

Abstract
Mechanisms that regulate tissue-specific progenitors for maintenance and differentiation during development are poorly understood. Here, we demonstrate that the co-repressor protein Sin3a is crucial for lung endoderm development. Loss of Sin3a in mouse early foregut endoderm led to a specific and profound defect in lung development with lung buds failing to undergo branching morphogenesis and progressive atrophy of the proximal lung endoderm with complete epithelial loss at later stages of development. Consequently, neonatal pups died at birth due to respiratory insufficiency. Further analysis revealed that loss of Sin3a resulted in embryonic lung epithelial progenitor cells adopting a senescence-like state with permanent cell cycle arrest in G1 phase. This was mediated at least partially through upregulation of the cell cycle inhibitors Cdkn1a and Cdkn2c. At the same time, loss of endodermal Sin3a also disrupted cell differentiation of the mesoderm, suggesting aberrant epithelial-mesenchymal signaling. Together, these findings reveal that Sin3a is an essential regulator for early lung endoderm specification and differentiation.
KEY WORDS: Sin3a, Foregut endoderm, G1 arrest, p21/Cdkn1a, Progenitor cell fate, Epithelial-mesenchymal signaling, Mouse
Highlighted article: Loss of Sin3a in the mouse early foregut endoderm leads to defects in lung development and cell cycle arrest of epithelial progenitor cells.
INTRODUCTION
Chromatin remodeling and epigenetic regulation have been shown to play important roles in specification and differentiation of the developing lung. Hdac1 and Hdac2 in particular are thought to play an essential role in the regulation of Sox2+ proximal lung endoderm progenitors by repressing Bmp4 signaling and the cell cycle inhibitors Cdkn2a (also known as p16), Cdkn1a (also known as p21) and Rb1 (also known as Rb and pRB) (Wang et al., 2013). Hdac3 is crucial for the proper remodeling and expansion of the distal lung saccules into primitive alveoli (Wang et al., 2016). Furthermore, loss of the histone H3 lysine 27 (H3K27) methyltransferase Ezh2 from early lung endoderm led to ectopic and premature appearance of basal cells and reduced secretory cell differentiation during development (Galvis et al., 2015; Snitow et al., 2015). The fate of developing lung mesoderm is similarly dependent upon these epigenetic factors; loss of Ezh2 from early mesoderm resulted in the formation of ectopic smooth muscle cell differentiation from the mesothelium in the developing lung (Snitow et al., 2016). Furthermore, evidence showed that these epigenetic factors are also important for postnatal tissue homeostasis, regeneration and disease. Hdac2 expression is decreased in chronic obstructive pulmonary disease (COPD) (Ito et al., 2005, 2006) and the combination of Hdac1 and Hdac2 is required for epithelial regeneration after naphthalene-induced lung injury (Wang et al., 2013). Although Hdac activity is clearly central to chromatin remodeling, the full activity and genome targeting of Hdacs requires their assembly into multi-subunit complexes. For example, the recruitment of the Hdac1/2 complex into the vicinity of specific target genes is determined by the association of Hdac1/2 with specific co-repressor proteins/complexes such as NuRD, REST/CoREST or Sin3, which mediates the interaction with other subunits and sequence-specific transcription factors (Thomas, 2014). However, the role of specific co-repressor proteins in cell fate decisions during embryonic development and postnatal tissue homeostasis remains poorly understood.
The Sin3-Hdac co-repressor complex is believed to be one of a family of multi-protein complexes that inhibit or stall the basal transcription machinery through on-site recruitment of chromatin remodeling enzymes leading to localized deacetylation of histones (Burke and Baniahmad, 2000; Silverstein and Ekwall, 2005). The core Sin3-Hdac complex consists of at least eight subunits, including the scaffold protein Sin3 (Sin3a, Sin3b), the histone deacetylases Hdac1 and Hdac2, the histone-binding proteins RBBP1 (Arid4a), RBBP4 and RBBP7, and the Sin3-associated proteins SAP30, SDS3 (Suds3) and the CpG-methylated binding protein MeCP2 (Hassig et al., 1997; Laherty et al., 1997; Lai et al., 2001; Nan et al., 1998; Silverstein and Ekwall, 2005; Zhang et al., 1997). This core complex also transiently associates with other regulatory proteins including Rb1 and chromatin regulatory enzymes to control gene expression (Kadamb et al., 2013; Silverstein and Ekwall, 2005). In addition to the transcriptional repression activity of the Sin3-Hdac complex, growing evidence suggests that Sin3 might also function to activate transcription of certain target genes in different organisms, including yeast and Drosophila, as well as mammalian systems such as mouse muscle development, mouse embryonic fibroblast and mouse embryonic stem cells (Baltus et al., 2009; Dannenberg et al., 2005; Das et al., 2013; Lin et al., 2005; Ruiz-Roig et al., 2010; van Oevelen et al., 2010; Yoshimoto et al., 1992), which would be consistent with the observation that Sin3 could also serve as a scaffold protein for the histone demethylase dKDM5/LID (Gajan et al., 2016). Thus, these studies suggest that Sin3 modulates transcriptional activity by serving as a scaffold protein able to coordinate multiple histone modification activities, including deacetylation and demethylation in a context-dependent manner.
In this article, we focus on the role of Sin3a during development of foregut endoderm-derived organs. We show that loss of Sin3a from early Shh-expressing foregut endoderm resulted in early postnatal mortality owing to respiratory insufficiency. Sin3a mutant mouse embryos generated all foregut-derived organs with the exception of lung tissue, which showed a profound growth defect with complete failure of epithelial branching morphogenesis. This was at least in part due to permanent cell cycle arrest of epithelial progenitor cells in G1 phase via upregulation of the cell cycle inhibitors Cdkn1a and Cdkn2c (also known as p18) (Seshadri and Campisi, 1990; Stein et al., 1990, 1991; Tominaga, 2015). Together, these studies show that the Sin3a chromatin-modifying complex is essential for lung endoderm development and that loss of Sin3a results in the induction of a senescence-like state in lung epithelial progenitor cells.
RESULTS
Loss of Sin3a leads to specific lung developmental defects
Analysis of Sin3a expression during lung development from embryonic day (E) 14.5 to postnatal stages by immunohistochemistry revealed robust Sin3a protein expression in both the endoderm and mesenchyme throughout all stages of development and postnatally (Fig. S1A). To investigate the importance of Sin3a during lung development, we generated a foregut endoderm-specific deletion of Sin3a using a Sin3aflox/flox allele, Rosa-mTmG reporter line and ShhCre line, which efficiently drives Cre-mediated recombination in anterior foregut endoderm from approximately E8.75 (Goss et al., 2009; Montgomery et al., 2007). Hereafter, animals from this line will be referred to as Sin3af/f (ShhCre/+;Sin3aflox/flox;Rosa-mTmG) or Sin3af/+ (ShhCre/+;Sin3aflox/+;Rosa-mTmG). Sin3af/f mutants are devoid of Sin3a expression in the lung epithelium (Fig. S1A) and died at birth as a result of respiratory failure. Gross and histological inspection of embryos showed an almost complete absence of lung tissue at birth (P0) (Fig. 1A). Further analysis of fetal lung development revealed that Sin3af/f mutants do form early lung buds at E10.5 but exhibit major developmental defects thereafter (Fig. 1B-F, Fig. S1B). Whole-mount fluorescence imaging of embryos and microdissected lung and gastrointestinal tracts showed GFP reporter expression in all foregut endoderm-derived organs, with major developmental defects restricted to the Sin3af/f lung. Development of all other foregut endoderm-derived tissue was comparable to control Sin3af/+ littermates (Fig. 1B,C, Fig. S1B). Evaluation of the temporal sequence of Sin3a-dependent developmental defects revealed that after separation from the foregut, an early lung bud forms and bifurcates to generate left and right primary lung buds. However, at this point development of mutant Sin3af/f lungs are arrested, whereas control Sin3af/+ lungs continue to undergo branching morphogenesis. Rather, after formation of the primary lung buds, Sin3af/f lungs appear to undergo epithelial degeneration with progressive loss of the trachea observed from E11.5 (Fig. 1D-F, Fig. S1B). In contrast, depletion of Sin3a in lung mesenchyme using the lung mesoderm-specific Tbx4Cre has no apparent effect on lung development (Fig. S1C,D). These data indicate that epithelial expression of Sin3a is essential for branching morphogenesis and growth of the primordial lung bud, but is dispensable for development of other foregut endoderm-derived tissues and lung mesoderm.
Fig. 1.
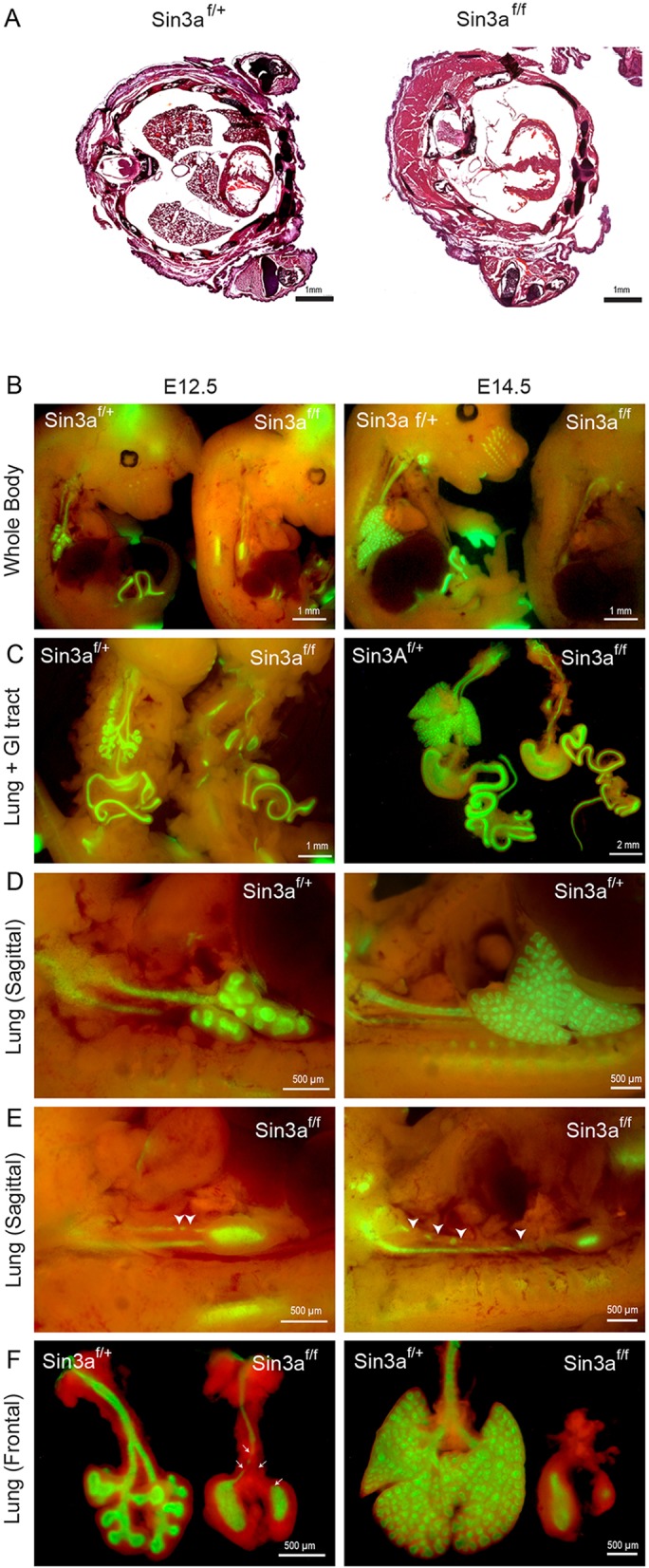
Loss of Sin3a leads to specific lung developmental defects. (A) Hematoxylin and Eosin staining of thoracic transverse section of neonatal (P0) Sin3af/f (ShhCre/+;Sin3aflox/flox;Rosa-mTmG) mutant and Sin3af/+ (ShhCre/+;Sin3aflox/+;Rosa-mTmG) littermate. (B-F) Whole-mount fluorescence imaging of whole embryo (B), lung and gastrointestinal tract (C) and lung (D-F) of Sin3af/f mutants and Sin3af/+ littermates at E12.5 and E14.5. Domain of Cre activity is indicated by GFP expression (green). Arrowheads in E indicate disruption of tracheal integrity.
Endodermal Sin3a is required for activation of gene expression programs associated with epithelial cell fate and lung development
To assess the molecular consequences of loss of Sin3a in early lung endoderm, RNA-seq analysis was performed on Sin3af/+ control and Sin3af/f mutant lungs at E12.5. We found that 678 genes were significantly upregulated and 636 genes were significantly downregulated. Within the top 100 differentially expressed genes we observed an over-representation of endodermal genes in the downregulated set and of mesodermal genes in the upregulated group (Fig. 2A). This was confirmed using the LungGENS database (Du et al., 2015) to cross-reference differentially expressed genes with cell-specific expression of genes involved in lung development, which showed that 64.9% of genes downregulated in Sin3af/f lungs mapped to genes that are expressed in epithelial cells during development (Fig. 2B). These include genes that code for the transcription factors Nkx2.1 (Nkx2-1), Foxa1, Foxa2 and Sox2, which are important for lung epithelial lineage specification; Shh, which is a key factor involved in epithelial-mesenchymal interactions; the alveolar type II marker Sftpc; and the epithelial adhesion proteins Epcam, Cldn1, Cldn4, Cldn6, Cldn10 and Cldn18 (Fig. 2C). Moreover, gene ontology (GO) analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID; Huang et al., 2009a,b) showed that the top GO terms (biological processes) enriched in genes downregulated more than twofold (149 genes) include processes involved in lung and respiratory system development, cell fate specification and cell-cell adhesion (Fig. 2D). Confirming the RNA-seq results, quantitative real-time PCR (Fig. S2A) showed differential expression of selected genes (Shh, Wnt2, Wnt7b, Bmp2, Bmp4, Fgf9, Fgf10 and Fgfr2) that are known to be important for regulating lung development (Bellusci et al., 1997). These data suggest that endodermal Sin3a is necessary for activation of gene expression programs associated with epithelial cell fate and branching morphogenesis. In contrast, the majority (57.1%) of genes upregulated in Sin3af/f lungs mapped to genes expressed in mesodermal cells during development, with only 15.2% of upregulated genes mapped to epithelial cells (Fig. 2E). These included Fgf10, which is known to be expressed in mesenchymal progenitor cells, and the mesoderm-specific extracellular matrix genes Col1a1, Col1a2, Col6a1 and Col6a2 (Fig. 2F). These data suggest that changes in epithelial cell fate caused by loss of endodermal Sin3a result from cell-autonomous changes in addition to non-cell-autonomous effects on lung mesoderm, probably as a result of aberrant epithelial-mesenchymal interactions.
Fig. 2.

Endodermal Sin3a is required for activation of gene expression programs associated with epithelial cell fate and lung development. (A) Heatmap of the top 100 differentially expressed genes from RNA-seq. (B,E) Pie charts of downregulated (B) and upregulated (E) genes mapped to fetal lung cells using LungGENS database. (C,F) Normalized fold change of RNA-Seq gene expression values for selected downregulated (C) and upregulated (F) genes. (D) Top GO terms for genes downregulated more than twofold. Significance determined by Cufflinks software; ***P<0.001,****P<0.0001.
Pathway analysis of differentially expressed genes by Ingenuity Pathway Analysis (IPA) predicts a number of canonical pathways to be altered by loss of Sin3a, including ‘Sonic Hedgehog Signaling’ and ‘Tight Junction Signaling’ (Fig. 3A). Additional network interaction analysis using IPA (Fig. 3B) confirmed downregulation of gene networks associated with lung epithelial development and upregulation of mesenchymal gene networks. Moreover, we identified an upregulation of gene networks involved in cell cycle inhibition (i.e. Cdkn1a and Cdkn2c).
Fig. 3.
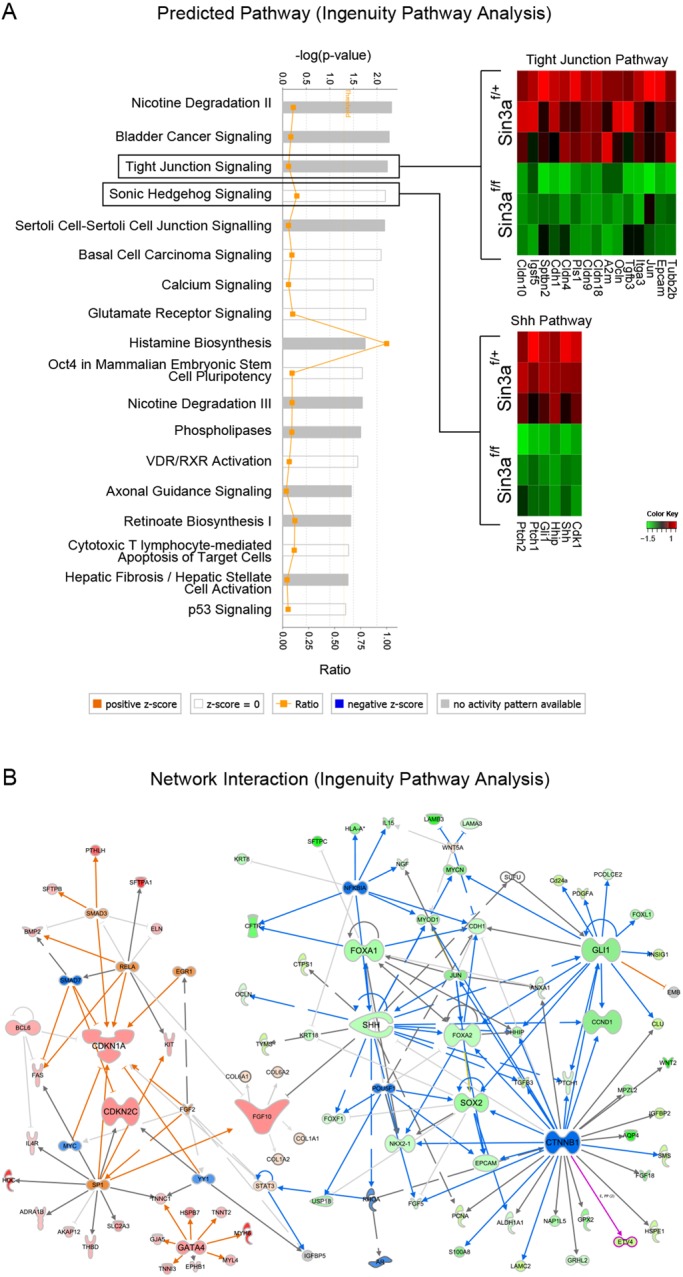
Multiple pathways are regulated by Sin3a loss of function. (A) Signaling pathways regulated by Sin3a predicted by Ingenuity Pathway Analysis with corresponding heatmaps of differentially expressed genes for ‘Tight Junction Signaling’ and ‘Sonic Hedgehog Signaling’. (B) Predicted network interaction of differentially expressed genes. Warm and cold colors represent up- and downregulated gene networks, respectively.
Given that deacetylation of histone tails by the Sin3-Hdac complex is one of the mechanisms utilized by cells to regulate gene transcription, we assessed whether loss of Sin3a would lead to histone deacetylation defects. Strikingly, immunostaining for histone H3 acetylation, a marker for histone acetylation level, showed no significant changes of histone H3 acetylation level, as determined by relative fluorescence intensity, between Sin3a-deficient epithelial cells and Sin3a-sufficient non-epithelial cells (Fig. S2B,C). This indicates that loss of Sin3a in epithelial cells has no effect on global histone H3 acetylation level. However, this does not exclude the possibility that the acetylation levels of other histones or individual target genes might have be affected by Sin3a loss of function (LOF). It is also plausible that redundancy between Sin3a and the other Sin3 family member Sin3b, which is unaltered in Sin3af/f mutant lung epithelial cells (Fig. S2D), might compensate some functions such as restoration of the functional Sin3-Hdac complex, in these mice. Interestingly, when we compared the significantly differentially expressed genes from our RNA-seq data with those of previously published Hdac1/2 double knockout microarray data (Wang et al., 2013), there was only a very small fraction of overlapping genes (Fig. S2E, Table S3). This suggests that the effects of Sin3a deficiency that we observed during lung development could be independent of the deacetylation activity of the Sin3-Hdac complex. This possibility is supported by our finding that the acetyltransferase inhibitor anacardic acid was unable to rescue the phenotype of Sin3af/f mutants both in vivo (Fig. S5A) and in vitro (Fig. S5B). Furthermore, our data suggest that Sin3a LOF cannot be functionally compensated for by presence of the related Sin3 family member Sin3b. These findings are consistent with previous publications suggesting that Sin3a also serves as scaffold protein for other histone modification complexes such as those involved in histone demethylation (Gajan et al., 2016) and that Sin3b has functions that only partially overlap with those of Sin3a, but are not redundant (Das et al., 2013; McDonel et al., 2012; van Oevelen et al., 2010).
Sin3a is required for lung branching morphogenesis and determination of epithelial cell fate in the developing lung
During early embryonic lung development in mouse, the respiratory tree develops in a distinct proximal-distal manner with Sox2 expression marking both progenitors and specialized cell types of the developing conducting airway (Arnold et al., 2011; Que et al., 2007; Sommer et al., 2009; Tompkins et al., 2011, 2009). In contrast, Sox9 expression demarks distal epithelial progenitors and a subpopulation of mesodermal cells associated with proximal lung endoderm (Chen et al., 2014; Rawlins et al., 2009; Rockich et al., 2013). To assess further the molecular effects of Sin3a LOF on lung branching morphogenesis, immunofluorescence co-staining of Sox2, Sox9 and the epithelial adherens junction marker E-cadherin (cadherin 1) was performed on lung sections from E12.5 Sin3af/f mutant and Sin3af/+ littermate control embryos. Compared with the Sin3af/+ lungs, which exhibit robust Sox2 immunoreactivity in the proximal airways of the developing lung bud, lungs from Sin3af/f mice were devoid of Sox2 protein immunoreactivity in the proximal airway remnant (Fig. 4A). All E-cadherin-positive cells in lungs of Sin3af/f mice were positive for both Sox9 and Nkx2.1 immunoreactivity, similar to observations in distal epithelial cells of control Sin3af/+ lungs (Fig. 4B). Differential abundance of Sox2, Sox9 and Nkx2.1 immunoreactivity was confirmed at the mRNA expression level by quantitative real-time PCR (Fig. 4C). At the pseudoglandular stage of lung development (E14.5), control Sin3af/+ lungs show immunoreactivity for the alveolar type II cell marker proSP-C, which marks early stages in the differentiation of Sox9-positive distal epithelial progenitors into functional distal epithelial cell types (Fig. 4D). In contrast, no proSP-C was detected in Sin3af/f mutant epithelial cells (Fig. 4D). These data suggest that loss of endodermal Sin3a arrests maturation of distal lung endoderm at the early Sox9− Sftpc− progenitor cell stage. As predicted by the transcriptome analysis, changes in epithelial cell fate due to loss of endodermal Sin3a also affected the development of the Sin3a-sufficient lung mesoderm. Despite the complete lack of epithelial branching in Sin3af/f mutant lung, the mesoderm did show evidence of branching and early lobe formation (Fig. 4E). However, immunostaining of the myofibroblast/smooth muscle marker alpha-smooth muscle actin (α-SMA) at E12.5 and E14.5 revealed a lack of lung mesenchymal differentiation (Fig. 4F). Notably, α-SMA expression in the esophagus, which was also deficient for endodermal Sin3a, was comparable to that of Sin3af/+ littermate control embryos (Fig. 4F). These data suggest that loss of endodermal Sin3a results in early distalization of the embryonic lung. However, subsequent differentiation into mature alveolar epithelial cells is inhibited and maturation of the lung mesenchyme is disrupted.
Fig. 4.

Sin3a is required for branching morphogenesis and cell fate in the developing lung. (A,B) Representative immunofluorescence staining of E12.5 embryonic lungs from Sin3af/f mutant and Sin3af/+ littermates showing Sox2 and E-cadherin (A), Sox9, E-cadherin (B, top) and Nkx2.1 (B, top). (C) Quantitative real-time PCR expression data for Sox2, Sox9 and Nkx2.1 for E12.5 embryonic lungs of Sin3af/f mutant and Sin3af/+ littermates. (D) Representative immunofluorescence staining of E14.5 embryonic lungs from Sin3af/f mutant and Sin3af/+ littermates showing proSP-C and E-cadherin. (E) Whole-mount confocal image of E12.5 embryonic lungs from Sin3af/f mutant and Sin3af/+ littermates. Dashed line delineates the mesoderm border of the right lobes. (F) Representative immunofluorescence staining of E12.5 and E14.5 embryonic lungs from Sin3af/f mutant and Sin3af/+ littermates showing spatial localization of α-SMA. Significance determined by Student's t-test; ns, not significant; **P<0.01, ****P<0.0001.
Loss of endodermal Sin3a results in epithelial cells adopting a senescence-like state with permanent cycle arrest in G1 phase via upregulation of Cdkn1a and Cdkn2c
Previous studies have shown that a common effect of the loss of Sin3a expression in stem cells and cancer cells is decreased cell proliferation coupled with increased apoptosis (Ellison-Zelski and Alarid, 2010; Heideman et al., 2014; McDonel et al., 2012). In this study, immunostaining for the apoptotic marker cleaved caspase 3 revealed that there are, albeit very few, apoptotic epithelial cells in Sin3af/f mutant lungs at E12.5, whereas Sin3af/+ control lungs were largely devoid of cleaved caspase 3 staining (Fig. 5A). Interestingly, cleaved caspase 3-positive cells were found in the lumen of Sin3af/f mutant lungs (Fig. 5A). With only very weak immunoreactivity for E-cadherin, it is unclear whether these cells represent exfoliated epithelial cells or cellular debris of other cell lineages. To determine the extent to which increased apoptosis contributed to the developmental defect in Sin3af/f mutant lungs, we performed in vitro lung explant (E11.5) cultures and treated them with the pan-caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-(O-methyl)- fluoromethylketone (Z-VAD-FMK) or DMSO for 48 h. In DMSO control cultures, Sin3af/+ control lungs underwent branching morphogenesis, whereas Sin3af/f mutant lungs exhibited a complete failure of branching morphogenesis, equivalent to that observed in vivo during development (Fig. 5B). In Z-VAD-FMK-treated cultures the inhibition of caspase activity was insufficient to restore branching morphogenesis, but did halt the rapid death of epithelial cells and spread of mesenchymal cells (Fig. 5B). These data suggest that the action of Sin3a in lung development is not mediated through apoptosis alone.
Fig. 5.
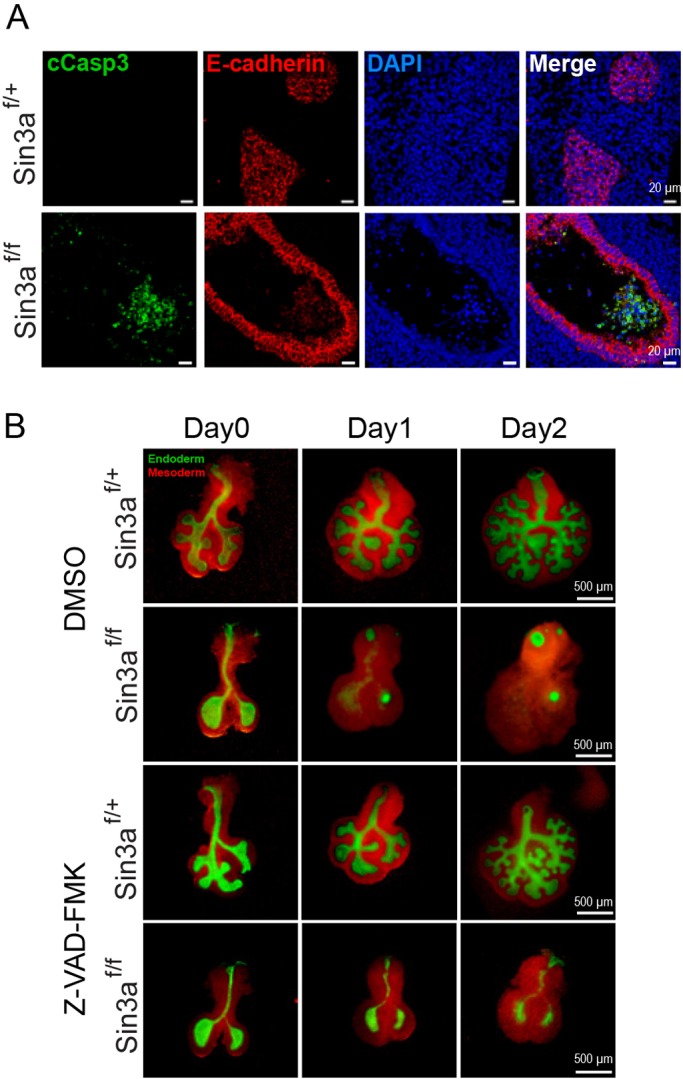
Disruption of lung development in Sin3af/f mutant mice is only partially mediated by apoptosis. (A) Representative immunofluorescence staining of E12.5 embryonic lungs from Sin3af/f mutant and Sin3af/+ littermates showing cleaved caspase 3 (cCasp3) and E-cadherin. (B) Whole-mount images of E11.5 lung explant cultures supplemented with the caspase inhibitor Z-VAD-FMK or vehicle (DMSO) control.
To investigate the role of cell cycling, we began by analyzing all the genes that were significantly downregulated in Sin3af/f mutant lungs in the context of cell cycle-related functions. This revealed an enrichment of GO terms associated with positive regulation of cell cycle (Fig. 6A), which included 46 key cell cycle regulatory genes that were downregulated due to loss of Sin3a (Fig. 6B). Notably, however, two of the key G1/S transition cell cycle inhibitors, Cdkn1a and Cdkn2c, were significantly upregulated (Fig. 6B). To clarify further the effect of Sin3a deficiency on cell cycling in the embryonic lung at E12.5 (Fig. 6C-G) and E14.5 (Fig. S3) we performed immunofluorescence staining of markers expressed at various stages of the cell cycle. Ki-67 (Mki67), which is present during all active phases of the cell cycle (G1, S, G2 and mitosis), was abundantly expressed in almost every cell (epithelial and mesenchymal) in both Sin3af/+ control and Sin3af/f mutant lungs (Fig. 6C,G, Fig. S3A). This is as expected for cells during normal lung development where cells are highly proliferative, but implies that Sin3af/f mutant cells are also actively cycling despite the failure of branching morphogenesis and lack of lung growth. At the same time, phospho-histone-H3-Serine10 (pH3S10) staining, which labels mitotic cells from G2 phase through to anaphase, also showed no significant difference in the abundance of pH3S10-immunolabeled epithelial cells in Sin3af/+ control (mean±s.e.m.: 31.76±2.724%) and Sin3af/f mutant (27.613±3.818%) lungs at E12.5 (Fig. 6D,G) and only a slight, albeit significant, decrease in pH3S10-immunolabeled epithelial cells in Sin3af/f mutant lungs at E14.5 (Fig. S3B,D). This suggests that about 70% of epithelial cells should be in either G1 or S phase of the cell cycle. However, when we performed a 2 h bromodeoxyuridine (BrdU) labeling to mark actively proliferating cells, there was a complete lack of BrdU uptake by E12.5 Sin3af/f mutant lung epithelial cells compared with Sin3af/+ control lung epithelial cells, of which 50.908±6.667% were positive for BrdU uptake by immunostaining (Fig. 6E,G). A similar lack of BrdU uptake was observed in E14.5 Sin3af/f mutant lungs following a 2 h BrdU labeling (Fig. S3C,D). Moreover, after 2 days of continual BrdU labeling from E12.5 to E14.5, which resulted in every Sin3af/+ control lung epithelial cell taking up BrdU, Sin3af/f mutant lung epithelial cells again showed no evidence of BrdU uptake (Fig. S3E,F). In contrast, there was no significant difference in BrdU uptake by lung mesenchymal cells or stomach epithelial and mesenchymal cells (Fig. S4). This is consistent with observations that discernable morphological abnormalities in Sin3af/f mutant mice are lung specific (Fig. 1).
Fig. 6.

Loss of endodermal Sin3a results in epithelial cell cycle arrest at G1 via upregulation of Cdkn1a and Cdkn2c. (A) Gene ontology analysis of significantly downregulated genes related to cell cycle. (B) Heatmap of differentially expressed cell cycle genes from RNA-seq data. (C-F) Representative immunofluorescence staining of E12.5 embryonic lungs from Sin3af/f mutant and Sin3af/+ littermates showing Ki-67 and E-cadherin (C), pH3S10 and E-cadherin (D), BrdU (after 2 h labeling) and E-cadherin (E) and Cdkn1a and Sin3a (F). (G) Mitotic index of Ki-67, BrdU and pH3S10 as determined by relative percentage of immunopositive epithelial cells. (H) Quantitative real-time expression data for the genes encoding the G1/S checkpoint proteins Cdkn1a and Cdkn2c, the G1/S transition-promoting protein Cdk1, Ccne1 and Ccnd1 and the DNA replication licensing factors Mcm7 and Mcm8. (I) CHIP qPCR using Sin3a antibody on Cdkn1a and Cdkn2c promoters. Significance determined by Student's t-test; ns, not significant; *P<0.05,***P<0.001,****P<0.0001.
Given that BrdU incorporates in newly synthesized DNA, which occurs during S phase, these data suggest that although Sin3af/f mutant lung epithelial cells enter the cell cycle (i.e. are positive for Ki-67 and pH3S10) the same as littermate controls, the majority of Sin3af/f mutant lung epithelial cells appear to be arrested in G1 phase. This is likely to be due to cell cycle arrest at the G1/S checkpoint with the upregulation of the G1/S transition cell cycle inhibitors Cdkn1a and Cdkn2c, as indicated by the RNA-seq data (Fig. 6B).
In order to verify an increase in G1/S checkpoint activity, we next performed immunostaining of Cdkn1a on E12.5 embryos, which showed that Cdkn1a is expressed at detectable levels in Sin3af/f mutant lung epithelial cells only (Fig. 6F). Cdkn1a was not detected in Sin3af/+ control lung epithelial cells or mesenchymal cells from mutant and control mice. In addition, quantitative real-time PCR analysis of cell cycle checkpoint and regulatory genes confirmed an upregulation of the genes encoding the G1/S checkpoint proteins Cdkn1a and Cdkn2c, along with downregulation of the G1/S transition-promoting factors Ccnd1, Ccne1 and Cdk1 and the DNA replication licensing factors Mcm7 and Mcm8. To ascertain whether Cdkn1a and Cdkn2c are direct targets of the Sin3a repressor complex, we performed chromatin immunoprecipitation (ChIP) assays using chromatin extracts from E14.5 lungs. These studies showed that Sin3a bound robustly to the proximal promoters of Cdkn1a and Cdkn2c (Fig. 6I). These data suggest that Sin3a regulates the fate of lung epithelial progenitor cells, at least in part, through direct targeting of cell cycle inhibitors, including Cdkn1a and Cdkn2c, during lung development. This is indicative of cells entering a senescence-like state, which is defined as cell cycle arrested at G1 phase with the inability to enter S phase, an outcome that is typically associated with upregulation of Cdkn1a (Seshadri and Campisi, 1990; Stein et al., 1990, 1991; Tominaga, 2015). Furthermore, senescence-associated β-galactosidase staining (Fig. S6A) and γH2AX staining (Fig. S6B) further support the notion that the endodermal lining of proximal lung tubules of E12.5 Sin3af/f mutant embryos include numerous senescent cells that were not observed in wild-type embryos.
DISCUSSION
Here, we use Sin3a conditional LOF mouse models to define roles for this transcriptional co-repressor in development of the foregut endoderm. We show that the consequences of Sin3a loss are benign among all endodermal tissues in the domain of ShhCre activity with the exception of the developing lung. Developing lung endoderm was completely arrested at the early embryonic bud stage in Sin3a LOF embryos, which led to non-cell-autonomous changes to lung mesoderm. Developing lungs of Sin3a LOF embryos established distinct proximal and distal endodermal fates, yet failed to undergo normal programs of lung branching morphogenesis and epithelial cell differentiation. Instead, lung endoderm of Sin3a LOF embryos showed significant downregulation of genes involved in formation of tight and adherens junctions typical of a normal epithelium, and showed enhanced expression of cell cycle inhibitory and pro-apoptotic genes, leading to atrophy of the developing endoderm. The defect in endoderm formation in Sin3a mutant embryos was associated with reduced expression of key regulators of developing lung mesoderm, including sonic hedgehog, and a corresponding increase in the expression of mesodermally derived endodermal growth factors including FGF10. These data suggest that the developmental requirement for Sin3a is lung specific among foregut-endoderm-derived tissues, wherein it is required for establishment of epithelial cell identity and function.
Previous work has shown that conditional loss of Hdac1/2 within early lung endoderm leads to defects in lung development including a branching defect and loss of the Sox2+ endodermal progenitors that normally generate airway epithelium (Wang et al., 2013). Histone deacetylases 1 and 2 usually form a heterodimer in multi-protein chromatin remodeling complexes with other co-factors, such as Sin3a, NuRD and REST/CoREST, which are important both for enzymatic activity and specificity of DNA/chromatin interactions (Dovey et al., 2010; Ji et al., 2014; Jia et al., 2012; Lagger et al., 2002; Mielcarek et al., 2013; Reichert et al., 2012; Trivedi et al., 2007; Tsai and Seto, 2002; Zimmermann et al., 2007). A surprise finding from our study was that Sin3a LOF not only recapitulated many of the defects observed with Hdac1/2 LOF, but led to more profound defects at an earlier stage of lung development. This was particularly notable as both the Sin3a LOF mice evaluated herein and the Hdac1/2 LOF mice evaluated by Wang et al. (2013) used the same ShhCre driver line to achieve efficient recombination of the respective floxed alleles in developing foregut endoderm. Given that the Sin3 complex represents only one of multiple potential chromatin remodeling complexes that include Hdac1/2 enzymes, these data suggest either that the Sin3a-containing Hdac1/2 complex is the predominant driver of Hdac1/2-dependent effects on lung development and/or that Sin3a fulfills Hdac1/2-independent functions to regulate the fate of early lung endoderm. We attribute the surprisingly limited overlap between altered lung transcriptomes of Sin3a LOF and Hdac1/2 LOF embryos to the much earlier developmental defects observed in the former. Sin3a-dependent defects in lung endoderm in our study are consistent with the recent results of Heideman et al. (2014) in the hematopoietic system, where Sin3a LOF phenocopied the effects of Hdac1/2 LOF on renewal and differentiation of hematopoietic stem and multipotent progenitor cells. Furthermore, cell type-specific differences in Sin3a function within breast cancer cells, in which estrogen receptor alpha status correlated with Sin3a-dependent tumor growth and survival (Ellison-Zelski and Alarid, 2010), shares similarities with our observation of cell/tissue-specific differences in the requirement for Sin3a within the foregut endoderm.
Lung endoderm-specific defects associated with Sin3a LOF included cell cycle inhibition and apoptosis. Sin3a has been reported to be essential for the viability and cell cycle regulation of pluripotent cells (McDonel et al., 2012), and together with Cul4B regulate cell cycle G1/S transition by targeting Cdkn1a and Cdkn1c (also known as p57) in mouse embryonic fibroblast cells (Ji et al., 2014). Here, we show that loss of Sin3a leads to upregulation of the cell cycle checkpoint inhibitors Cdkn1a and Cdkn2c accompanied by G1 cell cycle arrest. Sin3a bound directly to proximal promoter regions of both Cdkn1a and Cdkn2c, a finding consistent with its role as a transcriptional repressor. Cdkn1a immunoreactivity was elevated among Sin3a-deficient epithelial cells but not Sin3a-sufficient epithelial or mesenchymal cells, further supporting the notion that Cdkn1a is a direct downstream target of Sin3a. Decreased expression of Cdk1, Ccnd1 and Ccne1, which are required for G1/S transition, and of Mcm7 and Mcm8, which are required for DNA synthesis, further support the crucial role played by Sin3a in regulating G1/S phase of the cell cycle. Furthermore, the lack of BrdU incorporation even after 2 days of BrdU continuous labeling suggested that this G1/S arrest is a senescence-like permanent arrest. However, Sin3a-dependent effects on cell cycle progression were not limited to regulators of G1/S transition. Interestingly, despite the complete block in S phase we were able to detect pH3S10 immunoreactivity within Sin3a-deficient E14.5 lung endoderm. This was associated with elevated cleaved caspase 3 immunoreactivity suggesting that arrested cell cycle progression, perhaps coupled with loss of cellular junctional communication associated with reduced expression of tight junction proteins, led to apoptosis of Sin3a-deficient lung endoderm and degeneration of developing airways.
Evidence to suggest Hdac1/2-independent functions for Sin3a include an earlier and more severe defect in developing lungs of Sin3a LOF embryos compared with Hdac1/2 LOF embryos and only partial overlap of altered lung transcriptomes of E12.5 embryos (herein and Wang et al., 2013). Furthermore, we show that Sin3a is co-expressed with the related Sin3b isoform in both developing lung endoderm and mesoderm, and that Sin3a LOF is not accompanied by gross changes in H3 acetylation levels in lung endoderm. These data suggest that expression of Sin3b might be sufficient to maintain the histone deacetylase activity of the Sin3-Hdac1/2 complex. It remains possible that subtle changes in H3 acetylation on individual genes could be present in Sin3a LOF embryos but could not be measured in the present study. However, our data suggest that Hdac1/2-independent functions of Sin3a might be involved in early stages of lung endoderm development. Our findings reinforce those of others suggesting that Sin3a regulates gene expression in Sin3a-dependent tissues through both Hdac1/2-dependent or -independent mechanisms (Baltus et al., 2009; Ellison-Zelski and Alarid, 2010; Lin et al., 2005).
Our findings highlight the crucial role played by Sin3a in regulating the fate of early lung endoderm progenitor cells via transcriptional repression of the cell cycle inhibitors Cdkn1a and Cdkn2c to prevent induction of a senescence-like state. It is thought that cellular senescence is involved in the pathogenesis of many chronic lung diseases including COPD and idiopathic pulmonary fibrosis, which suggests that Sin3a might also play a role in regulating epithelial progenitor cell fate in the postnatal lung. Future studies are needed to determine the activity of Sin3a in these diseases and whether the same transcriptional regulation is involved in cell fate decisions in the postnatal lung. In conclusion, these studies highlight a role for specific co-repressor proteins such as Sin3a in regulating tissue-specific transcriptional activity and cell fate decisions during lung development.
MATERIALS AND METHODS
Mouse strains
ShhCre; Sin3aflox/flox; Rosa26-mT/mG mice and ShhCre; Sin3aflox/+; Rosa26-mT/mG mice were generated by crossing ShhCre mice (The Jackson Laboratory, stock number 005622) with Rosa26-mT/mG (The Jackson Laboratory, stock number 007576) and Sin3aflox/flox mice (Dannenberg et al., 2005). Tbx4Cre; Sin3aflox/flox; Rosa26-mT/mG mice and Tbx4Cre; Sin3aflox/+; Rosa26-mT/mG mice were generated by crossing Tbx4Cre mice (Kumar et al., 2014; Xie et al., 2016) with Rosa26-mT/mG and Sin3aflox/flox mice. All mice were maintained and treatments were carried out according to Institutional Animal Care and Use Committee-approved protocols.
Immunofluorescence staining, imaging and quantification
Sections (7 μm thick) were collected from embryonic lung tissues fixed with 4% paraformaldehyde and embedded in Histogel. De-waxing and antigen retrieval was performed and sections incubated with primary antibodies at 4°C overnight. Sections were washed with PBS and incubated with fluorochrome-conjugated secondary antibody and DAPI for 90 min at room temperature. Sections were washed again and mounted in Fluoromount G. Primary antibodies used for immunofluorescence staining included: chicken anti-GFP (1:1000, Abcam, ab13970), goat anti-Sox2 (1:200, R&D Systems, AF2018), rabbit anti-Sox9 (1:200, Millipore, AB5535), mouse anti-Nkx2.1 (1:500, Seven Hills Bioreagents, WMAB-8G7G31), mouse anti-E-cadherin (1:1000, BD Transduction Laboratories, 610182), rat anti-BrdU (1:300, Accurate Chemical & Scientific Corporation, OBT0030), rabbit anti-Ki-67 (1:1000, Abcam, ab15580), rabbit anti-phospho-histone H3 serine10 (1:200, Cell Signaling, CST 9701), rabbit anti-cleaved caspase 3 (1:200, Cell Signaling, CST 9661), rabbit anti-proSP-C (1:1000, Millipore, AB3786), mouse anti-smooth muscle actin (1:10,000, Sigma-Aldrich, A2547), rabbit anti-Sin3a (1:100, Santa Cruz Biotechnologies, sc-767), mouse anti-p21 (1:100, Santa Cruz Biotechnologies, sc-6246), rabbit anti-acetyl-histone H3 (1:100, Millipore, 06-599), mouse anti-γH2AX[pS139] (1:500, Novus Biologicals, NB100-384). Secondary antibodies were: Alexa Fluor 647-conjugated donkey anti-mouse IgG(H+L) (1:1000, Life Technologies, A31571), Alexa Fluor 594-conjugated donkey anti-rat IgG(H+L) (1:1000, Life Technologies, A21209), Alexa Fluor 594-conjugated donkey anti-rabbit IgG(H+L) (1:1000, Life Technologies, A21207), Alexa Fluor 594-conjugated donkey anti-goat IgG(H+L) (1:1000, Life Technologies, a11058), Alexa Fluor 488-conjugated donkey anti-goat IgG(H+L) (1:1000, Life Technologies, A11055), Alexa Fluor 488-conjugated donkey anti-rabbit IgG(H+L) (1:1000, Life Technologies, A21206), Alexa Fluor 488-conjugated donkey anti-mouse IgG(H+L) (1:1000, Life Technologies, A21202), Alexa Fluor 488-conjugated goat anti-chicken IgG(H+L) (1:1000, Life Technologies, A11039) and Alexa Fluor 488-conjugated donkey anti-chicken IgY (IgG)(H+L) (1:1000, Jackson ImmunoResearch, 703-546-155). Visualization and image capture of immunofluorescence staining was performed using a Zeiss LSM 780 confocal microscope. Quantification was performed using tissue sections obtained from at least three independent mice per condition. Whole-mount embryonic lungs were imaged using a Zeiss Discovery V8 SteREO Microscope.
RNA-seq
RNA was extracted from lung tissue of ShhCre; Sin3aflox/flox; Rosa26-mT/mG mice (experimental group) and ShhCre; Sin3aflox/+; Rosa26-mT/mG mice (control group) using a miRNeasy Micro Kit (Qiagen). Library preparation and sequencing were performed by Cedars-Sinai Genomic Core using Illumina NextSeq 500 (Illumina) with paired-end 75bpx2 sequencing chemistry. On average, about 20 million reads were generated from each sample. Raw reads were aligned using TopHat (Bowtie2) aligner. Differential gene expression was determined by Cufflinks Assembly & DE, Version 2.0.0. Top 100 differential genes were determined based on fold change and test statistics. A full list of genes differentially expressed between groups is provided in Table S2. Overlap with genes dysregulated with Hdac1/2 LOF (Wang et al., 2013) are shown in Table S3.
Quantitative real-time PCR
Complementary DNA was synthesized from total RNA by using iScript Reverse Transcription Supermix (Bio-Rad). Quantitative RT-PCR was performed using the SYBR Green Master Mix (Bio-Rad) with the primers listed in Table S1. Gapdh expression values were used to control for RNA quality and quantity. Experiments were performed with at least five independent biological replicates and data shown represent the average ±s.e.m.
ChIP assays
Sin3a ChIP was performed using at least ten lungs from E14.5 mouse embryos. Lung tissue was crosslinked using 1% formaldehyde. Crosslinked tissue was sonicated to obtain genomic DNA fragments between 200 and 400 bp. Chromatin was prepared using a CHIP-IT High Sensitivity kit (Active Motif), immunoprecipitated using rabbit anti-Sin3a (Santa Cruz Biotechnologies, sc-767) and detected by quantitative real-time PCR with the primers listed in Table S1. Rabbit IgG was used as a negative control antibody in these assays (Snitow et al., 2015).
Embryonic lung explant culture
Lung buds were microdissected from E11.5 embryos and cultured as previously described (Goss et al., 2011) in the presence of 50 μg/ml Z-VAD-FMK (Bachem, N-1510), 20 μM of anacardic acid (Sigma-Aldrich) or DMSO as control. Images were captured by a Zeiss Discovery V8 SteREO Microscope.
BrdU labeling index
BrdU (Sigma-Aldrich) was injected at a concentration of 50 mg/kg body weight at 2 h before sacrifice or every 12 h for 2 days before sacrifice. Immunofluorescence staining with rat anti-BrdU primary antibody was carried out on 7-μm-thick sections as described above. Cells with positive or negative for BrdU staining were counted.
Anacardic acid treatment
Anacardic acid (Sigma-Aldrich) was administered by intraperitoneal injection at a dose of 5 mg/kg per day from E8.5 to E12.5; embryonic lungs were harvested and analyzed at E12.5.
Senescence-associated β-galactosidase detection
E12.5 embryonic lungs were dissected and stained using Senescence Cells Histochemical Staining Kit (Sigma-Aldrich, CS0030-1KT) to detect senescence-associated β-galactosidase detection following the manufacturer's recommendations.
Statistical analysis
Data were analyzed and compared between groups by one-way ANOVA with significance of differences determined by two-tailed, unpaired Student's t-test, (Prism, GraphPad). P<0.05 was considered statistically significant and significance levels are presented as *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Acknowledgements
We are grateful to Jordan Brown, and the Cedars-Sinai Genomics Core for their assistance with low-input RNA-Seq. We also thank the Stripp lab members for suggestions and critical reading of the manuscript. We also thank Dr Dianhua Jiang for feedback on experimental design.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: C.Y.; Methodology: C.Y., B.R.S.; Software: C.Y.; Validation: C.Y.; Formal analysis: C.Y., J.L.M.; Investigation: C.Y., G.C., B.K., X.G., T.M., N.C., M.K., A.K., J.L.M.; Resources: G.D.; Data curation: C.Y.; Writing - original draft: C.Y., J.L.M., B.R.S.; Writing - review & editing: C.Y., G.C., T.M., N.C., G.D., B.R.S.; Supervision: J.L.M., B.R.S.
Funding
This study was funded by the California Institute of Regenerative Medicine (LA1-06915) and the National Institutes of Health (LRRC-UO1HL111018 and 1T32HL134637-01). Deposited in PMC for release after 12 months.
Data availability
RNA-seq data have been deposited into the Gene Expression Omnibus database with accession number GSE94306.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.149708.supplemental
References
- Arnold K., Sarkar A., Yram M. A., Polo J. M., Bronson R., Sengupta S., Seandel M., Geijsen N. and Hochedlinger K. (2011). Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9, 317-329. 10.1016/j.stem.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltus G. A., Kowalski M. P., Tutter A. V. and Kadam S. (2009). A positive regulatory role for the mSin3A-HDAC complex in pluripotency through Nanog and Sox2. J. Biol. Chem. 284, 6998-7006. 10.1074/jbc.M807670200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellusci S., Grindley J., Emoto H., Itoh N. and Hogan B. L. (1997). Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124, 4867-4878. [DOI] [PubMed] [Google Scholar]
- Burke L. J. and Baniahmad A. (2000). Co-repressors 2000. FASEB J. 14, 1876-1888. 10.1096/fj.99-0943rev [DOI] [PubMed] [Google Scholar]
- Chen Z., Huang J., Liu Y., Dattilo L. K., Huh S.-H., Ornitz D. and Beebe D. C. (2014). FGF signaling activates a Sox9-Sox10 pathway for the formation and branching morphogenesis of mouse ocular glands. Development 141, 2691-2701. 10.1242/dev.108944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg J.-H., David G., Zhong S., van der Torre J., Wong W. H. and DePinho R. A. (2005). mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Gene Dev. 19, 1581-1595. 10.1101/gad.1286905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T. K., Sangodkar J., Negre N., Narla G. and Cagan R. L. (2013). Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 32, 3184-3197. 10.1038/onc.2012.326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey O. M., Foster C. T. and Cowley S. M. (2010). Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc. Natl. Acad. Sci. USA 107, 8242-8247. 10.1073/pnas.1000478107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y. N., Guo M. Z., Whitsett J. A. and Xu Y. (2015). ‘LungGENS’: a web-based tool for mapping single-cell gene expression in the developing lung. Thorax 70, 1092-1094. 10.1136/thoraxjnl-2015-207035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison-Zelski S. J. and Alarid E. T. (2010). Maximum growth and survival of estrogen receptor-alpha positive breast cancer cells requires the Sin3A transcriptional repressor. Mol. Cancer 9, 263 10.1186/1476-4598-9-263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajan A., Barnes V. L., Liu M., Saha N. and Pile L. A. (2016). The histone demethylase dKDM5/LID interacts with the SIN3 histone deacetylase complex and shares functional similarities with SIN3. Epigenet. Chromatin 9, 4 10.1186/s13072-016-0053-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvis L. A., Holik A. Z., Short K. M., Pasquet J., Lun A. T. L., Blewitt M. E., Smyth I. M., Ritchie M. E. and Asselin-Labat M.-L. (2015). Repression of Igf1 expression by Ezh2 prevents basal cell differentiation in the developing lung. Development 142, 1458-1469. 10.1242/dev.122077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss A. M., Tian Y., Tsukiyama T., Cohen E. D., Zhou D., Lu M. M., Yamaguchi T. P. and Morrisey E. E. (2009). Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev. Cell 17, 290-298. 10.1016/j.devcel.2009.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss A. M., Tian Y., Cheng L., Yang J. F., Zhou D., Cohen E. D. and Morrisey E. E. (2011). Wnt2 signaling is necessary and sufficient to activate the airway smooth muscle program in the lung by regulating myocardin/Mrtf-B and Fgf10 expression. Dev. Biol. 356, 541-552. 10.1016/j.ydbio.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassig C. A., Fleischer T. C., Billin A. N., Schreiber S. L. and Ayer D. E. (1997). Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 89, 341-347. 10.1016/S0092-8674(00)80214-7 [DOI] [PubMed] [Google Scholar]
- Heideman M. R., Lancini C., Proost N., Yanover E., Jacobs H. and Dannenberg J.-H. (2014). Sin3a-associated Hdac1 and Hdac2 are essential for hematopoietic stem cell homeostasis and contribute differentially to hematopoiesis. Haematologica 99, 1292-1303. 10.3324/haematol.2013.092643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T. and Lempicki R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1-13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T. and Lempicki R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44-57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Ito K., Ito M., Elliott W. M., Cosio B., Caramori G., Kon O. M., Barczyk A., Hayashi S., Adcock I. M., Hogg J. C. et al. (2005). Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 352, 1967-1976. 10.1056/NEJMoa041892 [DOI] [PubMed] [Google Scholar]
- Ito K., Yamamura S., Essilfie-Quaye S., Cosio B., Ito M., Barnes P. J. and Adcock I. M. (2006). Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappa B suppression. J. Exp. Med. 203, 7-13. 10.1084/jem.20050466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Q., Hu H., Yang F., Yuan J., Yang Y., Jiang L., Qian Y., Jiang B., Zou Y., Wang Y. et al. (2014). CRL4B interacts with and coordinates the SIN3A-HDAC complex to repress CDKN1A and drive cell cycle progression. J. Cell Sci. 127, 4679-4691. 10.1242/jcs.154245 [DOI] [PubMed] [Google Scholar]
- Jia H. Q., Pallos J., Jacques V., Lau A., Tang B., Cooper A., Syed A., Purcell J., Chen Y., Sharma S. et al. (2012). Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington's disease. Neurobiol. Dis. 46, 351-361. 10.1016/j.nbd.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadamb R., Mittal S., Bansal N., Batra H. and Saluja D. (2013). Sin3: Insight into its transcription regulatory functions. Eur. J. Cell Biol. 92, 237-246. 10.1016/j.ejcb.2013.09.001 [DOI] [PubMed] [Google Scholar]
- Kumar M. E., Bogard P. E., Espinoza F. H., Menke D. B., Kingsley D. M. and Krasnow M. A. (2014). Defining a mesenchymal progenitor niche at single-cell resolution. Science 346, 827 10.1126/science.1258810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger G., O'Carroll D., Rembold M., Khier H., Tischler J., Weitzer G., Schuettengruber B., Hauser C., Brunmeir R., Jenuwein T. et al. (2002). Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 21, 2672-2681. 10.1093/emboj/21.11.2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laherty C. D., Yang W.-M., Sun J.-M., Davie J. R., Seto E. and Eisenman R. N. (1997). Histone deacetylases associated with the mSin3 corepressor mediate Mad transcriptional repression. Cell 89, 349-356. 10.1016/S0092-8674(00)80215-9 [DOI] [PubMed] [Google Scholar]
- Lai A., Kennedy B. K., Barbie D. A., Bertos N. R., Yang X. J., Theberge M.-C., Tsai S.-C., Seto E., Zhang Y., Kuzmichev A. et al. (2001). RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol. Cell. Biol. 21, 2918-2932. 10.1128/MCB.21.8.2918-2932.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T. X., Chao C., Saito S., Mazur S. J., Murphy M. E., Appella E. and Xu Y. (2005). P53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 7, U165-U180. 10.1038/ncb1211 [DOI] [PubMed] [Google Scholar]
- McDonel P., Demmers J., Tan D. W. M., Watt F. and Hendrich B. D. (2012). Sin3a is essential for the genome integrity and viability of pluripotent cells. Dev. Biol. 363, 62-73. 10.1016/j.ydbio.2011.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek M., Landles C., Weiss A., Bradaia A., Seredenina T., Inuabasi L., Osborne G. F., Wadel K., Touller C., Butler R. et al. (2013). HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 11, e1001717 10.1371/journal.pbio.1001717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery R. L., Davis C. A., Potthoff M. J., Haberland M., Fielitz J., Qi X. X., Hill J. A., Richardson J. A. and Olson E. N. (2007). Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790-1802. 10.1016/j.ydbio.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X. S., Ng H. H., Johnson C. A., Laherty C. D., Turner B. M., Eisenman R. N. and Bird A. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386-389. 10.1038/30764 [DOI] [PubMed] [Google Scholar]
- Que J., Okubo T., Goldenring J. R., Nam K.-T., Kurotani R., Morrisey E. E., Taranova O., Pevny L. H. and Hogan B. L. M. (2007). Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 134, 2521-2531. 10.1242/dev.003855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlins E. L., Clark C. P., Xue Y. and Hogan B. L. M. (2009). The Id2+ distal tip lung epithelium contains individual multipotent embryonic progenitor cells. Development 136, 3741-3745. 10.1242/dev.037317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert N., Choukrallah M.-A. and Matthias P. (2012). Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell. Mol. Life Sci. 69, 2173-2187. 10.1007/s00018-012-0921-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockich B. E., Hrycaj S. M., Shih H. P., Nagy M. S., Ferguson M. A. H., Kopp J. L., Sander M., Wellik D. M. and Spence J. R. (2013). Sox9 plays multiple roles in the lung epithelium during branching morphogenesis. Proc. Natl. Acad. Sci. USA 110, E4456-E4464. 10.1073/pnas.1311847110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Roig C., Viéitez C., Posas F. and de Nadal E. (2010). The Rpd3L HDAC complex is essential for the heat stress response in yeast. Mol. Microbiol. 76, 1049-1062. 10.1111/j.1365-2958.2010.07167.x [DOI] [PubMed] [Google Scholar]
- Seshadri T. and Campisi J. (1990). Repression of c-fos transcription and an altered genetic program in senescent human fibroblasts. Science 247, 205-209. 10.1126/science.2104680 [DOI] [PubMed] [Google Scholar]
- Silverstein R. A. and Ekwall K. (2005). Sin3: a flexible regulator of global gene expression and genome stability. Curr. Genet. 47, 1-17. 10.1007/s00294-004-0541-5 [DOI] [PubMed] [Google Scholar]
- Snitow M. E., Li S., Morley M. P., Rathi K., Lu M. M., Kadzik R. S., Stewart K. M. and Morrisey E. E. (2015). Ezh2 represses the basal cell lineage during lung endoderm development. Development 142, 108-117. 10.1242/dev.116947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitow M., Lu M. M., Cheng L., Zhou S. and Morrisey E. E. (2016). Ezh2 restricts the smooth muscle lineage during mouse lung mesothelial development. Development 143, 3733-3741. 10.1242/dev.134932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C. A., Stadtfeld M., Murphy G. J., Hochedlinger K., Kotton D. N. and Mostoslavsky G. (2009). Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells 27, 543-549. 10.1634/stemcells.2008-1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein G. H., Beeson M. and Gordon L. (1990). Failure to phosphorylate the retinoblastoma gene product in senescent human fibroblasts. Science 249, 666-669. 10.1126/science.2166342 [DOI] [PubMed] [Google Scholar]
- Stein G. H., Drullinger L. F., Robetorye R. S., Pereira-Smith O. M. and Smith J. R. (1991). Senescent cells fail to express cdc2, cycA, and cycB in response to mitogen stimulation. Proc. Natl. Acad. Sci. USA 88, 11012-11016. 10.1073/pnas.88.24.11012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas E. A. (2014). Involvement of HDAC1 and HDAC3 in the pathology of polyglutamine disorders: therapeutic implications for selective HDAC1/HDAC3 inhibitors. Pharmaceuticals 7, 634-661. 10.3390/ph7060634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga K. (2015). The emerging role of senescent cells in tissue homeostasis and pathophysiology. Pathobiol. Aging Age Relat. Dis. 5, 27743 10.3402/pba.v5.27743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins D. H., Besnard V., Lange A. W., Wert S. E., Keiser A. R., Smith A. N., Lang R. and Whitsett J. A. (2009). Sox2 is required for maintenance and differentiation of bronchiolar clara, ciliated, and goblet cells. PLoS ONE 4, e8248 10.1371/journal.pone.0008248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompkins D. H., Besnard V., Lange A. W., Keiser A. R., Wert S. E., Bruno M. D. and Whitsett J. A. (2011). Sox2 activates cell proliferation and differentiation in the respiratory epithelium. Am. J. Resp. Cell Mol. 45, 101-110. 10.1165/rcmb.2010-0149OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi C. M., Luo Y., Yin Z., Zhang M., Zhu W., Wang T., Floss T., Goettlicher M., Noppinger P. R., Wurst W. et al. (2007). Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat. Med. 13, 324-331. 10.1038/nm1552 [DOI] [PubMed] [Google Scholar]
- Tsai S.-C. and Seto E. (2002). Regulation of histone deacetylase 2 by protein kinase CK2. J. Biol. Chem. 277, 31826-31833. 10.1074/jbc.M204149200 [DOI] [PubMed] [Google Scholar]
- van Oevelen C., Bowman C., Pellegrino J., Asp P., Cheng J. M., Parisi F., Micsinai M., Kluger Y., Chu A., Blais A. et al. (2010). The mammalian Sin3 proteins are required for muscle development and sarcomere specification. Mol. Cell. Biol. 30, 5686-5697. 10.1128/MCB.00975-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Tian Y., Morley M. P., Lu M. M., DeMayo F. J., Olson E. N. and Morrisey E. E. (2013). Development and regeneration of Sox2+ endoderm progenitors are regulated by a HDAC1/2-Bmp4/Rb1 regulatory pathway. Dev. Cell 24, 345-358. 10.1016/j.devcel.2013.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Frank D. B., Morley M. P., Zhou S., Wang X., Lu M. M., Lazar M. A. and Morrisey E. E. (2016). HDAC3-dependent epigenetic pathway controls lung alveolar epithelial cell remodeling and spreading via miR-17-92 and TGF-beta signaling regulation. Dev. Cell 36, 303-315. 10.1016/j.devcel.2015.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T., Liang J., Liu N., Huan C., Zhang Y., Liu W., Kumar M., Xiao R., D'Armiento J., Metzger D. et al. (2016). Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J. Clin. Invest. 126, 3063-3079. 10.1172/JCI85328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto H., Ohmae M. and Yamashita I. (1992). The saccharomyces cerevisiae Gam2/Sin3 protein plays a role in both activation and repression of transcription. Mol. Gen. Genet. 233, 327-330. 10.1007/BF00587597 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Iratni R., Erdjument-Bromage H., Tempst P. and Reinberg D. (1997). Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell 89, 357-364. 10.1016/S0092-8674(00)80216-0 [DOI] [PubMed] [Google Scholar]
- Zimmermann S., Kiefer F., Prudenziati M., Spiller C., Hansen J., Floss T., Wurst W., Minucci S. and Gottlicher M. (2007). Reduced body size and decreased intestinal tumor rates in HDAC2-mutant mice. Cancer Res. 67, 9047-9054. 10.1158/0008-5472.CAN-07-0312 [DOI] [PubMed] [Google Scholar]