

Abstract
Toxoplasma gondii is a highly successful Apicomplexan protozoan capable of infecting any warm-blooded animal worldwide. In humans, Toxoplasma infections are life-long with approximately one-third of the world’s population chronically infected. Although normally controlled by the host immune system, T. gondii infection can lead to a variety of clinical outcomes in individuals with immature or suppressed immune systems. After penetrating the intestine, parasites rapidly disseminate throughout the body and stimulate production of the cytokines interleukin (IL)-12, IL-18, and interferon (IFN)-γ by immune cells. These cytokines play a key role in host resistance to T. gondii by promoting a strong Th1 response. Recent reports show that gut commensal bacteria can act as molecular adjuvants during T. gondii infection. Thus, T. gondii is an excellent model system to study host-pathogen interactions. This unit outlines the protocols for in vitro and in vivo maintenance and growth of T. gondii.
Keywords: Toxoplasma gondii, Human foreskin fibroblast (HFF) cells, D10, mice infection, passage and maintenance, storage
INTRODUCTION
Toxoplasma gondii is a ubiquitous, single cell protozoan parasite that infects any nucleated cell in vertebrates and birds worldwide. One-third of the world’s human population are chronically infected with T. gondii (Dubey 2010). T. gondii was isolated for the first time from tissues of an African hamster-like desert rodent known as gundi, Ctenodactylus gundi, which was used as an animal model to study Leishmania in the laboratory of Charles Nicolle at the Pasteur Institute in Tunis (Nicolle and Manceaux 1908). During the same time, Splendore also discovered the same parasite in a rabbit in Brazil (Splendore 1908). T. gondii has three predominant life cycle stages: 1) tachyzoites (Greek tachos = speed); the rapidly multiplying stage, 2) bradyzoite (Greek brady = slow); the encysted transmissible stage found in tissues, 3) sexual stages found in feline intestine (Frenkel 1973; Frenkel, Dubey, and Miller 1970). Tachyzoites undergo proliferation to form two daughter cells through a process termed endodyogeny (Goldman, Carver, and Sulzer 1958). In the laboratory, tachyzoites can be maintained and cultured indefinitely, and this chapter will illustrate the routine method for maintaining and growing tachyzoites in a laboratory setting.
The genetic population structure of T. gondii is very unique and consists of at least 16 distinct haplogroups (Lorenzi et al. 2016). North American and European strains are clonal, whereas South American strains are highly divergent and non-clonal (Sibley and Ajioka 2008). Interestingly, different T. gondii strains do not only vary genotypically, but also differ extensively in their ability to induce pathology in animal infections, and in their replication rates across different cell lines (Saeij, Boyle, and Boothroyd 2005). Tachyzoites are obligate intracellular parasites that replicate with a duplication time of 6 to 9 hours during in vitro growth, depending on strain type. Infected cells typically rupture when they reach 64 to 128 parasites/cell. At rupture, the freed tachyzoites then rapidly infect neighboring cells (Radke and White 1998). A variety of cell lines, including transformed cell lines, (CHO, HeLa, LM, MDBK, Vero, 3T3, etc.) and culturing methods have been used to maintain tachyzoites in vitro (Evans et al. 1999; Saadatnia et al. 2010; Cook and Jacobs). Various unsuccessful attempts have also been made to propagate T. gondii extracellularly using a variety of nutrient-rich media (Cook and Jacobs ; Macfarlane and Ruchman 1948; Hughes, Hudson, and Fleck 1986). Although T. gondii strains have different growth rates in various cell lines (Evans et al. 1999), human foreskin fibroblast (HFF) cells (ATCC CRL-1634TM) have been utilized widely as the primary cell line to maintain in vitro cultures of T. gondii (Sibley and Howe 1996; Roos et al. 1994). T. gondii can be maintained in a variety of media, including Dulbecco’s Modified Eagle (DMEM) and RPMI 1640 medium (Invitrogen, USA) supplemented with 1 to 10% fetal bovine serum, 2 mM glutamine (Invitrogen), 10ug/ml gentamicin (Invitrogen) and 1% penicillin/streptomycin at (Invitrogen) pH 7.2 with 5% CO2. High pH and low CO2 will affect how the parasites grow, including stage conversion from tachyzoites to bradyzoites. Thus, DMEM at pH 7.2 with 5% CO2 should be used to maintain and culture the tachyzoites of T. gondii.
CAUTION: Several T. gondii strains are highly virulent and can propagate in any human tissue. Certain individuals, particularly those with weakened immune systems such as AIDS patients and pregnant women, are at high risk and should be cautious. Follow all preventive measures of biosafety level 2 laboratories for using and handling of T. gondii (Section VIII-c: Parasitic Agents). Guidelines for BSL2 practices can be obtained from the latest edition of Biosafety in Microbiological and Biomedical Laboratories (BMBL, 5th Edition) via the following CDC Web link: http://www.cdc.gov/biosafety/publications/bmbl5/. See also biosafety guidelines presented in UNIT 1A.1 (Burnett, et al., 2009).
BASIC PROTOCOL 1: Culturing HFF cells
Human foreskin fibroblast (HFF) cells have been widely used to culture and maintain T. gondii tachyzoites. HFF cells can be cryopreserved in 95% culture medium with 5% DMSO in liquid nitrogen or −150°C freezer for many years. Viability of the HFF cells after cryopreservation depends on several factors, including the stresses imposed on HFF cells during the freezing and thawing procedure. After thawing, HFF cells should not be sub-cultured for more than 25 passages. After 25 passages, new cryovials with frozen HFF cells should be thawed.
Materials
Frozen stock of HFF cells (ATCC CRL-1634TM)
1X phosphate buffered saline (PBS) without calcium
Complete culture medium (D10): Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, USA) supplemented with 10% heat inactivated feral bovine serum (FBS; Sigma F0926)), 2 mM glutamine (Gibco 25030-081), 10ug/ml gentamicin (Gibco 15750-060), and 1% penicillin/streptomycin (Gibco 15140-122)
0.25% trypsin (Gibco 25200-056) with 0.03% EDTA solution (final concentration)
Freezing medium: 20% DMSO (Sigma D5879) and 50% FBS
Ice bucket
70% Ethanol
37°C water bath
37°C laboratory incubator with 5% CO2
Laminar hood
Inverted microscope equipped with phase contrast optics
175 cm2 (T175) cell culture flasks
25 cm2 (T25) cell culture flasks
Sterile cryovials
Sterile latex exam gloves
Sterile transfer pipets
Sterile graduated pipettes (5ml, 10ml, and 25ml)
Sterile polystyrene conical centrifuge tubes (15ml and 50ml)
Sterile aspirators
Thawing HFF cells
Work in laminar hood while wearing a protective laboratory coat, disposable latex exam gloves, and eye goggles.
-
Warm D10 medium in a 37°C water bath and place a T175 flask in the sterile laminar hood and label properly with the passage number and current date.
It is critical to heat-inactivate the FBS added to D10 medium for at least 45 mins at 56°C. This step inactivates complement in the serum which may interfere with parasite growth in vitro.
Take out container of the warm D10 medium from the water bath and place in laminar hood after sterilizing the bottle with 70% ethanol.
Dispense 35 ml of warm D10 medium into T175 flask with a sterile 50 ml pipette.
-
Remove a frozen cryovial of HFF cells from freezer/liquid nitrogen tank, quickly place into a 37°C water bath and shake until it is completely thawed.
Caution: the presence of liquid nitrogen in the cryovial can cause an explosion; liquid nitrogen will expand very quickly, up to ~700 times, during the warming process. Safety measures will be needed, including the use of a covered and insulated bucket or box during the transportation and warming processes
Place the thawed cryovial of HFF cells in the laminar hood after sterilizing with 70% ethanol in order to avoid transfer of any microorganisms from water bath.
Resuspend the material from cryovial and transfer it into T175 flask. Mix well but slowly to avoid frothing.
-
Place the T175 flask with D10 medium and HFF cells in a 37°C laboratory incubator with 5% CO2.
Ensure that the cap on the T175 flask is loose, to facilitate gas exchange. Once the flask is gassed overnight, firmly close the lid on the flask to decrease the risk of culture contamination. This procedure needs to be done every time new “ungassed” media is added to a cell culture flask. Alternatively, tissue culture flasks with filters built into the cap can be used, in which case the cap can be closed tightly.
Discard empty cryovial and used pipets in the biohazardous waste.
The next day, warm D10 medium in 37°C water bath and place the warm D10 medium in laminar hood after sterilizing the outside of the bottle with 70% ethanol.
-
Remove the T175 flask with HFF cells from the incubator and examine under microscope. Place it in the laminar hood after spraying with 70% ethanol.
Caution: in order to avoid physical damage to the HFF cells, the flask should not be touched for at least 16 hours.
Aseptically aspirate the medium from the T175 flask into a biohazardous waste container, and aliquot 35ml of warm D10 into the T175 flask. Place the flask back into the 37°C laboratory incubator with 5% CO2 and examine the growth of HFF cells microscopically every day until they achieve a confluent monolayer (typically 4–7 days).
Sub-culturing HFF cells
Place complete D10 medium, 1X phosphate buffered saline without calcium (1X PBS), Trypsin-EDTA solution in the 37°C water bath.
Place 20 T25 or 4 T175 flasks in sterile laminar hood.
Take a T175 flask with confluent monolayer of HFF cells from the incubator and place it in the hood after sterilizing the outside of the flask with 70% ethanol.
Place the warm D10 medium, 1X PBS, and trypsin-EDTA solution in the hood after spraying the outside of each container with 70% ethanol.
Aspirate the medium from the T175 flask into a biohazardous liquid waste container. Wash the monolayer of HFF cells with 25 ml of 1X PBS and aspirate into biohazardous waste container.
Incubate the monolayer with 3 ml of trypsin-EDTA solution. Allow the flask to sit at room temperature or at 37°C in the incubator until the cells detach from the flask (approximately 2–3 mins).
Add 150 ml of D10 media into the flask and mix well to ensure all the cells have been detached. Dispense 5 ml or 25 ml of the resultant cell suspension in each labeled T25 or T175 flask, respectively.
Swirl the flasks to cover the entire bottom of the flask and place them in the 37°C laboratory incubator with 5% CO2.
Discard used flask and pipets into a biohazardous waste box.
Changing Medium
Place complete D10 medium in the 37°C water bath. After warming, sterilize the D10 container with 70% ethanol and place the medium in the hood.
Take the T25 or T175 flasks from the incubator and place them in the laminar hood after spraying with 70% ethanol.
Remove the medium with aspirator from the flasks and replace it with fresh warm D10 medium.
Exam flasks for the confluency of the HFF monolayer microscopically and place the flasks in the Incubator.
Discard used pipets into the biohazardous waste box.
Medium Renewal: 1 to 2 times per week.
Freezing HFF cells
Place 1X PBS, Trypsin-EDTA solution at room temperature.
Place an ice bucket in the laminar hood after sterilizing with 70% ethanol.
-
Label sterile cryovials with freezing date, passage number and place them in the sterile ice bucket in the hood.
Caution: only freeze HFF cells with very low passage numbers. It is recommended to sub-passage these primary cell lines no more than 25 passages
Place freezing medium (50% FBS and 20% DMSO) in the ice bucket.
Take the T175 flask with a confluent monolayer of HFF cells from the incubator and place it in the hood after sterilizing the outside of the flask with 70% ethanol.
Place the 1X PBS and trypsin-EDTA solution in the hood after spraying with 70% ethanol.
Aspirate the medium from the T175 flask into a biohazardous liquid waste container. Wash the monolayer of HFF cells with 25 ml of 1X PBS and aspirate into biohazardous waste container.
Trypsinize the monolayer with 3 ml of trypsin-EDTA solution at 37°C incubator until the cells detach from the flask.
Add 4 ml of ice cold 50% FBS in the flask and swirl well to detach maximum number of HFF cell from the flask.
Remove the mixed solution from the flask and transfer to 15 ml polystyrene tube. Place the 15 ml tube with solution in the ice bucket in the hood.
Add 4 ml of 20% ice cold DMSO in the 15 ml tube and mix three times by pipetting up and down. Dispense 1 ml aliquots in each pre-cooled cryovial and transfer them into a cooler where the cooling rate is 1°C per minute in the range from ice cold to below −50°C. Place the cryovials in the −150°C freezer or liquid nitrogen tank after they have reached a temperature of −80°C for long term cryopreservation.
BASIC PROTOCOL 2: In vitro maintenance and growth of T. gondii
T. gondii is an obligate intracellular parasite that belongs to the phylum Apicomplexa which includes several important human and animal pathogens such as Plasmodium, Cryptosporidium, Eimeria, and Neospora. Among these medically important eukaryotic pathogens, T. gondii has emerged as the most successful genetically-tractable model system because of its ease to maintain and grow in vitro, The parasite is naturally infectious in rodent animal models for pathogenesis studies, and a large suite of genetic tools exist that support both forward and reverse genetic experiments to facilitate parasite biology studies. Although both the tachyzoite and bradyzoite stages can be readily cultured in vitro, the tachyzoite stage is typically utilized to maintain and culture the parasites. Thus, this section discusses the maintenance and culture methods for T. gondii tachyzoites.
Materials
Frozen stock of T. gondii strains (see ATCC [www.atcc.org] for list of available strains)
1X phosphate buffered saline (PBS) without calcium
Complete culture medium (D10): Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, USA) supplemented with 10% heat inactivated feral bovine serum (FBS; Sigma F0926)), 2 mM glutamine (Gibco 25030-081), 10ug/ml gentamicin (Gibco 15750-060), and 1% penicillin/streptomycin (Gibco 15140-122)
0.25% trypsin (Gibco 25200-056) with 0.03% EDTA solution (final concentration)
Freezing medium: 20% DMSO (Sigma D5879) and 50% FBS Ice bucket
70% Ethanol
37°C water bath
37°C laboratory incubator with 5% CO2
3-μm pore size polycarbonate filters
Laminar hood
Inverted microscope equipped with phase contrast optics
Eppendorf 5810R centrifuge (or equivalent) with appropriate 15 and 50 ml centrifuge tubes adaptors
T175 cell culture flasks
T25 cell culture flasks
Cell Scraper
Sterile cryovials
Sterile latex exam gloves
Sterile transfer pipets
Sterile graduated pipettes (5ml, 10ml, and 25ml)
Sterile polystyrene conical centrifuge tubes (15ml and 50ml)
Sterile aspirators
Sterile 10 ml syringes
Sterile 5 ml syringes with 25 G needles
Thawing T. gondii cells
Work in laminar hood while wearing a protective laboratory coat, disposable latex exam gloves, and eye goggles.
Place complete D10 medium in the 37°C water bath. After warming, sterilize the outside of the D10 container with 70% ethanol and place the medium in the hood.
-
Remove a T25 flask with confluent monolayer of HFF cells from the incubator, examine under microscope for confluency and place in the hood after sterilizing the outside of the flask with 70% ethanol. Change the medium of the flask if the color of the medium looks yellow. Label the T25 flask with current date and strain name.
It is advisable to use a confluent flask inoculated with HFF cells that is between 7 and 21 days old for routine growth and maintenance of T. gondii tachyzoites
Remove a frozen cryovial of T. gondii from −80°C freezer/liquid nitrogen tank and quickly place into a 37°C water bath and shake until it is completely thawed.
Place the thawed cryovial of T. gondii cells in the laminar hood after spraying with 70% ethanol to avoid transferring any microorganisms from water bath.
Transfer the material from cryovial using sterile transfer pipette and dispense it into the T25 flask. Shake carefully to mix and avoid frothing.
-
Recap the inoculated T25 flask and place in the 37°C laboratory incubator with 5% CO2
Ensure that the cap on the T25 flask is loose, to facilitate gas exchange. Once the flask is gassed overnight, firmly close the lid on the flask to decrease the risk of culture contamination. This procedure needs to be done every time new “ungassed” media is added to a cell culture flask. Alternatively, tissue culture flasks with filters built into the cap can be used, in which case the cap can be closed tightly.
Discard the empty cryovial and used pipets into the biohazardous waste.
Next day, warm D10 medium in 37°C water bath and place the warm D10 medium after sterilization with 70% ethanol into the laminar hood.
Remove the T25 flask infected with HFF cells from the incubator and examine under microscope. Place in the laminar hood after spraying the outside of the flask with 70% ethanol.
Aspirate the medium from the T25 flask into a biohazardous waste container and pipette 5–7ml of warm D10 into the T25 flask. Place the flask into the 37°C laboratory incubator with 5% CO2. Examine for T. gondii tachyzoite growth and vacuole formation within HFF cells microscopically every day (Fig. 1A).
Figure 1.
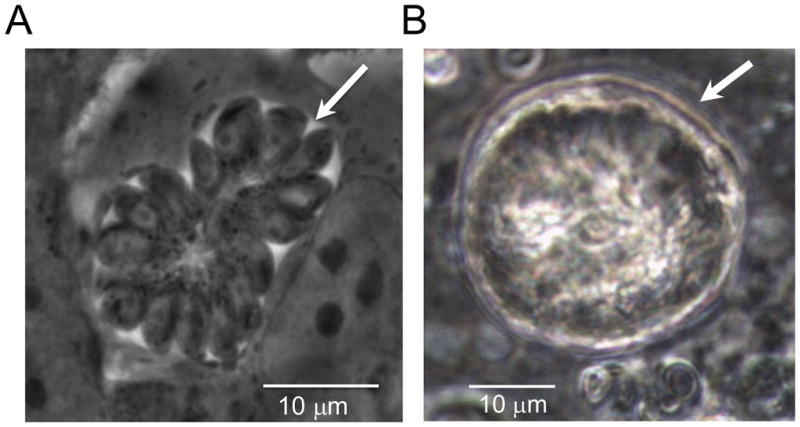
Life cycle stages of T. gondii. A) Rosette of intracellular tachyzoites (arrowhead) is multiplying in HFF cells in vitro after 24 hours of post-infection. B) A mature tissue cyst of T. gondii (arrowhead) is developed in CD1 mice after one month of post intraperitoneal infection.
Passaging T. gondii cells
Work in a laminar hood while wearing a protective laboratory coat, disposable latex exam gloves, and eye goggles.
Place complete D10 medium in the 37°C water bath. After warming, sterilize the outside of the D10 container with 70% ethanol and place the medium in the hood.
Remove the T. gondii infected T25 flask from the incubator. Examine the HFF monolayer under the microscope to check for contamination, parasite growth, and lysis of the HFF monolayer. Typically, release of ~70% of parasites from HFF cells generally takes 2–3 days of infection for most of the T. gondii strains.
Remove a T25 flask with a confluent monolayer of HFF cells from the incubator, examine under microscope for confluency and contamination, and place in the hood after sterilizing the outside of the flask with 70% ethanol. Change the medium in the flask if the medium has turned yellow in color. Label the T25 flask with current date and strain name.
Remove 1.0 ml of supernatant from the lysed T25 flask using a sterile transfer pipette and transfer into the new T25 flask with confluent HFF cells.
Recap and place the newly T. gondii infected HFF cells in the 37°C laboratory incubator with 5% CO2. Examine for growth and vacuole formation of T. gondii within HFF cells microscopically every day (Fig. 1A).
Repeat steps 1 to 6 after lysis of HFF cells to maintain the parasites in vitro..
-
Discard the empty used T25 flask and used transfer pipets in the biohazardous waste.
Note: The difference in growth rate of various T. gondii strains is based on the relative lengths of chromosomal replication and segregation phases. Type I T. gondii strains can complete one round of cell cycle within 6 to 8 hours, whereas type II and III T. gondii strains generally take 8 to 12 hours (Radke et al. 2001). Thus, infections with 1 ml (~5 × 106) of type I and II/III strains will lyse a confluent monolayer of HFF cells in a T25 flask within 2 and 3 days, respectively. Parasites can be maintained for a long period of time by serial passage in vitro culture, however, it has been suggested that parasites maintained in vitro culture for a long period of time can become defective in traversing the sexual cycle or developing tissue cysts in mice (Frenkel, Dubey, and Hoff 1976).
Harvesting tachyzoites of T. gondii
Place complete D10 medium in the 37°C water bath. After warming, sterilize the outside of the D10 container with 70% ethanol and place the medium in the hood.
Place 1X PBS solution at room temperature in the hood after spraying the outside of the container with 70% ethanol.
Remove the T. gondii infected T25 flask from the incubator then examine under microscope to check for contamination and lysis of the HFF monolayer, which is typically the release of ~70% of parasites from HFF cells.
-
Scrape the monolayer of infected HFF cells with a cell scarper and transfer into a 15 ml polystyrene conical centrifuge tube
T. gondii tachyzoites will stick to polypropylene tubes, therefore all parasite preps should be prepared using plastic that has been treated for tissue culture, or is made out of polystyrene.
Syringe pass the cell suspension 3 times through a 25-gauge needle to ensure that most of the host cells are lysed and the parasites are outside of the host cells.
Filter the syringe lysed cell suspension through 3-μm pore size polycarbonate filter then centrifuge at 400xg for 10 mins at 16°C in a benchtop centrifuge.
After centrifugation, discard the supernatant and resuspend the pellet with 5 ml of D10 medium and make a 1:10 dilution of an aliquot of this cell suspension by adding 100 μl of the cell suspension into 900 μl of PBS.
-
Count the number of parasites present in cell suspension by transferring 10 μl of the 1:10 diluted cell suspension into a standard hemocytometer and by examining through an inverted microscope.
Note: It is not required to harvest tachyzoites of T. gondii for routine passage. 3-μm pore size filter will filter out most of the host cell debris and aggregated parasites.
Freezing T. gondii cells
T. gondii is an intercellular parasite and dies rapidly in the extracellular environment. Thus, it is preferable to freeze parasites when they are still within host cells. However, large numbers of extracellular parasites from freshly lysed host cells can be cryopreserved efficiently using a small volume of freezing medium.
A) Intracellular freezing
Place 1X PBS and Trypsin-EDTA solution at room temperature.
Place an ice bucket after sterilizing with 70% ethanol in the laminar hood.
Label sterile cryovials with freezing date, T. gondii strain name, and passage number, and then place them in sterile ice bucket in the hood.
Place freezing mediums (50% FBS and 20% DMSO) in the ice bucket.
Take the T25 flask infected with T. gondii strain from the incubator and check under microscope for infectivity. For optimum survival of T. gondii strains during preservation, take those flasks where ~70% of HFF cells have T. gondii vacuoles containing ~8 parasites per vacuole.
Place the optimum T25 flasks for preservation in the hood after sterilizing with 70% ethanol.
Place the room temperature 1X PBS, and trypsin-EDTA solution in the hood after spraying with 70% ethanol.
Aspirate the medium from the T25 flask into a biohazardous waste container. Wash the monolayer of HFF cells with 5 ml of 1X PBS twice and aspirate using an aspirator into biohazardous waste container.
Trypsinize the monolayer with 0.5 ml of trypsin-EDTA solution at 37°C incubator until the cells detach from the flask. Tap the bottom of the flasks vigorously to detach the HFF cells from the flask. Medium will become opaque as a result of detachment of HFF cells from the flask.
Add 1 ml of ice cold 50% FBS in the flask and swirl well to detach maximum number of HFF cell from the flask.
Add 1 ml 20% ice cold DMSO in the flask and mix three times by pipetting up and down slowly. Dispense 1 ml of the aliquots in each pre-cooled cryovial and transfer them into a cooler where the cooling rate is 1°C per minute, in the range from ice cold to below −50°C. Place the cryovials in the −150°C freezer or liquid nitrogen tank after they reach a temperature of −80°C for long term cryopreservation.
B) Extracellular freezing
Place 1X PBS and Trypsin-EDTA solution at room temperature.
Place an ice bucket after steriling with 70% ethanol in the laminar hood.
Label sterile cryovials with freezing date, T. gondii strain name, and passage number then place them in sterile ice bucket in the hood.
Place freezing medium (50% FBS and 20% DMSO) in the ice bucket.
Place the T25 flask infected with T. gondii strain from the incubator and check under microscope to ensure that ~70% of the host cell monolayer has been lysed.
-
Swirl the flasks vigorously to detach all parasites and host cells from the bottom of the flask and transfer the lysed HFF cells containing parasites into 15 ml polystyrene centrifuge tubes.
Filtration through a 3-μm pore size polycarbonate filter is not necessary
Centrifuge the parasites in suspension for 10 mins at 800xg at 16°C using a benchtop Eppendorf centrifuge or equivalent, and aspirate the supernatant using an aspirator.
-
Resuspend the parasite pellet with 1 ml of ice cold 50% FBS and 1 ml 20% ice cold DMSO in the 15 ml polystyrene tube and mix three times by pipetting up and down slowly. Dispense in 1 ml aliquots into each precooled cryovial and transfer them into a cooler where the cooling rate is 1°C per minute, in the range from ice cold to below −50°C. Place the cryovials in the −150°C freezer or liquid nitrogen tank after they have reached a temperature of −80°C for long term cryopreservation.
Recommendation: T. gondii cells within 2 days of passage are optimum for freezing the next day after infection. Mycoplasma testing needs to be done before freezing of T. gondii strains. Two cryovials can be frozen for each T25 flask. Direct freezing in either liquid nitrogen and/or −150°C is detrimental to parasite survival. A slow freezing protocol is always recommended for higher viability of parasites. It is always recommended to examine the viability of the frozen stock before discarding the parental parasite culture.
BASIC PROTOCOL 3: In vivo maintenance and growth of T. gondii
T. gondii strains can be maintained in vivo in a wide range of laboratory mice, including Swiss-Webster, CBA/Ca, and C57BL/6 mice. Mice are infected either perorally with tissue cysts, or intraperitoneally with tachyzoites or tissue cysts. To generate tissue cysts in the brain, mice need to be infected chronically for a minimum of 15 days to a month. However, a serial passage in mice can be achieved within 4 to 5 days of interperitoneal infection, followed by recovery of the parasites from the peritoneal exudate. This preparation can be reinfected intraperitoneally to maintain the parasite culture.
Materials
6 to 8 weeks old mice (Swiss-Webster, CBA/Ca, and C57BL/6 mice)
T. gondii tachyzoites propagated in vitro, or mice chronically infected with T. gondii
Inverted microscope equipped with phase contrast optics
Microscope slides and 22 × 22 mm coverslips
1ml syringes with 27 G needle
5 ml syringes with 25 G needle
5 ml syringes with 21 G needle
5 ml syringes with 19 G needle
5 ml syringes with 16 G needle
18 G, 1.5 in. (3.8 cm) gavage needle
Scissors
Forceps
1X phosphate buffered saline
70% Ethanol
Sterile latex exam gloves
Sterile transfer pipets
Sterile aspirators
Benchtop centrifuge
Eppendorf 5810R centrifuge (or equivalent) with appropriate 15 and 50 ml centrifuge tubes adaptors
Ice bucket
Styrofoam block
Sterile polystyrene conical centrifuge tubes (15ml and 50ml)
Additional reagents and equipment for intraperitoneal and oral infection of mice, euthanasia, in vitro culture of T. gondii.
Mice infection with T. gondii strains
Work in a laminar hood while wearing a protective laboratory coat, disposable latex exam gloves, and eye goggles.
-
Place the 1X PBS solution in the hood after spraying the outside of the bottle with 70% ethanol.
Tachyzoite infection
Remove a frozen cryovial of T. gondii from freezer/liquid nitrogen tank and quickly place into a 37°C water bath and shake until it is completely thawed.
Centrifuge tachyzoite suspension using benchtop centrifuge at 400xg for 10 mins at 4°C.
Place the cryovial of T. gondii cells in the laminar hood and aspirate the supernatant, followed by resuspending the parasite pellet in 400 μl of PBS.
-
Take the 400 μl of parasite suspension into a 1ml syringe with a 27 G needle and inject 200 μl each into two mice intraperitoneally.
Alternatively, T. gondii tachyzoites expanded by in vitro culture can be substituted. Inject mice with either 100, 1000, or 10,000 tachyzoites that have been washed into PBS in an injection volume of no more than 0.5ml. PBS is used to ensure that no FBS is injected into the mice peritoneum.
Tissue cysts infection
Take mice infected with T. gondii at 30 days post infection. Euthanize the mice by CO2 asphyxiation. Spray the mouse with 70% ethanol to sterilize the head and incise the skin to expose the skull.
Open the skull to expose the brain with scissor and remove the brain with forceps and transfer it into a 50 ml conical centrifuge tube.
Add 1 ml of PBS per brain and homogenize the brain by repeated syringing through first a 16 G, then 19 G, then 21 G needle attached to 5 ml syringes.
Take 20 μl of the brain suspension and squash onto a microscope slide using a 22 × 22 mm coverslip to cover the sample. Count the number of tissue cysts by examining under a microscope using either 20× or 40 × magnification (see Fig. 1B).
Inject a 200μl cell suspension containing 20 to 50 tissue cysts per mice either orally or intraperitoneally using either an 18 G gavage needle or a 27 G injection needle, respectively.
Check the mice once every day after infection with either tachyzoites or tissue cysts for illness. Weight loss is an indication of infection, and mice can lose up to 20% of their bodyweight prior to showing any signs of infection.
Euthanize infected animals by CO2 asphyxiation as soon as they show any sickness, including ruffled fur and lethargy.
Spray the mice with 70% ethanol and mount them on a Styrofoam block on their back and incise the abdominal wall.
-
Inject 5 ml of 1 × PBS into the peritoneal cavity using 25 G needle.
Note: Do not rupture any organs.
Massage the peritoneum gently to detach any T. gondii cells.
Withdraw the peritoneal cavity lavage as much as possible by inserting a 25 G needle attached with a 5 ml syringe.
Transfer the peritoneal fluid into a 15 ml polystyrene centrifuge tube and centrifuge the cell suspension in a benchtop Eppendorf 5810R centrifuge at 400xg for 10 minutes at 16°C
Resuspend the pellet in 10 ml of PBS and make a 1:10 dilution of this cell suspension by adding 100 μl of the cell suspension into 900 μl of PBS.
Count the number of parasites present in cell suspension by transferring 10 μl of the 1:10 diluted cell suspension into a hemocytometer and by examining through inverted microscope.
-
Inject 100 μl to 200 μl of the original cell suspension into each mice using 1ml syringes with 27 G needles. The number of parasites that need to be injected will depend on strain type, which should be standardized by the investigator.
Note: Growth rates of T. gondii in the mouse model are completely dependent upon the strain type and the virulence property of the strains. The steps described above need to be standardized by the investigator based on T. gondii strain type, days of transfer, mice strain, etc. Parasites can be maintained for a long period of time by serial passage using the mouse model by repeating steps 11 to 15 every 3 to 5 days, or by developing a chronic infection. Chronically infected mice can be kept for 6 months to a year. Parasites can be maintained by chronic infection for a long time by repeating steps 7 to 12.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see APPENDIX 5.
D10
Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat inactivated (Δ56°C for 45 mins) fetal bovine serum, 2 mM glutamine, 10ug/ml gentamicin, and 1% penicillin/streptomycin.
Freezing medium
20% DMSO: 40 ml of D10 + 10 ml of DMSO
50% FBS: 25 ml D10 + 25 ml heat inactivated FBS
COMMENTARY
Background information
T. gondii, an obligate intracellular pathogen infects approximately one third of the human population by ingestion of contaminated food or water (Dubey and Beattie 1988). Infection with T. gondii in healthy adults typically results in a mild and temporary flu-like syndrome known as toxoplasmosis (Frenkel 1973). However, immunocompromised patients can become seriously ill and infection may result in death due to parasite-induced encephalitis (Israelski and Remington 1993; Denkers and Gazzinelli 1998). Recent studies have also shown that T. gondii is the causative agent of several ocular diseases (Jones, Alexander, and Roberts 2006; Holland 2003, 2004) that are associated with unusual or exotic genotypes of T. gondii present in South America (SA) (Grigg et al. 2001).
Unlike other species in the phylum Apicomplexa, T. gondii is arguably the most successful because the parasite can be transmitted among all carnivorous hosts asexually by ingestion of tissue cysts present in all infected animal hosts (Dubey 1988; Dubey and Frenkel 1976). Furthermore, cats are the definitive hosts of T. gondii and once infected, can excrete >107 environmentally stable oocyts per day (Dubey and Frenkel 1976) which can contaminate water supplies, vegetation and infect any warm-blooded vertebrate. Although it possesses a meiotic life cycle phase, the population structure of T. gondii is unusual in that it is dominated by three clonal lineages in North America and Europe (NA/E) know as types I, II, and III (Howe and Sibley 1995; Ajzenberg et al. 2004). Whereas SA strains are very divergent and highly recombinogenic (Su et al. 2012). Recently, whole genome sequencing of 62 strains not only depicted the extensive genetic diversity and admixture profile among the T. gondii strains but also shed light on genetic basis for the wide range of phenotypic differences (Lorenzi et al. 2016).
T. gondii provides a valuable model for studying the evolution of pathogens because it is widespread and infects virtually all warm-blooded vertebrates (Dubey 2010). T. gondii is also considered as a model organism for cellular, biochemical, molecular and genetic studies for other clinically important apicomplexan organisms such as malaria parasites (Plasmodium spp.) because of the ease of doing genetic manipulation of the T. gondii genome (Roos et al. 1994). Molecular tools for both forward and reverse genetics including transfection, transformation, and gene knockouts are regularly used to manipulate this parasite (Ajioka and Sibley 2007; Striepen et al. 2007; Meissner, Schluter, and Soldati 2002; Huynh and Carruthers 2009; Shen et al. 2014; Sidik et al. 2014).
Detection and Isolation
Various tests for detecting the presence of T. gondii have been used, including serological tests for detecting T. gondii specific IgG and IgM antibodies and PCR-based tests (polymerase chain reaction) for detecting the presence of genomic DNA. The most commonly used serological methods include: the Sabin-Feldman dye test (DT), indirect hemagglutination (IHA) assay, indirect fluorescent antibody (IFA) techniques, modified agglutination tests (MAT), latex agglutination (LA), enzyme-linked immunoabsorbent assay (ELISA), and complement fixation (CF) test (Dubey 2010). Of these serological tests, the modified MAT has been used extensively for diagnosis of toxoplasmosis in both animals and humans (Dubey and Desmonts 1987). Although serological tests have been widely employed, recently developed quantitative real time PCR techniques (qRT-PCR) take the advantage of repetitive regions within the T. gondii genome. This methodology can provide not only a rapid, sensitive, and specific detection of gDNA, but can also quantify the parasites burden in the infected samples. The most frequently used repetitive regions are the 35-copy B1 gene (Burg et al. 1989) and 300-copy 529 bp element to detect the presence of gDNA of T. gondii (Bourdin et al. 2014; Fekkar et al. 2008). To isolate live parasites from either serological or PCR positive samples, heparinized blood or cerebrospinal fluid of acutely or chronically infected tissues can be inoculated into mice subcutaneously, intraperitoneally or orally (Dubey 2010). If the sample is contaminated with bacteria, it is recommended that the sample be treated with antibiotics in 0.9% NaCl solution with a concentration of 1,000 units of penicillin and 100μg of streptomycin per milliliter (Dubey 2010). Acutely or chronically infected tissues need to be homogenized before infecting mice. Pepsin or trypsin treatment of chronically infected tissues has also been suggested in order to increase the probability of DNA isolation (Jacobs, Remington, and Melton 1960; Sharma and Dubey 1981). To isolate parasites from low infectious samples, cats have been used successfully to isolate non-virulent T. gondii strains from mice as well as from food samples that contain very few tissues cysts (Dubey 2010; Dubey, Swan, and Frenkel 1972). However, there are several disadvantages to utilize the cat model over the mice model: A) Oocysts are highly resistant in both soil and water and to most chemical and physical disinfectants (Frenkel and Dubey 1973). B) Cats are the definitive hosts for T. gondii and if the samples are infected with two different parasites, then a cat infection will increase the probability of generating recombinants (Frenkel and Dubey 1972). C) Only 50% of cats shed oocysts after ingesting bradyzoites or oocysts (Dubey and Frenkel 1972; Dubey 1996). D) Cats are much more costly to maintain than the mouse model.
Critical Parameters and Troubleshooting
T. gondii can infect any nucleated cell. However, the in vitro growth rate of T. gondii is significantly dependent on the type of primary cell lines used as host cells for infection. Hela cells reportedly support the growth of tachyzoites of T. gondii significantly better than LLC cells (P<0.00005) or Vero cells (P<0.05) (Evans et al. 1999). Previous studies have also demonstrated that the in vitro growth rate differs significantly between different genotype strains. Type I strains grow substantially faster than type II and type III strains (Radke and White 1999). Interestingly, one of the most commonly used laboratory type I strains RH (ATCC Number: 50839), which has been adapted for in vitro culture since 1977 (Pfefferkorn and Pfefferkorn 1976), not only has significant higher in vitro growth rate (Khan et al. 2009) but also can survive much longer times extracellularly than any other known T. gondii strains. The viability of this strain is only diminished 50% over 24 hours after egress from the host cell at 37°C (Khan et al. 2009). Thus, extended periods of in vitro culture may change some of the phenotypic characteristics of the parasites. Extended periods of in vivo serial passages in laboratory mice can also alter the capacity of the parasite to go through its sexual cycle (Frenkel, Dubey, and Hoff 1976). It is important to avoid repeated serial passages of intermediately virulent strains of T. gondii, such as Type II strains in mice as this may increase the virulence of these parasites.
T. gondii undergoes stage conversion between quickly replicating tachyzoites to slowly replicating bradyzoite stage in response to any kind of stress, including immune response, alkaline pH and/or high temperature (Skariah, McIntyre, and Mordue 2010). Tachyzoites can also differentiate to bradyzoites if HFF cells or other host cells are not healthy. HFF cells have a limited replicative lifespan and it has been suggested that HFF cells should not be sub-cultured over 25 passages. It has also been observed that freshly isolated sporozoites or bradyzoites inoculated into HFF cells have a tendency to spontaneously differentiate into bradyzoites rather than be maintained as tachyzoites (Jerome et al. 1998; Radke et al. 2003). In this situation, repetitive syringe passages (up to 20 division cycles) will be required to keep the parasites in the tachyzoite stage.
FBS must be heat-inactivated by incubating at a temperature of 56°C for at least 30 mins before preparing D10 medium in order to reduce the parasite exposure to serum antibodies and complement proteins. For immunological studies, define ultrapure FBS should be employed. Dialyzed FBS has been suggested for drug studies.
Mycoplasma contamination of HFF stock cells and culture medium needs to be examined at least once a month, as this will dramatically alter the results output of the vast majority of laboratory experiments associated with host pathogen interactions. The most common sources of mycoplasma contamination in the laboratory are infected cells sent from another lab and laboratory personnel infected with Mycoplasma orale or Mycoplasma fermentas. Thus, any strains received from another laboratory should be screened for the presence of Mycoplasma. Should Mycoplasma be detected, the best practice is to discard the contaminated flask in order to reduce the spread of contamination. However, if the T. gondii strains are precious, it is possible to salvage the strain by passing them through mice, which will eliminate the mycoplasma infection.
Anticipated Results
HFF cells have been widely used to culture and maintain of T. gondii because their morphology is large and flat so they can hold multiple rounds of parasite replication (~256 tachyzoites) and they are strongly contact inhibited. A T175 flask of HFF cells will be confluent within a week after thawing a cryovial from liquid nitrogen. Depending on the requirement, HFF cells require subculturing. Up to 30 T25 flasks can be prepared from a T175 flask and they will be ready to use within a week.
Type I strain RH grows rapidly with a replication cycle of 6 to 8 hours and after one day post-infection, parasites form “rosettes” with the parasites apical ends directed towards parasitophorous vacuole membrane. Parasites will lyse the whole monolayer within 36 to 48 hrs. Freshly lysed parasites are motile and squirm vigorously. A completely lysed T25 flask will yield 1×107 tachyzoites/ml whereas a fully lysed T175 flask will yield 1–2 × 109 parasites. Type II strain Me49 (ATCC Number: 50611) and type III strain CTG (ATCC Number: 50842) are slow growing parasites and a T175 flask can produce 5–8 ×108 parasites after complete lysis of HFF monolayer.
Type I strains exhibit a consistent growth rate within 1 to 2 weeks in the mouse model after thawing and infecting them from cryovial. Type I strains are highly virulent in laboratory mice, whereas the type II and III strains are intermediate and non-virulent, respectively (Saeij et al. 2006; Taylor et al. 2006; Sibley and Boothroyd 1992). Maintenance and yield of parasites using type II and III strains in mice model are highly variable and dependent upon the mouse strain used for passage as well as the type of parasites. Thus, it is highly recommended that continuous growth and maintenance of type II and III parasites should be standardized by the investigator.
Time Considerations
Thawing HFF cell requires 30 to 60 mins. Subculture of HFF cells can be accomplished within 30 to 60 minutes. Changing media of HFF requires 2 to 3 minutes per flask. Freezing of HFF can be completed within 30 to 60 mins. Thawing parasites from −150°C freezer or liquid nitrogen tank requires 30 to 60 minutes. Serial passage of T. gondii strains can be completed as little as 2 to 3 mins per strain. Complete lysis of host cells requires ~2 to 3 days depending on the strain type and infection dose. Harvesting of tachyzoites requires 30 to 45 minutes. Freezing parasites in −150°C freezer or liquid nitrogen tank can be accomplished within 30 to 60 minutes. Serial passage of tachyzoites in mice can be completed within 1 to 2 hours, however harvesting tissue cysts and reinjecting mice can take 3 to 4 hours.
Acknowledgments
Authors are supported by Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID) at National Institute of Health. M.E.G. is a scholar of the Canadian Institute for Advanced Research (CIFAR) Program for Integrated Microbial Biodiversity.
Literature Cited
- Ajioka JW, Sibley LD. Development and application of classical genetics in Toxoplasma gondii. In: Weiss LM, Kim K, editors. Toxoplasma gondii The Model Apicomplexan: Perspectives and Methods. Academic Press, Elsevier; New York: 2007. [Google Scholar]
- Ajzenberg D, Bañuls AL, Su C, Dumètre A, Demar M, Carme B, Dardé ML. Genetic diversity, clonality and sexuality in Toxoplasma gondii. Intl J Parasitol. 2004;34:1185–96. doi: 10.1016/j.ijpara.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Bourdin C, Busse A, Kouamou E, Touafek F, Bodaghi B, Le Hoang P, Mazier D, Paris L, Fekkar A. PCR-based detection of Toxoplasma gondii DNA in blood and ocular samples for diagnosis of ocular toxoplasmosis. J Clin Microbiol. 2014;52:3987–91. doi: 10.1128/JCM.01793-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg JL, Grover CM, Pouletty P, Boothroyd JC. Direct and sensitive detection of a pathogenic protozoan, Toxoplasma gondii, by polymerase chain reaction. Journal of Clinical Microbiology. 1989;27:1787–92. doi: 10.1128/jcm.27.8.1787-1792.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett LC, Lunn G, Coico R. Biosafety: Guidelines for Working with Pathogenic and Infectious Microorganisms. Current Protocols in Microbiology. 2009;13:1A.1.1–1A.1.14. doi: 10.1002/9780471729259.mc01a01s13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook MK, Jacobs L. Cultivation of Toxoplasma gondii in tissue cultures of various derivations. Journal Parasitology. :172–82. [PubMed] [Google Scholar]
- Denkers EY, Gazzinelli RT. Regulation and function of T-cell mediated immunity during Toxoplasma gondii infection. Clin Micro Rev. 1998;11:569–88. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey JP, Beattie CP. Toxoplasmosis of animals and man. CRC Press; Boca Raton, Florida: 1988. [Google Scholar]
- Dubey JP, Swan GV, Frenkel JK. A simplified method for isolation of Toxoplasma gondii from the feces of cats. Journal of Parasitology. 1972;58:1005–06. [PubMed] [Google Scholar]
- Dubey JP. Infectivity and pathogenicity of Toxoplasma gondii oocysts for cats. Journal of Parasitology. 1996;82:957–61. [PubMed] [Google Scholar]
- Dubey JP. Toxoplasmosis of animals and humans. CRC Press; Boca Raton: 2010. [Google Scholar]
- Dubey JP, Beattie CP. Toxoplasmosis of animals and man. CRC Press; Boca Raton: 1988. [Google Scholar]
- Dubey JP, Desmonts G. Serological responses of equids fed Toxoplasma gondii oocysts. Equine Vet J. 1987;19:337–39. doi: 10.1111/j.2042-3306.1987.tb01426.x. [DOI] [PubMed] [Google Scholar]
- Dubey JP, Frenkel JF. Cyst-induced toxoplasmosis in cats. Journal of Protozoology. 1972;19:155–77. doi: 10.1111/j.1550-7408.1972.tb03431.x. [DOI] [PubMed] [Google Scholar]
- Dubey JP, Frenkel JK. Feline toxoplasmosis from acutely infected mice and the development of Toxoplasma cysts. Journal of Protozoology. 1976;23:537–46. doi: 10.1111/j.1550-7408.1976.tb03836.x. [DOI] [PubMed] [Google Scholar]
- Evans R, Chatterton JM, Ashburn D, Joss AW, Ho-Yen DO. Cell-culture system for continuous production of Toxoplasma gondii tachyzoites. Eur J Clin Microbiol Infect Dis. 1999;18:879–84. doi: 10.1007/s100960050423. [DOI] [PubMed] [Google Scholar]
- Fekkar A, Bodaghi B, Touafek F, Le Hoang P, Mazier D, Paris L. Comparison of immunoblotting, calculation of the Goldmann-Witmer coefficient, and real-time PCR using aqueous humor samples for diagnosis of ocular toxoplasmosis. J Clin Microbiol. 2008;46:1965–7. doi: 10.1128/JCM.01900-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel JK, Dubey JP. Effects of freezing on the viability of Toxoplasma oocysts. Journal of Parsitology. 1973;59:587–8. [PubMed] [Google Scholar]
- Frenkel JK. Toxoplasma in and around us. Bio Science. 1973;23:343–52. [Google Scholar]
- Frenkel JK, Dubey JP. Toxoplasmosis and its prevention in cats and man. Journal of Infectious Diseases. 1972;126:664–73. doi: 10.1093/infdis/126.6.664. [DOI] [PubMed] [Google Scholar]
- Frenkel JK, Dubey JP, Hoff RL. Loss of stages after continuous passage of Toxoplasma gondii and Besnoitia jellisoni. Journal of Protozoology. 1976;23:421–24. doi: 10.1111/j.1550-7408.1976.tb03799.x. [DOI] [PubMed] [Google Scholar]
- Frenkel JK, Dubey JP, Miller NL. Toxoplasma gondii in cats: fecal stages identified as coccidian oocysts. Science. 1970;167:893–96. doi: 10.1126/science.167.3919.893. [DOI] [PubMed] [Google Scholar]
- Goldman M, Carver RK, Sulzer AJ. Reproduction of Toxoplasma gondii by internal budding. J Parasitol. 1958;44:161–71. [PubMed] [Google Scholar]
- Grigg ME, Ganatra J, Boothroyd JC, Margolis TP. Unusual abundance of atypical strains associated with human ocular toxoplasmosis. J Infect Dis. 2001;184:633–39. doi: 10.1086/322800. [DOI] [PubMed] [Google Scholar]
- Holland GN. Ocular toxoplasmosis: a global reassessment. Part I: epidemiology and course of disease. American Journal of Ophthalmology. 2003;136:973–88. doi: 10.1016/j.ajo.2003.09.040. [DOI] [PubMed] [Google Scholar]
- Holland GN. Ocular toxoplasmosis: a global reassessment. Part II: disease manifestations and management. American Journal of Ophthalmology. 2004;137:1–17. [PubMed] [Google Scholar]
- Howe DK, Sibley LD. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J Infect Dis. 1995;172:1561–66. doi: 10.1093/infdis/172.6.1561. [DOI] [PubMed] [Google Scholar]
- Hughes HP, Hudson L, Fleck DG. In vitro culture of Toxoplasma gondii in primary and established cell lines. Int J Parasitol. 1986;16:317–22. doi: 10.1016/0020-7519(86)90109-8. [DOI] [PubMed] [Google Scholar]
- Huynh MH, V, Carruthers B. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot Cell. 2009;8:530–39. doi: 10.1128/EC.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelski DM, Remington JS. Toxoplasmosis in the non-AIDS immunocompromised host. Current Clinical Topics in Infectious Diseases. 1993;13:322–56. [PubMed] [Google Scholar]
- Jacobs L, Remington J, Melton ML. The resistance of the encysted form of Toxoplasma gondii. J Parasitol. 1960;46:11– 21. [PubMed] [Google Scholar]
- Jerome ME, Radke JR, Bohne W, Roos DS, White MW. Toxoplasma gondii bradyzoites form spontaneously during sporozoite-initiated development. Infection and Immunity. 1998;66:4838–44. doi: 10.1128/iai.66.10.4838-4844.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LA, Alexander J, Roberts CW. Ocular toxoplasmosis: in the storm of the eye. Parasite Immunol. 2006;28:635–42. doi: 10.1111/j.1365-3024.2006.00874.x. [DOI] [PubMed] [Google Scholar]
- Khan A, Behnke MS, Dunay IR, White MW, Sibley LD. Phenotypic and gene expression changes among clonal type I strains of Toxoplasma gondii. Euk Cell. 2009;8:1828–36. doi: 10.1128/EC.00150-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi H, Khan A, Behnke MS, Namasivayam S, Swapna LS, Hadjithomas M, Karamycheva S, Pinney D, Brunk BP, Ajioka JW, Ajzenberg D, Boothroyd JC, Boyle JP, Darde ML, Diaz-Miranda MA, Dubey JP, Fritz HM, Gennari SM, Gregory BD, Kim K, Saeij JP, Su C, White MW, Zhu XQ, Howe DK, Rosenthal BM, Grigg ME, Parkinson J, Liu L, Kissinger JC, Roos DS, Sibley LD. Local admixture of amplified and diversified secreted pathogenesis determinants shapes mosaic Toxoplasma gondii genomes. Nat Commun. 2016;7:10147. doi: 10.1038/ncomms10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane JO, Ruchman I. Cultivation of toxoplasma in the developing chick embryo. Proc Soc Exp Biol Med. 1948;67:1–4. doi: 10.3181/00379727-67-16185. [DOI] [PubMed] [Google Scholar]
- Meissner M, Schluter D, Soldati D. Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science. 2002;298:837–40. doi: 10.1126/science.1074553. [DOI] [PubMed] [Google Scholar]
- Nicolle C, Manceaux LH. Sur une infection a corp de Leishman (ou organismes voisins) du gondi. Comptes Rendus de l Academie des Sciences Serie III, Sciences de la Vie. 1908;147:763–66. [Google Scholar]
- Pfefferkorn ER, Pfefferkorn LC. Toxoplasma gondii: isolation and preliminary characterization of temperature sensitive mutants. Experimental Parasitology. 1976;39:365–76. doi: 10.1016/0014-4894(76)90040-0. [DOI] [PubMed] [Google Scholar]
- Radke JR, Striepen B, Guerini MN, Jerome ME, Roos DS, White MW. Defining the cell cycle for the tachyzoite stage of Toxoplasma gondii. Molec Biochem Parasitol. 2001;115:165–75. doi: 10.1016/s0166-6851(01)00284-5. [DOI] [PubMed] [Google Scholar]
- Radke JR, White MW. A cell cycle model for the tachyzoite of Toxoplasma gondii using the Herpes simplex virus thymidine kinase. Molec Biochem Parasitol. 1998;94:237–47. doi: 10.1016/s0166-6851(98)00074-7. [DOI] [PubMed] [Google Scholar]
- Radke JR, White MW. Expression of herpes simplex virus thymidine kinase in Toxoplasma gondii attenuates tachyzoite virulence in mice. Infect Immun. 1999;67:5292–7. doi: 10.1128/iai.67.10.5292-5297.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radke JR, Guerini MN, Jerome ME, White MW. A change in the premitotic period of the cell cycle is associated with bradyzoite differentiation in Toxoplasma gondii. Mol Biochem Parasitol. 2003;131:119–27. doi: 10.1016/s0166-6851(03)00198-1. [DOI] [PubMed] [Google Scholar]
- Roos DS, Donald RGK, Morrissette NS, Moulton AL. Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods in Cell Biology. 1994;45:28–61. doi: 10.1016/s0091-679x(08)61845-2. [DOI] [PubMed] [Google Scholar]
- Saadatnia G, Haj Ghani H, Khoo BY, Maimunah A, Rahmah N. Optimization of Toxoplasma gondii cultivation in VERO cell line. Trop Biomed. 2010;27:125–30. [PubMed] [Google Scholar]
- Saeij JP, Boyle JP, Boothroyd JC. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol. 2005;21:476–81. doi: 10.1016/j.pt.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Saeij JPJ, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, Boothroyd JC. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science. 2006;314:1780–83. doi: 10.1126/science.1133690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SP, Dubey JP. Quantitative survival of Toxoplasma gondii tachyzoites and bradyzoites in pepsin and in trypsin solutions. Am J Vet Res. 1981;42:128–30. [PubMed] [Google Scholar]
- Shen B, Brown KM, Lee TD, Sibley LD. Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. MBio. 2014;5:e01114–14. doi: 10.1128/mBio.01114-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley LD, Ajioka JW. Population structure of Toxoplasma gondii: Clonal expansion driven by infrequent recombination and selective sweeps. Ann Rev Microbiol. 2008;62:329–51. doi: 10.1146/annurev.micro.62.081307.162925. [DOI] [PubMed] [Google Scholar]
- Sibley LD, Boothroyd JC. Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature (Lond) 1992;359:82–85. doi: 10.1038/359082a0. [DOI] [PubMed] [Google Scholar]
- Sibley LD, Howe DK. Genetic basis of pathogenicity in toxoplasmosis. Current Topics Microbiology Immunology. 1996;219:1–15. doi: 10.1007/978-3-642-51014-4_1. [DOI] [PubMed] [Google Scholar]
- Sidik SM, Hackett CG, Tran F, Westwood NJ, Lourido S. Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One. 2014;9:e100450. doi: 10.1371/journal.pone.0100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skariah S, McIntyre MK, Mordue DG. Toxoplasma gondii: determinants of tachyzoite to bradyzoite conversion. Parasitol Res. 2010;107:253–60. doi: 10.1007/s00436-010-1899-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splendore A. Un nuovo protozoa parassita de’ conigli. Incontrato nelle lesioni anatomiche d’une malattia che ricorda in molti punti il Kala-azar dell’uomo. Nota preliminare pel. Rev Soc Scient Sao Paulo. 1908;3:109–12. [Google Scholar]
- Striepen B, Jordan CN, Reiff S, van Dooren GG. Building the perfect parasite: cell division in Apicomplexa. PLoS Pathog. 2007;3:691–98. doi: 10.1371/journal.ppat.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striepen B, Soldati D. Genetic manipulation of Toxoplasma gondii. In: Weiss LM, Kim K, editors. Toxoplasma gondii The Model Apicomplexan: Perspectives and Methods. Academic Press, Elsevier; New York: 2007. [Google Scholar]
- Su CL, Khan A, Zhou P, Majumdar D, Ajzenberg D, Dardé ML, Zhu XQ, Ajioka JW, Rosenthal B, Dubey JP, Sibley LD. Globally diverse Toxoplasma gondii isolates comprise six major clades originating from a small number of distinct ancestral lineages. Proc Natl Acad Sci (USA) 2012;109:5844–49. doi: 10.1073/pnas.1203190109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S, Barragan A, Su C, Fux B, Fentress SJ, Tang K, Beatty WL, Haijj EL, Jerome M, Behnke MS, White M, Wootton JC, Sibley LD. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science. 2006;314:1776–80. doi: 10.1126/science.1133643. [DOI] [PubMed] [Google Scholar]
- Weiss LM, Kim K. Bradyzoite development. In: Weiss LM, Kim K, editors. Toxoplasma gondii the model apicomplexan: perspectives and methods. Academic Press; New York: 2007. [Google Scholar]