

Abstract
Background
Nutritional excess of vitamin A, a precursor for retinoic acid (RA), causes premature epiphyseal fusion, craniosynostosis, and light-dependent retinopathy. Similarly, homozygous loss-of-function mutations in CYP26B1, one of the major RA-metabolizing enzymes, cause advanced bone age, premature epiphyseal fusion, and craniosynostosis. In this paper, a patient with markedly accelerated skeletal and dental development, retinal scarring, and autism-spectrum disease is presented and the role of retinoic acid in longitudinal bone growth and skeletal maturation is reviewed.
Method
Genetic studies using SNP array and exome sequencing. RA isomers were measured in the patient, family members, and in 18 age-matched healthy children using high-performance liquid chromatography coupled to tandem mass spectrometry.
Results
A genomic SNP array identified a novel 8.3 megabase microdeletion on chromosome 10q23.2-23.33. The 79 deleted genes included CYP26A1 and C1, both major RA-metabolizing enzymes. Exome sequencing did not detect any variants that were predicted to be deleterious in the remaining alleles of these genes or other known retinoic acid-metabolizing enzymes. The patient exhibited elevated serum total RA (16.5 vs. 12.6 ± 1.5 nM, mean ± SD, subject vs controls) and 13-cisRA (10.7 nM vs. 6.1 ± 1.1).
Conclusion
The findings support the hypothesis that elevated RA concentrations accelerate bone and dental maturation in humans. CYP26A1 and C1 haploinsufficiency may contribute to the elevated retinoic acid concentrations and clinical findings of the patient, although this phenotype has not been reported in other patients with similar deletions, suggesting that other unknown genetic or environmental factors may also contribute.
At key terms: Retinoic acid, Bone age, Growth plate, Growth
Introduction
At the growth plate, chondrocyte proliferation, chondrocyte hypertrophy, and cartilage matrix secretion contribute to chondrogenesis and thus to longitudinal bone growth [1]. The newly formed cartilage is continuously remodeled into new bone tissue by invading blood vessels and bone cells. The net result is bone elongation [2].
Growth slows dramatically with age, primarily due to declines in chondrocyte proliferation and hypertrophy [3]. These functional changes are accompanied by structural changes, including a gradual decline in the overall height and cellularity of the growth plate [3]. Collectively, these functional and structural changes have been termed growth plate senescence [3]. Clinically, skeletal age, assessed during childhood with wrist and hand radiography, reflects the extent to which the cartilaginous skeleton has been remodeled into bone and is an indirect marker for progression through the growth plate senescence program [4]. This program is driven by growth rather than time [3]. Consequently, growth-inhibiting conditions, such as malnutrition, hypothyroidism, and growth hormone deficiency, tend to delay growth plate senescence and growth-accelerating conditions, such as obesity and hyperthyroidism tend to advance bone age [4–6]. This developmental program is also accelerated by estrogen, leading to advanced bone age in children with precocious children [7–9]. In addition, bone age is accelerated in genetic conditions causing accelerated growth plate chondrocyte differentiation, e.g. pseudohypoparathyroidism 1A due to GNAS mutations [10,11], Acrodysostosis due to PRKAR1A mutations [12], and other rare genetic disorders such as autosomal short stature due to heterozygous mutations in Aggrecan [13].
The process of longitudinal bone growth is governed by a complex network of endocrine, paracrine, and intracellular mechanisms, including retinoic acid (RA) signaling [14–16]. Deficiency of vitamin A, a precursor for retinoic acid (RA), is associated with growth failure [17], whereas excess of RA causes accelerated bone maturation, premature epiphyseal fusion, craniosynostosis, as well as light-dependent retinopathy [18–20]. RA metabolizing CYP26 enzymes have crucial roles during organ and limb development where they primarily act to spatially restrict RA signaling [18]. Consequently, homozygous loss-of-function mutations in CYP26B1 cause advanced bone maturation, bone fusion, and craniosynostosis [21].
We report here a patient with markedly accelerated skeletal and dental development, retinal scarring, and autism-spectrum disorder associated with elevated concentrations of total RA and 13-cisRA in light-protected serum and a 8.3 Mb microdeletion on chromosome 10q23.2-23.33 that included the genes CYP26A1 and C1, which encode two major RA metabolizing enzymes. In addition, the literature on the role of retinoic acid in longitudinal bone growth and skeletal maturation is reviewed.
Methods
Subjects
This study was approved by the Institutional Review Boards of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and written informed assent and consent was obtained from all participants or their legal guardians as appropriate. Height and weights were plotted on the Centers for Disease Control growth charts (www.cdc.gov/growthcharts) and Z-scores were calculated using NHANES III growth data (U.S. Department of Health and Human Services (DHHS), National Center for Health Statistics NCHS. Third National Health and Nutrition Examination Survey, 1988–1994. Hyattsville: National Center for Health Statistics, 1996).
Control group
In order to establish the normal range for concentrations of total RA, all-trans-RA (atRA), and 13-cisRA, healthy, age-matched (age 8–12 years) boys and girls (n = 9, each sex) were recruited and consented. Morning (8:00–10:00 am) fasting, light-protected serum samples were collected and RA metabolites measured as described below.
Genetic studies
In order to detect possible chromosomal aberrations and uniparental disomy, a high-resolution genome-wide DNA microarray analysis using Human SNP Array 6.0 (Affymetrix Inc, Santa Clara, CA) that includes 1.8 million genetic markers was performed on the patient, the patient’s sister, and both parents.
Exome sequencing of the patient using genomic DNA isolated from blood was performed at the Broad Institute (Cambridge, MA). Hybrid selection was performed using Agilent’s SureSelect Human All Exon Kit v2 (Agilent Technologies, Santa Clara, CA). The samples were sequenced using the Illumina HiSeq 2000 platform (Illumina Inc, San Diego, California) and aligned the resulting reads were aligned to the human genome version 19 reference genome with Burrows-Wheeler Aligner (http://biobwa.sourceforge.net/) [22]. We then applied the Genome Analysis Toolkit (http://www.broadinstitute.org/gatk) [23], base quality score recalibration, and indel realignment, and performed single nucleotide polymorphism and indel discovery and genotyping across all samples simultaneously using variant quality score recalibration [24]. The exome sequencing achieved >20X coverage for 88% of the targeted bases. Variants were annotated for functional effect using SnpEff 2.0.5 (http://snpeff.sourceforge.net/) [25].
Retinoic acid measurements
RA and 13-cisRA concentrations were measured in light-protected plasma from collected in the morning from fasting subjects (patient, family members, and control group) as previously described (Arnold et al J. Lipid Res 2012) using an AB Sciex 5500 qtrap mass spectrometer coupled with an Agilent UHPLC using 0.1mL of plasma with proteins precipitated using 0.2 mL of acetonitrile. Deuterium labeled RA and 13-cisRA were used as internal standards and all concentrations were determined according to linear calibration curves prepared in DC mass spect Gold (Golden West biologics) blank serum.
Case Report
The patient was a 10-yr old prepubertal male with a history of markedly advanced skeletal and dental development, left eye exudative vitrioretinopathy and right eye chorioretinal scarring, as well as microcephaly of postnatal onset, developmental delay and autism-spectrum disorder. He was the second child of healthy parents of northern European decent with a midparental target height of 177.1 cm. He was born full term after an uncomplicated pregnancy and delivery with a birth weight of 2.98 kg (−1.3 SDS) and birth length of 46 cm (−2.3 SDS). At the 2-month well baby visit, there was lack of eye contact and a small anterior fontanelle was noted. Ophthalmological evaluation revealed exudative vitreoretinopathy in the left eye and chorioretinal scarring in the right eye. At age 4 months, due to the retinopathy and retinal scarring, serum retinol levels were assessed and found to be low. Despite several courses of vitamin A supplementation during the first 4 years of life, his serum retinol levels never normalized and vitamin A supplements were discontinued due to lack of clinical improvement.
A markedly advanced dental age was noted including the presence of 16 primary teeth at age 12 months and shedding of primary teeth beginning prematurely at age 3 yrs. He showed poor weight gain, whereas his height continued at the 50th percentile (Fig 1). Extended evaluations for metabolic and gastroenterology disorders were negative, including negative serological celiac screening tests. At a chronological age of 3 years and 6 months, his bone age was assessed and found to be markedly advanced at approximately 7 years and 0 months. Without an evident etiology for his failure to thrive, he was empirically started on a gluten- and dairy-free diet consisting of meat, fruits, vegetables and supplemented with a hypoallergenic supplement (Neocate splash E028) at the age of 6 years and 9 months. This treatment was followed by improvement of his GI symptoms and a dramatic improvement in weight gain and continued linear growth along the 50th percentile (Fig 1).
Figure 1.
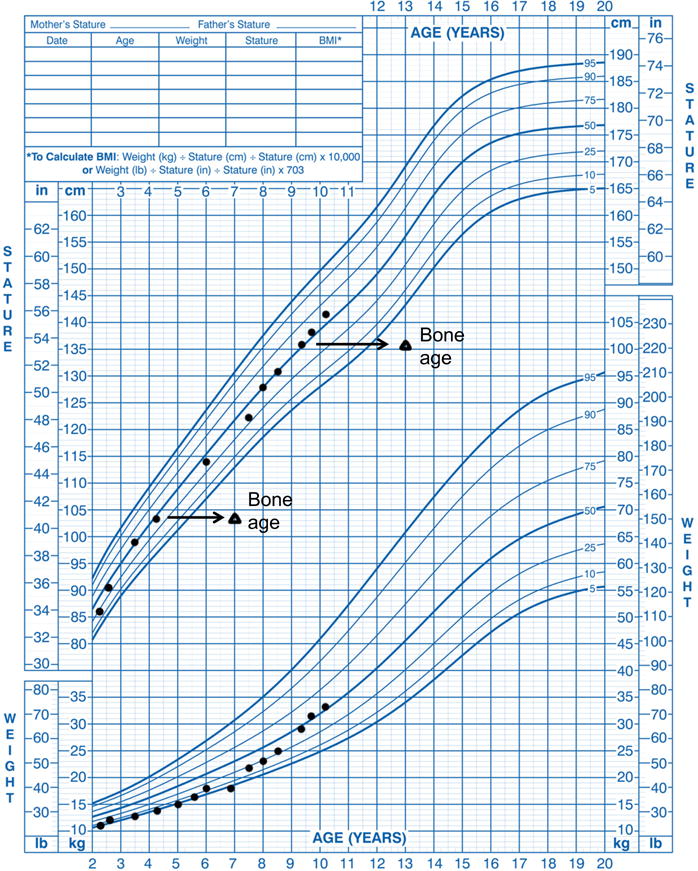
Height, weight, and bone age measurements from the proband plotted on the US Centers for Disease Control growth charts.
A bone age assessment at age 9 4/12 years was again markedly advanced at 12 9/12 years (Fig 1S). Endocrine evaluation at age 9 7/12 years was unremarkable including early Tanner II pubic hair, Tanner I genitalia, testicular size of 3–4 mL bilaterally, and no gynecomastia; thus the evaluation did not provide a clear explanation for his markedly advanced bone age.
Results
The family was evaluated at the NIH Clinical Center when the patient was 10 years and 2 months of age. His height was 141.7 cm (+0.4 SDS), weight 33.2 kg (+0.1 SDS), BMI 16.5 kg/m2 (−0.1 SDS), sitting height index 0.53 (+1.0 SDS), arm span 139.8 cm. There was left eye strabismus, with a dilated left pupil, and marked light sensitivity. Distal phalanges appeared short. Pubic hair was limited to straight, non-pigmented hair and remained at a Tanner stage II distribution. Testicular size was 4 ml bilaterally. Thyroid function tests were normal, serum estradiol undetectable, total testosterone 0.31 nmol/L (normal range for Tanner II: 0.30 – 2.30), and dehydroepiandrosterone sulfate 4.48 μmol/l (normal range for Tanner II: <0.40 – 9.00).
SNP microarray analysis
The patient, his parents, and unaffected older sister underwent DNA microarray analysis. This analysis detected a novel de novo 8.3 Mb interstitial deletion on 10q23.2 – 10q23.33 unique to the patient and not present in any of the other family members. This microdeletion contained 79 genes (Table 1S) and microRNAs, including PTEN, a tumor suppressor gene in which monoallelic mutations cause Cowden syndrome and PLCE1, which encodes a phospholipase, in which biallelic mutations cause nephrotic syndrome. Analysis of the biomedical literature as well as analyses of spatial and temporal expression patterns in growth plate cartilage for the deleted genes (Table 1S) identified several genes involved in retinoic acid (RA) metabolism, including 2 of the 3 major RA metabolizing enzymes CYP26A1 and C1 as well as the gene for retinol binding protein, RBP4, and RA metabolizing enzymes CYP2C8 and CYP2C9 [26].
Retinoic Acid concentrations in patient, unaffected family members and age-matched healthy controls
Because the deletion included genes involved in RA metabolism and because many of the patient’s phenotypic features might be explained by RA excess, we hypothesized that this disorder might be caused by increased RA signaling. We therefore next measured total RA, all-trans RA (atRA), and 13-cisRA in the patient, the unaffected family members, and age-matched, healthy boys and girls (9 of each sex) using high-performance liquid chromatography (HPLC) coupled to tandem mass spectrometry (MS/MS). In the control group, the concentrations of all RA metabolites measured were similar in male and female subjects and therefore are reported together. Compared to age-matched controls, the patient was found to have increased serum levels of total RA (patient: 16.5 nM vs. controls: 12.6 ± 1.5; mean ± SD) and 13-cisRA (patient: 10.7 nM vs. controls: 6.1 ± 1.1), but normal serum atRA (patient: 5.9 nM vs. controls: 6.4 ± 0.7; Fig 2, Table 2S).
Figure 2.
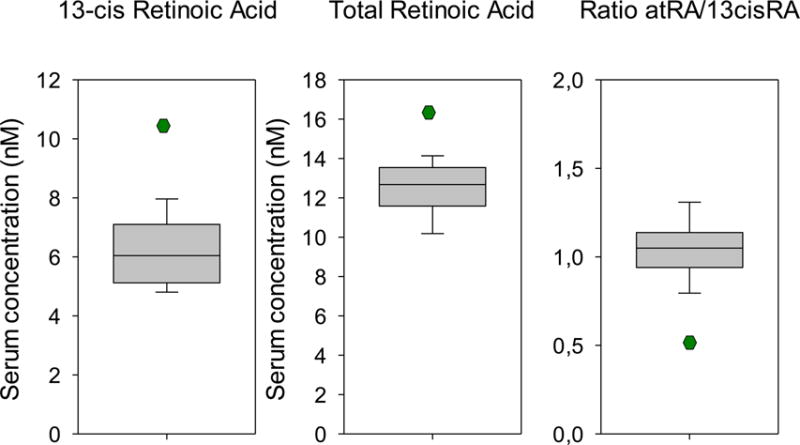
13-cis-retinoic acid (13-cisRA), total retinoic acid (RA), and the all-trans RA/13-cisRA ratio in healthy age-matched controls (n=20) and the proband were assessed by HPLC-MS/MS. The central line segments of the boxes represent the median of control values, while box margins indicate the 25th and 75th percentiles, and the error bars, the 5th and 95th percentiles. The single point in each plot indicates the proband’s value.
Exome sequencing
We next asked whether there might be additional genetic changes, such as point mutations in the non-deleted alleles of CYP26A1, CYP26C1, CYP2C8, or CYP2C9, that might contribute to the elevated RA concentrations and the other phenotypic features. We therefore performed exome sequencing and analyzed known genes that encode RA-metabolizing enzymes for variants. We did not detect any rare genomic variants predicted to affect function of RA synthesizing enzymes ALDH1A family (ALDH1A1, ALDH1A2, and ALDH1A3) or RA metabolizing enzymes, including CYP26A1, CYP26B1, CYP26C1, CYP2C8, and CYP2C9.
Discussion
We present a patient with a previously undiagnosed syndrome characterized by retinal scarring, photophobia, autism spectrum disease, markedly advanced bone age, and accelerated dental development. This syndrome was associated with an 8.3 Mb deletion on chromosome 10q23, low serum vitamin A and elevated total and 13-cisRA concentrations. The deletion was not present in either parent. Because the parents were unaffected, the de novo occurrence of the deletion was consistent with the possibility that the deletion and the syndrome were causally related.
The genomic microdeletion of our patient includes RBP4, the gene encoding retinol-binding protein, which is an important carrier of retinol in the circulation. Haploinsufficiency of this gene likely explains his low serum vitamin A; hypovitaminosis A is also observed in patients with isolated microphthalmia due to heterozygous RBP4 inactivating mutations [27]. Our patient did not have microphthalmia, perhaps because this abnormality seems to occur only in individuals whose mothers also have RBP4 haploinsufficiency, most likely due to a placental effect of RBP4 haploinsuffciency [27].
The microdeletion also included CYP26A1 and CYP26C1. These two genes encode enzymes that metabolize RA to polar metabolites including 4-hydroxy-RA, 4-oxo-RA and 18-hydroxy-RA [28,29]. Thus, the patient’s haploinsufficiency for these genes would be predicted to decrease RA metabolism to polar metabolites and therefore to increased tissue RA concentrations. Consistent with this prediction, we found that the patient had elevated circulating concentrations of total RA and 13-cisRA, although serum atRA was not elevated compared to controls. The specific increase in 13-cisRA is likely a consequence of increased isomerization of RA to the 13-cisRA in tissues following reduced oxidative metabolism. The deletion of the CYP2C8 gene may also decrease the clearance of 13-cisRA in this patients as this enzyme has been shown to metabolize 13-cisRA.
Elevated tissue RA concentrations may explain the accelerated skeletal maturation in this patient. He exhibited a radiographic bone age that was 3 years greater than chronological age. The bone age is a measure of the extent to which the fetal cartilaginous skeleton has been remodeled into a bony skeleton. In this patient, a common cause of accelerated skeletal maturation, estrogen excess, was excluded. The accelerated skeletal maturation involved not only endochondral bones but also the skull, resulting in early closure of the fontanelle. Retinoid excess has previously been implicated in accelerated skeletal maturation; vitamin A intoxication causes premature growth cessation and epiphyseal fusion as well as craniosynostosis [19]. Similarly, homozygous inactivating mutations in the gene encoding the RA metabolizing enzyme CYP26B1 was found to cause skeletal and craniofacial defects, fusion of long bones, and craniosynostosis [21]. Consistently, cartilage-specific targeting of CYP26B1 in mice results in reduced growth rate, at least in part due to reduced chondrocyte proliferation, as well as premature epiphyseal fusion presumably due to accelerated differentiation of resting and proliferative chondrocytes. The phenotype of these mice can be partially rescued by a diet deficient in vitamin A [30], again underscoring the role of the CYP26 enzymes as important quenchers of RA signaling.
Whether RA excess might explain the accelerated dental development is less clear. RA does appear to regulate tooth formation in the embryo. For example, an inhibitor of endogenous retinoic acid synthesis blocks the initiation of murine odontogenesis in vitro [31]. RA excess in embryonic mice delays tooth development [32]. The effects of RA on postnatal dental maturation are less well studied. Interestingly, during late gestation in the mouse, Cyp26C1 exhibits region-specific expression in the inner dental epithelium of the developing teeth [33].
The ophthalmological abnormalities in this patient may be related to the haploinsufficiency of RBP4 and the decreased circulating retinol levels. In the eye, retinoids both regulate expression of target genes and serves as the chromophore (11-cis-retinal) of the visual pigment. Deficiency of retinol causes night blindness because the steady loss of rhodopsin during the rod visual cycle cannot be sufficiently replaced. Severe, prolonged deficiency results in degeneration of retinal pigment epithelium (RPE) and photoreceptors.
In humans, heterozygote mutations in RBP4 cause a syndrome characterized by microphthalmia and colobomas [27], whereas homozygous mutations cause a more severe condition including visual impairment, retinal dystrophy, coloboma, and severe comedogenic acne [34,35]. In addition, retinol binding protein deficiency causes low vitamin A concentrations without clinical vitamin A deficiency [36]. Attempts to raise serum vitamin A in a patient with retinol binding protein deficiency will ultimately fail and could potentially cause vitamin A toxicity.
Excess RA might additionally be linked to our patient’s autism spectrum disorder. Exposure to excess vitamin A or treatment with isotretinoin in pregnant women causes abnormal brain development, typically involving malformation of the hindbrain, although cortical abnormalities have also been reported [37]. Even in the absence of structural brain malformations by imaging, excess retinoids can cause behavioral and cognitive impairments [38].
Other individuals with similar deletions have been listed in the DECIPHER database (https://decipher.sanger.ac.uk) and the listings did not include descriptions of accelerated skeletal maturation or eye disease. We therefore considered the possibility that the patient described in the current report might have another undetected genetic abnormality that contributed to the phenotype. For example, a point mutation in the undeleted alleles of CYP26A1 or CYP26C1 might cause biallelic insufficiency of either enzyme. We performed exome sequencing of the proband but did not find significant genetic variants in any of the major RA synthesizing or metabolizing enzymes that we examined. However, exome sequencing primarily provides sequence of exons and thus does not exclude pathogenic regulatory mutations or epigenetic changes.
The role of retinoic acid in longitudinal bone growth and bone maturation
Retinoic acid (RA) is an important regulator of cellular differentiation and proliferation during embryogenesis and development [18]. In particular, RA is an important local regulator of growth plate cartilage. Early indications of the important effects of retinoids on growth plate cartilage and longitudinal bone growth came from observations that deficiency of vitamin A, the precursor of all naturally occurring retinoids, including RA, was associated with short stature [17]. Later it was demonstrated that vitamin A intoxication causes premature growth cessation and epiphyseal fusion as well as craniosynostosis [19] and that retinoic acid has teratogenic effects in the limbs, craniofacial skeleton, and central nervous system [18,37]. CYP26A1, B1, and C1, which metabolize RA, are expressed in multiple different fetal and adult human tissues, each enzyme showing a distinct expression profile [28,39]. In general, the expression pattern of the CYP26 enzymes is similar in mice and humans. Homozygous inactivating mutations in the CYP26B1 gene, was found to cause skeletal and craniofacial defects, fusion of long bones, accelerated bone maturation, and craniosynostosis [21,40]. Recently, it was found that compound heterozygote mutations in retinol dehydrogenase 11 (RDH11), an enzyme that metabolizes retinaldehyde to retinol and therefore promotes the flux to RA, is associated with atypical, progressive retinitis pigmentosa, facial dysmorphology, and short stature and a growth pattern that is highly suggestive of accelerated bone maturation [41]. In addition, clinical observations of patients treated with RA derivatives also supports the hypothesis that RA accelerates bone maturation and thus promotes early growth cessation and adult short stature. This effect is likely dose-dependent and may thus not be as pronounced in acne patients treated with isotretinoin [42] as in oncology patients treated with high doses of RA [43,44].
In mice, cartilage-specific targeting of CYP26B1 results in reduced growth rate, at least in part due to reduced chondrocyte proliferation, as well as premature epiphyseal fusion presumably due to accelerated differentiation of resting and proliferative chondrocytes. The phenotype of these mice can be partially rescued by a diet deficient in vitamin A [30], again underscoring the role of the CYP26 enzymes as important quenchers of RA signaling.
Daily injections of atRA or 13-cisRA for ten days induce epiphyseal closure in young guinea pigs. In rats, a single oral dose of RA (300 mg/kg) reduce the height of the growth plate, presumably due to a direct effects on the growth plate [15]. Consistently, in rat metatarsal bone cultures, RA inhibits longitudinal bone growth by decreasing chondrocyte proliferation, matrix synthesis, and hypertrophic cell size [15]. The growth inhibition of RA are reversed by a retinoic acid receptor (RAR) antagonist, confirming that the effect is primarily mediated through the RAR [15]. Interestingly, in the abscence of exogenous RA, both RAR blockade and an RA-specific neutralizing antibody accelerate growth, suggesting that RA is produced locally in growth plate cartilage [45] and acts as a physiologic, negative regulator of growth plate chondrogenesis and thus of longitudinal bone growth [15,45]. The exact molecular mechanisms by which RA regulates chondrocyte proliferation and differentiation at the growth plate have not been determined, but the effects appear to be mediated primarily through RAR-gamma [46] and involve interaction with a number of paracrine factors, including fibroblast growth factors [47], Indian hedgehog [48,49], and bone morphogenetic proteins [50].
Summary
Here we describe a boy with markedly accelerated skeletal maturation, including an advanced bone age and craniosynostosis, accelerated dental development, retinal scarring, and autism-spectrum disorder. SNP array analysis identified a de novo 8.3 megabase deletion on chromosome 10q23, which included CYP26A1, CYP26C1, CYP2C8 and CYP2C9 which encode RA-metabolizing enzymes, as well as RBP4, which encodes retinol-binding protein, an important carrier of retinol in the circulation. Because RA has important development effects on the growth plate, skull, eye, and CNS, we hypothesized that his developmental abnormalities arose, at least in part, from RA excess. This hypothesis was supported by the finding of elevated circulating total and 13-cisRA. Because similar deletions have not been reported to cause a similar phenotype, using exome sequencing, we searched for, but did not find, additional contributing genetic abnormalities.
Supplementary Material
Figure 1S. Radiograph of the left hand and wrist of the patient at chronologic age 9 years and 4 months. The image was read as a bone age of 12 years and 9 months according to the method of Greulich and Pyle.
Table 1S. Expression and function of genes in the patients deletion.
Table 2S. Retinoic Acid concentrations in patient, unaffected family members and age-matched healthy controls.
Figure 3.
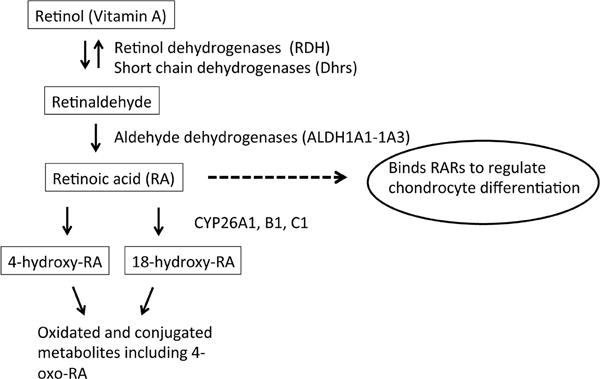
Diagram depicting retinoic acid (RA) biosynthesis, metabolism, and action on the growth plate. Boxed terms indicate RA, RA precursors, and RA metabolites. Solid arrows indicate chemical conversion, catalyzed by the indicated enzyme. Dashed arrow and oval indicate biological action of RA on growth plate. Interconversion of all-trans-RA, 9-cis-RA and 13-cis-RA isomers is not shown. RARs, retinoic acid receptors
Acknowledgments
We thank the patient, his family, and all healthy volunteers for their participation in the study.
The work of ON, YHJ, JCL, JAY, and JB was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health (Grant no. Z1AHD00641 and Z1AHD00640). NI was supported by extramural funding from the US National Institutes of Health (Grant no R01 GM111772). ON was supported by grants from the Swedish Research Council (Grant no. 521-2014-3063 and 2015-02227), the Swedish Governmental Agency for Innovation Systems (Vinnova) (2014-01438), Marianne and Marcus Wallenberg Foundation, the Stockholm County Council, the Swedish Society of Medicine, Byggmästare Olle Engkvist’s Foundation, HKH Kronprinsessan Lovisas förening för barnasjukvård, Sällskapet Barnavård, Stiftelsen Frimurare Barnhuset i Stockholm, and Karolinska Institutet.
This work was also supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development at the National Institutes of Health (1K23HD073351).
References
- 1.Hunziker EB. Mechanism of longitudinal bone growth and its regulation by growth plate chondrocytes. Microsc Res Tech. 1994;28:505–519. doi: 10.1002/jemt.1070280606. [DOI] [PubMed] [Google Scholar]
- 2.Abad V, Meyers JL, Weise M, Gafni RI, Barnes KM, Nilsson O, Bacher JD, Baron J. The role of the resting zone in growth plate chondrogenesis. Endocrinology. 2002;143:1851–1857. doi: 10.1210/endo.143.5.8776. [DOI] [PubMed] [Google Scholar]
- 3.Nilsson O, Baron J. Fundamental limits on longitudinal bone growth: growth plate senescence and epiphyseal fusion. Trends Endocrinol Metab. 2004;15:370–374. doi: 10.1016/j.tem.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Martin DD, Wit JM, Hochberg Z, Savendahl L, van Rijn RR, Fricke O, Cameron N, Caliebe J, Hertel T, Kiepe D, Albertsson-Wikland K, Thodberg HH, Binder G, Ranke MB. The use of bone age in clinical practice - part 1. Horm Res Paediatr. 2011;76:1–9. doi: 10.1159/000329372. [DOI] [PubMed] [Google Scholar]
- 5.Bossowski AT, Reddy V, Perry LA, Johnston LB, Banerjee K, Blair JC, Savage MO. Clinical and endocrine features and long-term outcome of Graves’ disease in early childhood. J Endocrinol Invest. 2007;30:388–392. doi: 10.1007/BF03346315. [DOI] [PubMed] [Google Scholar]
- 6.Klein KO, Newfield RS, Hassink SG. Bone maturation along the spectrum from normal weight to obesity: a complex interplay of sex, growth factors and weight gain. J Pediatr Endocrinol Metab: JPEM. 2016;29:311–318. doi: 10.1515/jpem-2015-0234. [DOI] [PubMed] [Google Scholar]
- 7.Nilsson O, Weise M, Landman EB, Meyers JL, Barnes KM, Baron J. Evidence that estrogen hastens epiphyseal fusion and cessation of longitudinal bone growth by irreversibly depleting the number of resting zone progenitor cells in female rabbits. Endocrinology. 2014;155:2892–2899. doi: 10.1210/en.2013-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weise M, De-Levi S, Barnes KM, Gafni RI, Abad V, Baron J. Effects of estrogen on growth plate senescence and epiphyseal fusion. Proc Natl Acad Sci U S A. 2001;98:6871–6876. doi: 10.1073/pnas.121180498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weise M, Flor A, Barnes KM, Cutler GB, Jr, Baron J. Determinants of growth during gonadotropin-releasing hormone analog therapy for precocious puberty. J Clin Endocrinol Metab. 2004;89:103–107. doi: 10.1210/jc.2002-021999. [DOI] [PubMed] [Google Scholar]
- 10.Chagin AS, Kronenberg HM. Role of G-proteins in the differentiation of epiphyseal chondrocytes. J Mol Endocrinol. 2014;53:R39–45. doi: 10.1530/JME-14-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mantovani G, Ferrante E, Giavoli C, Linglart A, Cappa M, Cisternino M, Maghnie M, Ghizzoni L, de Sanctis L, Lania AG, Beck-Peccoz P, Spada A. Recombinant human GH replacement therapy in children with pseudohypoparathyroidism type Ia: first study on the effect on growth. J Clin Endocrinol Metab. 2010;95:5011–5017. doi: 10.1210/jc.2010-1649. [DOI] [PubMed] [Google Scholar]
- 12.Silve C, Clauser E, Linglart A. Acrodysostosis. Horm Metab Res. 2012;44:749–758. doi: 10.1055/s-0032-1316330. [DOI] [PubMed] [Google Scholar]
- 13.Nilsson O, Guo MH, Dunbar N, Popovic J, Flynn D, Jacobsen C, Lui JC, Hirschhorn JN, Baron J, Dauber A. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J Clin Endocrinol Metab. 2014;99:E1510–1518. doi: 10.1210/jc.2014-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baron J, Savendahl L, De Luca F, Dauber A, Phillip M, Wit JM, Nilsson O. Short and tall stature: a new paradigm emerges. Nature reviews Endocrinology. 2015 doi: 10.1038/nrendo.2015.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Luca F, Uyeda JA, Mericq V, Mancilla EE, Yanovski JA, Barnes KM, Zile MH, Baron J. Retinoic acid is a potent regulator of growth plate chondrogenesis. Endocrinology. 2000;141:346–353. doi: 10.1210/endo.141.1.7283. [DOI] [PubMed] [Google Scholar]
- 16.Kwasigroch TE, Bullen M. Effects of isotretinoin (13-cis-retinoic acid) on the development of mouse limbs in vivo and in vitro. Teratology. 1991;44:605–616. doi: 10.1002/tera.1420440603. [DOI] [PubMed] [Google Scholar]
- 17.Wolbach SB. Vitamin-A deficiency and excess in relation to skeletal growth. J Bone Joint Surg Am. 1947;29:171–192. [PubMed] [Google Scholar]
- 18.Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol. 2015;16:110–123. doi: 10.1038/nrm3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pease CN. Focal retardation and arrestment of growth of bones due to vitamin A intoxication. JAMA. 1962;182:980–985. doi: 10.1001/jama.1962.03050490004002. [DOI] [PubMed] [Google Scholar]
- 20.Standeven AM, Davies PJ, Chandraratna RA, Mader DR, Johnson AT, Thomazy VA. Retinoid-induced epiphyseal plate closure in guinea pigs. Fundam Appl Toxicol. 1996;34:91–98. doi: 10.1006/faat.1996.0179. [DOI] [PubMed] [Google Scholar]
- 21.Laue K, Pogoda HM, Daniel PB, van Haeringen A, Alanay Y, von Ameln S, Rachwalski M, Morgan T, Gray MJ, Breuning MH, Sawyer GM, Sutherland-Smith AJ, Nikkels PG, Kubisch C, Bloch W, Wollnik B, Hammerschmidt M, Robertson SP. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet. 2011;89:595–606. doi: 10.1016/j.ajhg.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thatcher JE, Zelter A, Isoherranen N. The relative importance of CYP26A1 in hepatic clearance of all-trans retinoic acid. Biochem Pharmacol. 2010;80:903–912. doi: 10.1016/j.bcp.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou CM, Nelson C, Tarle SA, Pribila JT, Bardakjian T, Woods S, Schneider A, Glaser T. Biochemical Basis for Dominant Inheritance, Variable Penetrance, and Maternal Effects in RBP4 Congenital Eye Disease. Cell. 2015;161:634–646. doi: 10.1016/j.cell.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Topletz AR, Thatcher JE, Zelter A, Lutz JD, Tay S, Nelson WL, Isoherranen N. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem Pharmacol. 2012;83:149–163. doi: 10.1016/j.bcp.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Topletz AR, Tripathy S, Foti RS, Shimshoni JA, Nelson WL, Isoherranen N. Induction of CYP26A1 by metabolites of retinoic acid: evidence that CYP26A1 is an important enzyme in the elimination of active retinoids. Mol Pharmacol. 2015;87:430–441. doi: 10.1124/mol.114.096784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minegishi Y, Sakai Y, Yahara Y, Akiyama H, Yoshikawa H, Hosokawa K, Tsumaki N. Cyp26b1 within the growth plate regulates bone growth in juvenile mice. Biochem Biophys Res Commun. 2014;454:12–18. doi: 10.1016/j.bbrc.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Kronmiller JE, Beeman CS, Nguyen T, Berndt W. Blockade of the initiation of murine odontogenesis in vitro by citral, an inhibitor of endogenous retinoic acid synthesis. Arch Oral Biol. 1995;40:645–652. doi: 10.1016/0003-9969(95)00015-h. [DOI] [PubMed] [Google Scholar]
- 32.Jones DM, Fabian B, Kramer B. The effect of retinoic acid on mouse mandibular molar development in vitro, using alkaline phosphatase as a molecular indicator of differentiation. SADJ. 2008;63:276, 278–280. [PubMed] [Google Scholar]
- 33.Tahayato A, Dolle P, Petkovich M. Cyp26C1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first branchial arch and tooth buds during murine development. Gene Expr Patterns. 2003;3:449–454. doi: 10.1016/s1567-133x(03)00066-8. [DOI] [PubMed] [Google Scholar]
- 34.Cukras C, Gaasterland T, Lee P, Gudiseva HV, Chavali VR, Pullakhandam R, Maranhao B, Edsall L, Soares S, Reddy GB, Sieving PA, Ayyagari R. Exome analysis identified a novel mutation in the RBP4 gene in a consanguineous pedigree with retinal dystrophy and developmental abnormalities. PLoS One. 2012;7:e50205. doi: 10.1371/journal.pone.0050205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seeliger MW, Biesalski HK, Wissinger B, Gollnick H, Gielen S, Frank J, Beck S, Zrenner E. Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Invest Ophthalmol Vis Sci. 1999;40:3–11. [PubMed] [Google Scholar]
- 36.Biesalski HK, Frank J, Beck SC, Heinrich F, Illek B, Reifen R, Gollnick H, Seeliger MW, Wissinger B, Zrenner E. Biochemical but not clinical vitamin A deficiency results from mutations in the gene for retinol binding protein. Am J Clin Nutr. 1999;69:931–936. doi: 10.1093/ajcn/69.5.931. [DOI] [PubMed] [Google Scholar]
- 37.Adams J. The neurobehavioral teratology of retinoids: a 50-year history. Birth Defects Res A Clin Mol Teratol. 2010;88:895–905. doi: 10.1002/bdra.20721. [DOI] [PubMed] [Google Scholar]
- 38.Adams J, Lammer EJ. Neurobehavioral teratology of isotretinoin. Reprod Toxicol. 1993;7:175–177. doi: 10.1016/0890-6238(93)90273-a. [DOI] [PubMed] [Google Scholar]
- 39.Xi J, Yang Z. Expression of RALDHs (ALDH1As) and CYP26s in human tissues and during the neural differentiation of P19 embryonal carcinoma stem cell. Gene Expr Patterns. 2008;8:438–442. doi: 10.1016/j.gep.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Wen J, Lopes F, Soares G, Farrell SA, Nelson C, Qiao Y, Martell S, Badukke C, Bessa C, Ylstra B, Lewis S, Isoherranen N, Maciel P, Rajcan-Separovic E. Phenotypic and functional consequences of haploinsufficiency of genes from exocyst and retinoic acid pathway due to a recurrent microdeletion of 2p13.2. Orphanet J Rare Dis. 2013;8:100. doi: 10.1186/1750-1172-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie YA, Lee W, Cai C, Gambin T, Noupuu K, Sujirakul T, Ayuso C, Jhangiani S, Muzny D, Boerwinkle E, Gibbs R, Greenstein VC, Lupski JR, Tsang SH, Allikmets R. New syndrome with retinitis pigmentosa is caused by nonsense mutations in retinol dehydrogenase RDH11. Hum Mol Genet. 2014;23:5774–5780. doi: 10.1093/hmg/ddu291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Webster GF, Leyden JJ, Gross JA. Results of a Phase III, double-blind, randomized, parallel-group, non-inferiority study evaluating the safety and efficacy of isotretinoin-Lidose in patients with severe recalcitrant nodular acne. J Drugs Dermatol. 2014;13:665–670. [PubMed] [Google Scholar]
- 43.Hobbie WL, Mostoufi SM, Carlson CA, Gruccio D, Ginsberg JP. Prevalence of advanced bone age in a cohort of patients who received cis-retinoic acid for high-risk neuroblastoma. Pediatr Blood Cancer. 2011;56:474–476. doi: 10.1002/pbc.22839. [DOI] [PubMed] [Google Scholar]
- 44.Noyes JJ, Levine MA, Belasco JB, Mostoufi-Moab S. Premature Epiphyseal Closure of the Lower Extremities Contributing to Short Stature after cis-Retinoic Acid Therapy in Medulloblastoma: A Case Report. Horm Res Paediatr. 2016;85:69–73. doi: 10.1159/000441140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams JA, Kane M, Okabe T, Enomoto-Iwamoto M, Napoli JL, Pacifici M, Iwamoto M. Endogenous retinoids in mammalian growth plate cartilage: analysis and roles in matrix homeostasis and turnover. J Biol Chem. 2010;285:36674–36681. doi: 10.1074/jbc.M110.151878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams JA, Kondo N, Okabe T, Takeshita N, Pilchak DM, Koyama E, Ochiai T, Jensen D, Chu ML, Kane MA, Napoli JL, Enomoto-Iwamoto M, Ghyselinck N, Chambon P, Pacifici M, Iwamoto M. Retinoic acid receptors are required for skeletal growth, matrix homeostasis and growth plate function in postnatal mouse. Dev Biol. 2009;328:315–327. doi: 10.1016/j.ydbio.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cunningham TJ, Zhao X, Sandell LL, Evans SM, Trainor PA, Duester G. Antagonism between retinoic acid and fibroblast growth factor signaling during limb development. Cell Rep. 2013;3:1503–1511. doi: 10.1016/j.celrep.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu LN, Lu M, Genge BR, Guo GY, Nie D, Wuthier RE. Discovery of sonic hedgehog expression in postnatal growth plate chondrocytes: differential regulation of sonic and Indian hedgehog by retinoic acid. J Cell Biochem. 2002;87:173–187. doi: 10.1002/jcb.10285. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida E, Noshiro M, Kawamoto T, Tsutsumi S, Kuruta Y, Kato Y. Direct inhibition of Indian hedgehog expression by parathyroid hormone (PTH)/PTH-related peptide and up-regulation by retinoic acid in growth plate chondrocyte cultures. Exp Cell Res. 2001;265:64–72. doi: 10.1006/excr.2001.5161. [DOI] [PubMed] [Google Scholar]
- 50.Grimsrud CD, Rosier RN, Puzas JE, Reynolds PR, Reynolds SD, Hicks DG, O’Keefe RJ. Bone morphogenetic protein-7 in growth-plate chondrocytes: regulation by retinoic acid is dependent on the stage of chondrocyte maturation. J Orthop Res. 1998;16:247–255. doi: 10.1002/jor.1100160212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S. Radiograph of the left hand and wrist of the patient at chronologic age 9 years and 4 months. The image was read as a bone age of 12 years and 9 months according to the method of Greulich and Pyle.
Table 1S. Expression and function of genes in the patients deletion.
Table 2S. Retinoic Acid concentrations in patient, unaffected family members and age-matched healthy controls.