, Dandri M. 2016. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. J Hepatol 64:S17–S31. doi: 10.1016/j.jhep.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 70.Nassal M. 2015. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 71.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mao R, Zhang J, Jiang D, Cai D, Levy JM, Cuconati A, Block TM, Guo JT, Guo H. 2011. Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis B virus in human hepatocyte-derived cells. J Virol 85:1048–1057. doi: 10.1128/JVI.01998-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Summers J, Smith PM, Horwich AL. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol 64:2819–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zoulim F, Saputelli J, Seeger C. 1994. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol 68:2026–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu W, Goddard C, Clearfield E, Mills C, Xiao T, Guo H, Morrey JD, Motter NE, Zhao K, Block TM, Cuconati A, Xu X. 2011. Design, synthesis, and biological evaluation of triazolo-pyrimidine derivatives as novel inhibitors of hepatitis B virus surface antigen (HBsAg) secretion. J Med Chem 54:5660–5670. doi: 10.1021/jm200696v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang JM, Yao X, Wang YX, Liu F, Ma ZM, Weng XH, Wen YM. 2005. High replicative full-length lamivudine-resistant hepatitis B virus isolated during acute exacerbations. J Med Virol 77:203–208. doi: 10.1002/jmv.20453. [DOI] [PubMed] [Google Scholar]
- 77.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res 28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schrödinger, LLC. 2016. Small-Molecule Drug Discovery Suite 2016-3. Schrödinger LLC, New York, NY. [Google Scholar]
- 79.Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol 77:1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]