

Abstract
Monoglycerides play a central role in lipid metabolism and are important signaling metabolites. Quantitative analysis of monoglyceride molecular species has remained challenging due to rapid isomerization via α-hydroxy acyl migration. Herein, we describe a shotgun lipidomics approach that utilizes a single-phase methyl tert-butyl ether extraction to minimize acyl migration, a facile low temperature diacetyl derivatization to stabilize regiospecificity, and tandem mass spectrometric analysis to identify and quantify regioisomers of monoglycerides in biological samples. The rapid and robust diacetyl derivatization at low temperatures (e.g., −20 °C, 30 min) prevents postextraction acyl migration and preserves regiospecificity of monoglyceride structural isomers. Furthermore, ionization of ammonium adducts of diacetyl monoglyceride derivatives in positive-ion mode markedly increases analytic sensitivity (low fmol/μL). Critically, diacetyl derivatization enables the differentiation of discrete monoglyceride regioisomers without chromatography through their distinct signature fragmentation patterns during collision induced dissociation. The application of this approach in the analysis of monoglycerides in multiple biologic tissues demonstrated diverse profiles of molecular species. Remarkably, the regiospecificity of individual monoglyceride molecular species is also diverse from tissue to tissue. Collectively, this developed approach enables the profiling, identification and quantitation of monoglyceride regioisomers directly from tissue extracts.
Graphical abstract
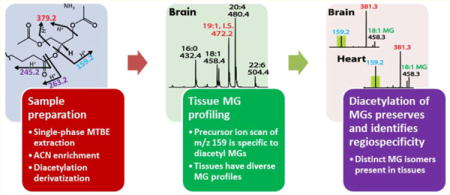
Monoglycerides (MGs) are essential lipid metabolites participating in cellular lipid synthesis, catabolism, and signaling. In biologic tissues, MGs are comprised of diverse subclasses that differ in the nature of their sn-1 linkages (i.e., ester, vinyl ether, or alkyl ether) and of diverse molecular species comprising different aliphatic chains with differing regiospecificities. Thus, the determination of the amount, flux, and roles of MG molecular species is critical to fully define alterations in lipid metabolism in health and disease. However, analysis of MGs has been complicated by their low abundance and facile α-hydroxy acyl migration during extraction and analysis.1–3 Therefore, development of sensitive and robust methods for the definitive identification and quantitation of MG regioisomers is an important goal of lipidomics.
Following the discovery of 2-arachidonoylglycerol (2-AG) as a physiological ligand of cannabinoid receptors in the 1990s, recent efforts have focused on the identification of other MGs that possess structural and functional similarities.1,2,4–9 Some of these compounds are known to produce “cannabimimetic effects” by directly binding cannabinoid receptors or “entourage effects” by modifying the activity of other endocannabinoids.1,8,10 Numerous methods have been reported to analyze these compounds, including HPLC coupled with UV or fluorescence detection, GC-based analysis (GC-MS, GC-MS/MS), and LC-based analysis (LC-MS, LC-MS/MS).1–5,7,10–20 Although the LC-MS/MS with positive electrospray ionization and selected reaction monitoring (SRM) has become the most frequently used technique for MG analysis due to its high sensitivity and specificity,3 its use has been limited to a relatively small number of compounds.4 In addition, SRM analysis is prone to false positive identifications.1 Another significant challenge is the isomerization of 2-AG into 1-AG by α-hydroxy acyl migration.1,2,21 Since protic solvents (e.g., methanol), high pH values, and high temperature promote the isomerization of 2-AG and AG degradation,1,3,10,13,22 aprotic solvents (e.g., toluene3,11), mild organic acids,1,3 or low temperatures have been utilized to minimize isomerization, but with limited success.
Previously we demonstrated the presence of a diverse array of diacylglycerols23 and ether-linked diglycerides24 in various biological samples. Thus, we anticipated that MGs also possess a diverse profile of subclasses and molecular species, including regioisomers derived from the diglyceride precursor pool.6,13,25 Although MG molecular species other than 2-AG, such as 2-linoleoylglycerol (2-LG) and 2-palmitoylglycerol (2-PG), have been detected in various biological samples,8,13,20,26–28 no current methods have comprehensively profiled a wide range of MG molecules. These MG molecular species presumably play important roles in cellular lipid metabolic flux and may be associated with cannabinoid system signaling by acting as 2-AG congeners or entourage compounds through as yet poorly understood pathways.1,8
Herein, we developed a shotgun lipidomics approach to comprehensively profile endogenous MG molecular species in multiple tissues. To address the analytical challenges inherent in the low abundance of endogenous MGs and spontaneous acyl migration, a rapid and facile low temperature diacetyl derivatization was performed following a simple single-phase aprotic solvent extraction. This simple and robust derivatization step promotes the high ionization efficiency, high sensitivity, and high specificity of tandem mass spectrometric analysis. More importantly, it stabilizes the constitutional (structural) isomers of MGs by occupying both free hydroxyl moieties present in glycerol. Furthermore, in contrast to derivatization (e.g., silylation, acylation, or urethane derivatization) generally employed in GC-based analysis,11,15–18,29–31 the derivatization reaction presented herein is: (1) rapid at low temperature (e.g., −20 °C) thus best preventing postextraction acyl migration; and (2) renders distinct signature fragmentation patterns for MG regioisomers via collision induced dissociation (CID) that differentiates individual regioisomers without chromatography.
EXPERIMENTAL SECTION
Detailed experimental procedures are provided in the Supporting Information.
Reagents
All reagents were purchased commercially and utilized without further purification.
Synthesis of Vinyl Ether Monoglycerides
Vinyl ether diglycerides were synthesized from plasmenylcholines by phospholipase C-catalyzed hydrolysis as described previously.24 The synthetic vinyl ether diglycerides were then subjected to base hydrolysis to hydrolyze the sn-2 ester bonds while the sn-1 vinyl ether bonds remained intact, as described previously.32
Extraction of MGs from Biological Samples
All steps were carried out at 4 °C or below to prevent isomerization or degradation of the MGs. Frozen tissue samples were subjected to a single-phase solvent extraction using methyl tert-butyl ether (MTBE). MGs were then enriched by liquid/liquid partitioning using hexane versus acetonitrile (1:1, v/v) containing 0.2 M acetic acid to prevent isomerization.
Derivatization of MGs
Dried MG residues were derivatized with acetic anhydride in chloroform in the presence of 4-(dimethylamino) pyridine (DMAP) at −20 °C for 30 min, extracted with hexane/methanol/water to enrich derivatized MGs into the hexane layer, and excess reagents and the majority of polar lipids were removed by partitioning into methanol layer.
Mass Spectrometric Analysis
Mass spectrometric analysis of derivatized MG standards and lipid extracts were performed using a TSQ Quantum Ultra mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an automated nanospray apparatus (i.e., Nanomate HD, Advion BioSciences, Ithaca, NY) as previously described.33,34 Product ion scanning or precursor ion scanning was performed in the positive ion mode in the presence of 10 mM ammonium acetate.
RESULTS AND DISCUSSION
Derivatization of Monoglycerides Enhances Ionization Efficiency and Enables Regioisomer Differentiation
Since MGs do not contain a charged moiety, their efficient ionization requires the addition of a charged adduct ion or the covalent attachment of a chargeable moiety through chemical derivatization. Most LC-based approaches directly analyze MG adduct cations such as protonated ions, ammonium adducts, or Ag+ adducts.11,35,36 However, since direct analysis of MG regioisomers (e.g., 1- and 2-AG) generates almost identical MS/MS spectra, complete chromatographic separation is therefore an absolute requirement for accurate analysis of MG regioisomers.11,21 In contrast to LC-based approaches, shotgun lipidomics is high-throughput and comprehensively profiles classes of lipid molecular species. However, this occurs at the expense of LC-based enrichment of low abundance species and separation of isomeric species. Strategically, this can be overcome by chemical derivatization that dramatically increases ionization efficiency while simultaneously providing signature fragmentation patterns to differentiate isomers. Furthermore, chemical derivatization of both free hydroxyl groups of a MG molecule stabilizes the isomeric integrity of MG regioisomers by eliminating intramolecular nucleophilic attack of carbonyl groups by hydroxyl groups (i.e., acyl chain migration).1
Diacylglycerol regioisomers have been previously analyzed after chemical derivatization by dimethylglycine (DMG).23 Therefore, we first examined DMG-derivatized MGs. Unfortunately, although ionization efficiency of the derivatized MG molecules was improved, tandem mass spectra of DMG-derivatized MG regioisomers (e.g., 1- and 2-LG) were almost identical using either H+ or Li+ adduct ions (Figure 1A).
Figure 1.
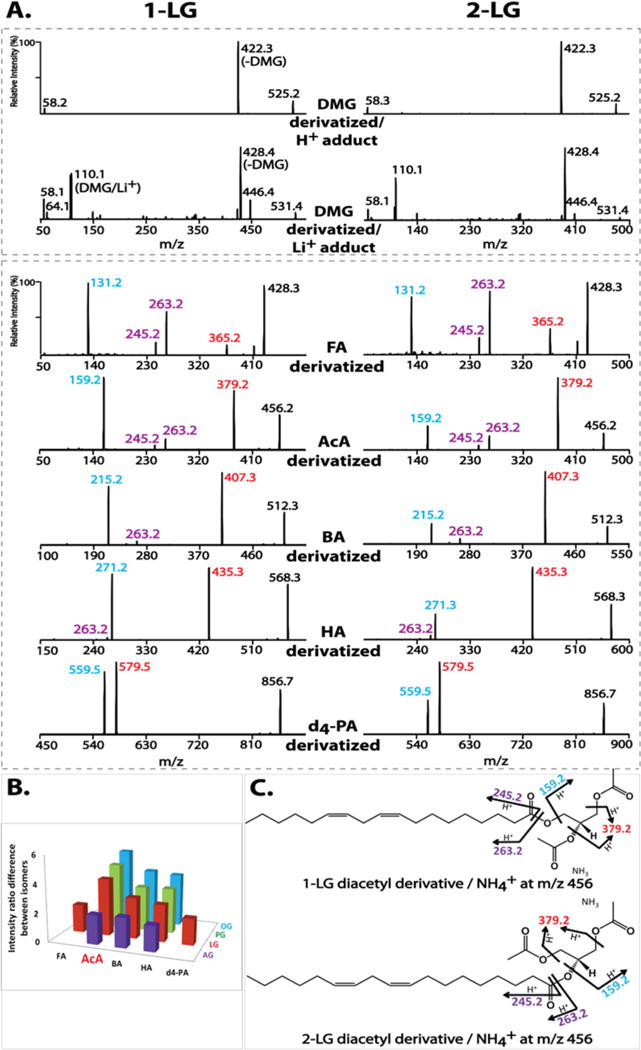
Optimization of derivatizing agents for monoglyceride (MG) analysis. (A) Tandem mass spectra of derivatized 1- and 2-linoleoylglycerol (LG) by dimethylglycine (DMG), d4-palmitic acid (d4-PA), or short-chain organic acids (formic (FA), acetic (AcA), butyric (BA) and hexanoic (HA)). The DMG derivatives were analyzed as their H+ and Li+ adducts at collision energies of 20 and 30 eV, respectively. The acid derivatives were analyzed as their NH4+ adducts at 15 eV. (B) Differences in fragmentation patterns of regioisomers of MG species (palmitoylglycerol (PG), oleoylglycerol (OG), LG, and arachidonoylglycerol (AG)) using different derivatizing acids. The differences are represented as the fold change in the intensity ratio of the two featured fragment ions for individual isomers (e.g., m/z 159 (blue) vs m/z 379 (red) for AcA-derivatized 1- and 2-LG). (C) The proposed fragmentation pathways for AcA-derivatized LGs. In addition to the m/z 159 (blue) resulting from loss of NH3 + linoleic acid and m/z 379 (red) from loss of NH3 + AcA, the ketene fragment ion and its dehydrated form (at m/z 263 and m/z 245) are shown in purple.
Since triacylglycerols have been successfully analyzed with either alkali metal (e.g., Na+ or Li+)37–41 or ammonium adducts,42–48 we next examined acylated MGs using long-chain fatty acids. As expected, the MS/MS spectra of ammonium adducts of d4-palmitic acid (d4-PA)-derivatized 1- and 2-LG both demonstrated two diagnostic fragment ions at m/z 579 and m/z 559 resulting from loss of NH3 + d4-PA and NH3 + linoleic acid, respectively (Figure 1A). The difference in the intensity ratio of the two fragment ions distinguished the regioisomers by the relatively less favorable loss of linoleic acid indicative of decreased fragmentation of the sn-2 acyl chain during CID. However, this difference was only modest.
We next examined whether alterations in the chain length of the derivatizing acids had an impact on MG isomer differentiation. Shorter-chain organic acids were examined including formic acid (FA), acetic acid (AcA), butyric acid (BA) and hexanoic acid (HA). These short-chain organic acid derivatized LG regioisomers demonstrated similar fragmentation patterns as d4-PA derivatives (Figure 1A), including the two featured fragment ions resulting from loss of NH3 + derivatizing acid or NH3 + linoleic acid. For example, the MS/MS spectra of AcA-derivatized 1- and 2-LG both displayed two major fragment ions at m/z 379 and m/z 159 from loss of AcA or linoleic acid, respectively. Two less abundant fragment ions were also observed at m/z 263 and m/z 245 originating from the linoleic acyl chain (i.e., protonated 18:2 ketene and its dehydration product). This is common for polyunsaturated acyl chain-containing species undergoing fragmentation in the positive ion mode.24 The proposed fragmentation pathway for AcA-derivatized LG regioisomers is shown in Figure 1C.
Similarly, these short-chain organic acid derivatized LGs also demonstrated distinct intensity ratios of the two featured fragment ions that differentiate LG regioisomers. For example, the intensity ratio of m/z 159 versus m/z 379 of AcA-derivatized 1-LG is 4-fold greater than that of AcA-derivatized 2-LG (i.e., a ratio of 1.2 for 1-LG indicating that the two fragment ions are paired in nearly equal intensities versus a ratio of 0.3 for 2-LG indicating that the loss of the sn-2 acyl chain occurs at a substantially lower rate). In comparison, the intensity ratio of m/z 559 versus m/z 579 of d4-PA-derivatized 1-LG is less than 2-fold of that of d4-PA-derivatized 2-LG (i.e., 0.87 and 0.47, respectively). In addition to LG isomers, other representative MG isomers (including AG, PG and oleoylglycerol (OG)) were also derivatized with short-chain organic acids to determine which acid derivatization method could most effectively discriminate between sn-1 and sn-2 isomers. Derivatization with AcA was the best reagent among all tested organic acids (Figure 1B).
Identification of Signature Fragmentation Patterns of Diacetyl MGs to Differentiate Monoacylglycerol Regioisomers
AcA derivatization of representative MG species was further examined in detail. A mixture of multiple MG species covering a wide range of acyl chains (e.g., 14 to 22 carbons in length and 0 to 6 double bonds) was derivatized under different reaction temperatures (−20 to 4 °C) and times (15 min to 1 h). The AcA derivatization not only was rapid and efficient but occurred at a very low reaction temperature for its completion (Figure S1). Low reaction temperatures (e.g., −20 °C) prevent isomerization, and the rapid reaction (e.g., 30 min) quickly stabilizes the regiospecificity.
Different adduct ions (Na+, H+ and NH4+) of diacetyl MGs were examined (Figure S2). Ammonium adducts of diacetyl MGs demonstrated superior ionization efficiencies and tandem MS discrimination for regioisomers. Ammonium adducts of diacetyl MGs containing varied acyl chain lengths and differential degrees of unsaturation (i.e., PG (16:0), OG (18:1), LG (18:2), and AG (20:4)) were next examined to verify the signature fragmentation patterns present for each pair of regioisomers (Figure 2A). All examined diacetyl MGs displayed the two featured fragment ions as expected, including the diacetyl glycerol derivative at m/z 159 resulting from loss of NH3 and the corresponding fatty acid, as well as the counterpart fragment ion at varied m/z values resulting from neutral loss of 77 amu (i.e., NH3 + AcA). All diacetyl MGs also displayed fragment ions corresponding to protonated ketenes (e.g., m/z 239 for 16:0, m/z 265 for 18:1, m/z 263 for 18:2 and m/z 287 for 20:4 protonated ketenes). The diacetyl MGs containing polyunsaturated acyl chains (e.g., 18:2 and 20:4) displayed additional fragment ions corresponding to the dehydrated form of protonated ketenes (e.g., m/z 245 for 18:2 or m/z 269 for 20:4 protonated ketene – H2O). Furthermore, the intensities of the ketene fragment ions increase with increasing unsaturation of the acyl chain. For example, diacetyl PGs had the least abundant ketene fragment ions (m/z 239) while diacetyl AGs had the most abundant ketene fragment ions (m/z 287 and m/z 269).
Figure 2.
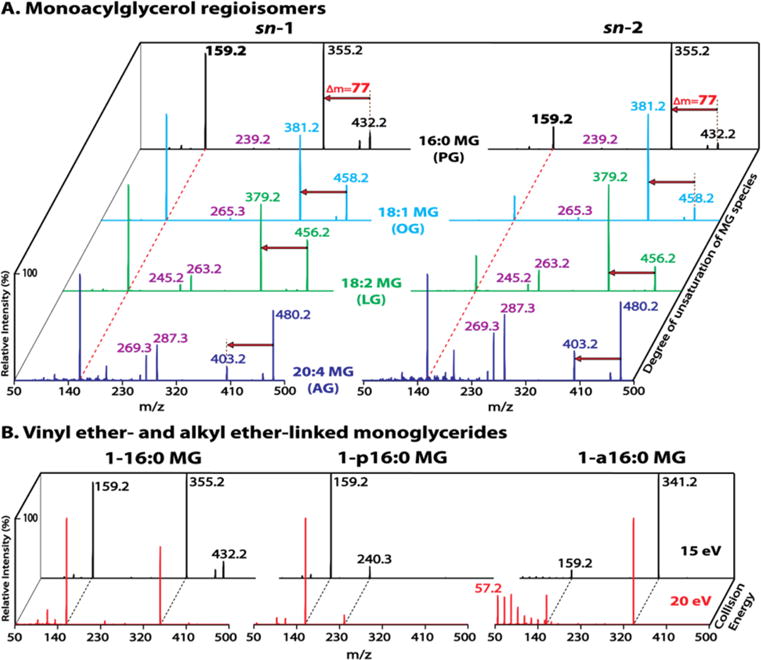
Identification of monoacylglycerol regioisomers and ether-linked monoglycerides (MGs). (A) Tandem mass spectra of acetic acid (AcA)-derivatized MG species (PG, OG, LG and AG) acquired in the presence of 10 mM ammonium acetate in the positive ion mode at a collision energy of 15 eV. The dashed red line indicates the first featured fragment ion at m/z 159 resulting from the loss of NH3 + the corresponding fatty acid. The red arrow indicates the second featured fragment ion resulting from the loss of NH3 + AcA (77 amu). (B) Tandem mass spectra of AcA-derivatized sn-1 16:0 MG species containing an ester bond (1–16:0 MG), a vinyl ether linkage (1-p16:0 MG), or an alkyl ether linkage (1-a16:0 MG). The spectra were acquired at collision energies of 15 eV (black trace) and 20 eV (red trace). A signature fragmentation pattern in the low mass range comprising a series of peaks separated by 14 Th is present exclusively for the alkyl ether-MG species at a higher collision energy. The complete absence of the fragment ion from loss of NH3 + AcA (77 amu), and the unique fragment ion at m/z 240 (16 carbon alkyne/NH4+) resulting from the cleavage of vinyl ether linkage are characteristic for vinyl ether-MG species.
Similar to diacetyl LG regioisomers, diacetyl PG or OG regioisomers were also readily differentiated by the intensity ratio of their two featured fragment ions (i.e., m/z 159 vs m/z 355 for PG isomers, or m/z 159 vs m/z 381 for OG isomers). The sn-1 isomers demonstrated an intensity ratio of ~1 while the sn-2 isomers displayed a much lower intensity ratio, which unambiguously differentiated the regioisomers (Figure 2A).
In contrast, the intensity ratio of the two fragment ions for diacetyl AGs (i.e., m/z 159 vs m/z 403) was much higher than 1 for both isomers (Figure 2A). Apparently this high intensity ratio was due to the very low abundance of m/z 403. Since the reduced intensity of m/z 403 (i.e., the monoacetyl-20:4-glycerol derivative) was accompanied by the increased 20:4 ketene fragment ions (m/z 287 and m/z 269), these 20:4 ketene fragments likely resulted from the sequential fragmentation of the monoacetyl-20:4-glycerol product ion during CID. Accordingly, the intensity of m/z 159 versus the sum intensity of m/z 403 and the ketene fragments were calculated. A much lower ratio was then obtained for the sn-2 isomer (i.e., 0.7 for 2-AG vs 1.4 for 1-AG), which thus differentiated the AG regioisomers.
Identification of Signature Fragmentation Patterns of Diacetyl MGs to Differentiate Acyl, Vinyl Ether-, and Alkyl Ether-Linked MG Species
Ether linked MG species were also examined to determine their characteristic fragmentation patterns (Figure 2B). Ammonium adducts of diacetyl 16:0 MGs containing an ester bond (1–16:0 MG), a vinyl ether bond (1-p16:0 MG), or an alkyl ether bond (1-a16:0 MG) were fragmented first under the same collision energy (15 eV) as above optimized for monoacylglycerol species. Fragmentation of 1-p16:0 MG generated a prominent product ion peak at m/z 159 as that of 1–16:0 MG, resulting from the neutral loss of the sn-1 moiety. In contrast, fragmentation of 1-a16:0 MG revealed only a very low abundance peak at m/z 159 (likely due to the unfavorable cleavage of alkyl ether bond) thereby discriminating the alkyl ether-linked MG species from their acyl- and vinyl ether-linked counterparts. To further validate the presence of the alkyl ether linkage, fragmentation was performed under a higher collision energy (20 eV). In contrast to the retained fragmentation patterns of 1–16:0 MG and 1-p16:0 MG during CID at the increased collision energy, the product ion spectrum of 1-a16:0 MG demonstrated a significant difference within the low mass range. A series of low mass product ion peaks, starting at m/z 57 (likely CH3(CH2)3+) with members separated by 14 Th (–CH2–), were exclusively present for 1-a16:0 MG, likely resulting from the higher collision energy-facilitated sequential fragmentation of monoacetyl-a16:0-glycerol product ion at m/z 341. This series of fragment ions therefore substantiate the presence of an alkyl ether linkage within the MG species (Figure 2B, red trace).
Furthermore, monoacyl- and vinyl ether-linked MG species were discriminated from their tandem MS spectra via two aspects: (1) the presence of the fragment ion resulting from neutral loss of NH3 + AcA (i.e., 77 amu) for 1–16:0 MG and its absence for 1-p16:0 MG (likely due to the instability of the vinyl ether linkage during CID); and (2) the presence of a unique fragment ion at m/z 240 corresponding to a 16 carbon alkyne/NH4+ product ion resulting from the cleavage of the vinyl ether linkage, similar to the characteristic tandem MS feature of the ammonium adducts of vinyl ether-linked diglyceride species.24
Collectively, these results demonstrate the signature fragmentation patterns for each of the three MG subclasses, therefore enabling the differentiation of isomeric vinyl ether- versus alkyl ether-linked species (e.g., 1-p16:0 MG vs 1-a16:1 MG), and the differentiation of isobaric monoacyl- versus ether-linked species (e.g., 1–15:1 MG vs 1-p16:0 MG or 1-a16:1 MG).
Quantitation of Coexisting MG Regioisomers in Mixtures
In biological samples, individual regioisomers might coexist as isomeric mixtures. For example, both 1-AG and 2-AG have been detected frequently in biological samples although it is still unclear whether 1-AG is an endogenous metabolite or an artifact.13 Moreover, previous studies have rarely investigated the coexistence of sn-1 isomers with other MG species. Since the highly efficient acetylation of the two hydroxyl groups of MGs immediately preserves their regiospecificity, it is possible to eliminate spontaneous isomerization during analysis and thereby provide convincing evidence for the presence of endogenous sn-1 MG isomers. Importantly, these results may provide insight into the metabolism and signaling functions of endogenous sn-1 isomers as well as interactions with their sn-2 counterparts.
The coexistence of MG regioisomers in a mixture can be identified by the distinct tandem MS spectrum of the mixture. We examined tandem MS spectra of various mixtures of different MG species, including LG (Figure 3A), AG (Figure 3B), PG (Figure S3A), and OG (Figure S3B). Each isomeric mixture at a defined composition clearly demonstrated a distinct fragmentation pattern from that of each single isomer, particularly in the intensity ratio of the featured fragment ions.
Figure 3.
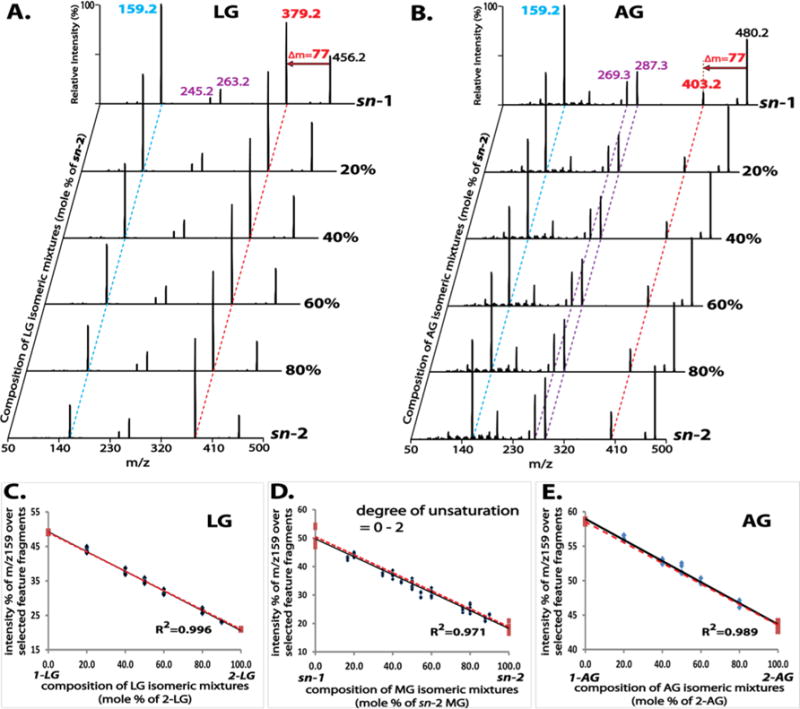
Tandem mass spectra of mixtures of monoglyceride (MG) regioisomers at defined compositions and the linear relationship between their tandem mass spectral features and isomeric compositions. Product ion spectra of these mixtures were acquired as described in Figure 2 and displayed in (A) for LG and in (B) for AG. The two featured fragment ions (m/z 159 and neutral loss of 77 amu) are indicated by the dashed blue and red lines, respectively. The intense ketene fragment ions for AG (m/z 287 and m/z 269) are indicated by the dashed purple lines. The linearity between the tandem mass spectral features (represented by the normalized intensity of m/z 159, as described in the text) and the molar compositions of isomeric mixtures is demonstrated in (C) for LG, in (E) for AG and in (D) for combined data from MG species containing acyl chains of 0–2 degree of unsaturation (i.e., PG, OG, and LG). Four replicates were analyzed for each mixture. Two types of linearity are displayed: the first established from a series of data points of isomeric mixtures at defined compositions (black solid line), and the second from the two data points of authentic 1- and 2-isomers (red dashed line).
To examine whether the composition of coexisting MG regioisomers in a mixture can be accurately quantified without relying on chromatographic isomer separation, the intensity ratios of the featured fragment ions were calculated from product ion spectra of isomeric mixtures at defined compositions. As described above, the intensities of the ketene fragment ions derived from the fatty acyl moieties are greater with increasing degrees of unsaturation. Moreover, the intensity ratios of the featured fragment ions calculated by considering these ketene fragment ions facilitate the regioisomer differentiation of AG. Accordingly, the intensity ratios of featured fragment ions calculated from product ion spectra were represented by the relative intensity (%) of m/z 159 after normalization to the sum of intensities of selected feature fragment ions including m/z 159 from the loss of the fatty acyl chain, the product ion from the neutral loss of 77 amu (e.g., m/z 379 in Figure 3A for LG, or m/z 403 in Figure 3B for AG), and the ketene fragment ions (e.g., m/z 263 and m/z 245 in Figure 3A for LG, or m/z 287 and m/z 269 in Figure 3B for AG).
Next, the calculated relative intensities were plotted against the compositions of the mixtures, which demonstrated a strong linear relationship (correlation coefficient of ≥0.99 for each examined MG species). Intriguingly, the linearity established using a series of isomeric mixtures was in agreement with that observed using only individual sn-1 and sn-2 isomers (i.e., black solid vs red dashed lines in Figure 3C for LG, Figure 3E for AG, Figure S3C for PG, or Figure S3D for OG), thus, implying that quantitation of isomeric compositions in mixtures can be simplified.
Furthermore, linearity was retained with a correlation coefficient of 0.97 when data points for PG, OG and LG were combined (Figure 3D). This result suggests that this linearity is largely independent of acyl chain length (Figure S4A) and degree of unsaturation (within 0 to 2). Additionally, product ion spectra of MG species containing acyl chains with 3 double bonds (e.g., 18:3 and 20:3 MG) demonstrated fragmentation features that were similar to those of MG species containing 0–2 double bonds but differed markedly from those of MG species with a higher degree of unsaturation (e.g., 20:4 MG) (Figures S4B and S4C). Collectively, this approach may provide good estimates for quantitation of isomeric compositions of numerous MG species containing acyl chains with 0 to 3 double bonds when their authentic isomers are not available.
Optimization of Tissue Extraction to Permit High Extraction Efficiency while Preventing Isomerization of MGs
Spontaneous isomerization of MG sn-2 isomers (particularly 2-AG) under experimental conditions has been a long-standing problem in lipidomics.3,11 Aprotic solvents4,11,20,49 and acidic conditions1,3,49 have proven to help prevent isomerization. We observed the nearly complete isomerization in methanol of 2-AG to 1-AG1,3,11 (Figure S5C), 2-LG to 1-LG (Figure S5A), and 2-OG to 1-OG (Figure S5B). The isomerization was largely prevented by the presence of 0.2 M acetic acid (pH ~ 3). Among numerous tested aprotic solvents, toluene was reported to best prevent 2-AG isomerization.3,11 However, in our experiments, isomerization of 2-LG, 2-OG and 2-AG was observed in toluene (Figure S5). Instead, we found that these sn-2 isomers were isomerized only marginally (LG and AG) or not at all (OG) in MTBE (Figure S5).
Various solvents were next tested as extraction solvents for biologic tissues in the presence of 0.2 M acetic acid. Before testing, the stability of a vinyl ether linkage under this acidic condition for the indicated times was confirmed (Figure S6). Both two-phase and single-phase extractions were examined (Figure 4). For two-phase approaches, a traditional Bligh–Dyer extraction using a chloroform/methanol/aqueous solution34 and a MTBE-based extraction using MTBE/methanol/aqueous solution developed more recently50,51 were both tested. The latter demonstrated significantly improved extraction efficiency (Figure 4A). In single-phase systems, extraction with ethyl acetate/hexane displayed a greater extraction efficiency with saturated MG species (e.g., 16:0 MG at m/z 432, and 18:0 MG at m/z 460). In contrast, extraction with acetonitrile demonstrated a dramatically enhanced extraction efficiency with unsaturated MG species (e.g., 18:2 MG at m/z 456, 18:1 MG at m/z 458, 20:4 MG at m/z 480, and 22:6 MG at m/z 504). Acetonitrile saturated with nonpolar hexane did not improve the extraction efficiency of saturated MG species. Remarkably, single-phase extraction with MTBE demonstrated the most pronounced and balanced extraction efficiency of both saturated and unsaturated MG species (Figure 4B).
Figure 4.
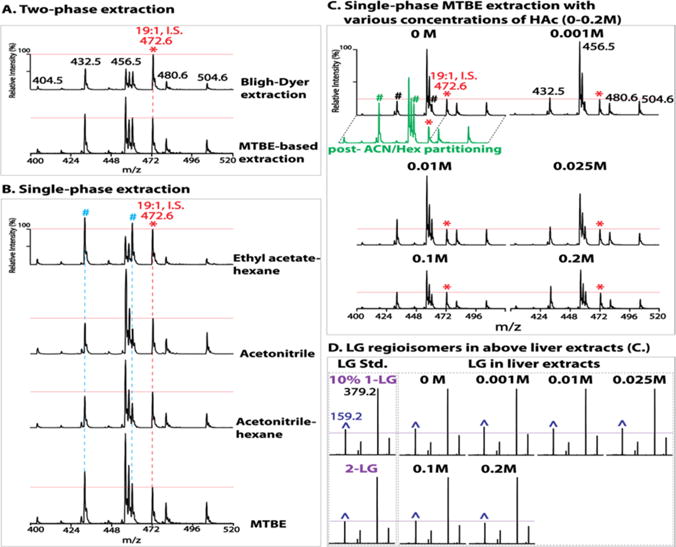
Optimization of extraction conditions for monoglyceride (MG) analysis in biologic tissues. Liver extracts were prepared by two-phase (A) or single-phase extraction (B) in the presence of 0.2 M acetic acid (HAc) as described in Experimental Section and in the text. Liver extracts were also prepared by a single-phase methyl tert-butyl ether (MTBE) extraction (C) in the presence of various concentrations of HAc (0–0.2 M). Extracts were analyzed by precursor ion scanning (PIS) of m/z 159 to profile MG species. “*” indicates the ion peak at m/z 472 for internal standard (I.S.) 1–19:1 MG (5 pmol/mg tissue). All spectra are displayed after normalization to the I.S. (whose peak height is indicated by the horizontal red line). “#” indicates the ion peaks of MG species containing saturated acyl chains (i.e., m/z 432 and m/z 460 for 16:0 and 18:0 MG, respectively). The green trace displays the spectrum of PIS of m/z 159 acquired from the liver MTBE extract after postextraction enrichment of MGs via acetonitrile/hexane (ACN/Hex) liquid/liquid partitioning as described in Experimental Section. Product ion spectra of LGs including authentic 1-LG or 2-LG standard (Std.) and endogenous LG species from the liver extracts in (C) are shown in (D). “^” indicates the m/z 159 fragment ion. The horizontal purple line indicates the peak height of m/z 159 for authentic 2-LG. Minimal (up to 10%) isomerization of endogenous 2-LG is present in the MTBE extract prepared without acid or in the presence of low concentrations (<0.1 M) of acetic acid; and no isomerization occurred in the presence of 0.1 M up to 0.2 M acetic acid.
Surprisingly, when the single-phase MTBE extraction was performed in the presence of varied concentrations of acetic acid (0 to 0.2 M), the extraction recovery was strikingly affected (Figure 4C). The extraction recovery decreased, by up to nearly one-third, with an increase in acid concentration from 0 to 0.2 M. This reduced extraction recovery of endogenous lipid species from biological samples, unfortunately, cannot be accounted for by the presence of internal standard since nearly 100% recovery of an internal standard occurs during a single-phase extraction. Next, the effect of acid concentration during MTBE extraction on the isomerization of sn-2 isomers of MGs in liver extracts was examined. Consistent with results obtained for authentic 2-LG (Figure S5A), the isomerization of endogenous 2-LG was also found to be minimal (less than 10%) during MTBE extraction without acid (Figure 4D). The effect of acidic conditions on the isomerization of other endogenous MG species (e.g., 16:0, 18:1, 20:4 MGs) in different tissues (e.g., brain, heart, and white adipose) was also confirmed to be minimal (Figure S7). It is worth noting that if definitively accurate MG isomeric compositions in biological samples are desired, mildly acidic MTBE extraction is recommended.
Furthermore, in order to enrich low abundance MG species and separate them from other relatively abundant glycerides (diglycerides and triglycerides), a postextraction MG enrichment using acetonitrile52 was performed by liquid/liquid partitioning with hexane versus acetonitrile in the presence of 0.2 M acetic acid to prevent postextraction isomerization. This enrichment step was advantageous from three perspectives. First, utilization of acetonitrile recovered MGs completely from the MTBE extract (Figure S8). Second, it reduced the complexity of the extract by complete removal of nonpolar triglycerides and cholesteryl esters and partial removal of diglycerides and cholesterol into the hexane phase. These lipid classes, although not overlapping with diacetyl-MGs in their m/z values, may have potential ion suppression effects on analysis of low abundance MGs by a shotgun approach if present in high abundance as in adipose, liver and muscle tissues. Finally, this step dramatically increased the recovery of saturated MG species (Figure 4C, green trace), which is likely due to the release of “trapped” saturated MG species from nonpolar lipid aggregates by nonpolar hexane.
Collectively, single-phase MTBE extraction followed by acetonitrile enrichment of MGs provides the best extraction efficiency of various MG species with only minimal isomerization (if any) of sn-2 isomers. Previously reported MG contents from acidic extractions likely have underestimated endogenous MGs due to markedly reduced extraction recoveries under acidic conditions. Extractions with various solvents with or without acids may also be responsible for the large discrepancies among published quantitation values.11,14
Identification and Quantitation of Differential MG Distribution and Isomeric Composition in Various Tissues
As described above, tandem MS spectra of diacetyl MGs demonstrated two featured fragment ions resulting from loss of NH3 + fatty acid (i.e., m/z 159), and from loss of NH3 + AcA (i.e., neutral loss of 77 amu). In theory, precursor ion scanning (PIS) of m/z 159 and neutral loss scanning (NLS) of 77 amu are both applicable to global profiling of the entire class of AcA-derivatized MG species. However, NLS of 77 amu is not specific to AcA-derivatized MGs since loss of one molecule of AcA during CID also occurs for other AcA-derivatized lipid classes such as cholesterol, diglycerides and ceramides (data not shown). By contrast, PIS of m/z 159 (i.e., diacetyl glycerol product ion) is unique and characteristic to diacetyl MG molecular ions. It was also highly sensitive as demonstrated by the extremely low background noise and impressive S/N ratios at concentrations as low as 1 fmol/μL (Figure S9).
Accordingly, PIS of m/z 159 was utilized to profile endogenous MG species in various biologic tissues. Individual MG species were specifically distributed in discrete tissues (mouse brain, liver, heart and white adipose) (Figure 5A). AG was present in all examined tissues with a wide range of concentrations from high abundance in brain to very low abundance in white adipose tissue. LG was present in extremely low abundance in brain while predominantly present in liver tissue. In contrast to the wide concentration range of unsaturated MGs (AG, LG and OG), saturated MG species (PG and 18:0 MG) demonstrated a relatively constant concentration among examined tissues.
Figure 5.
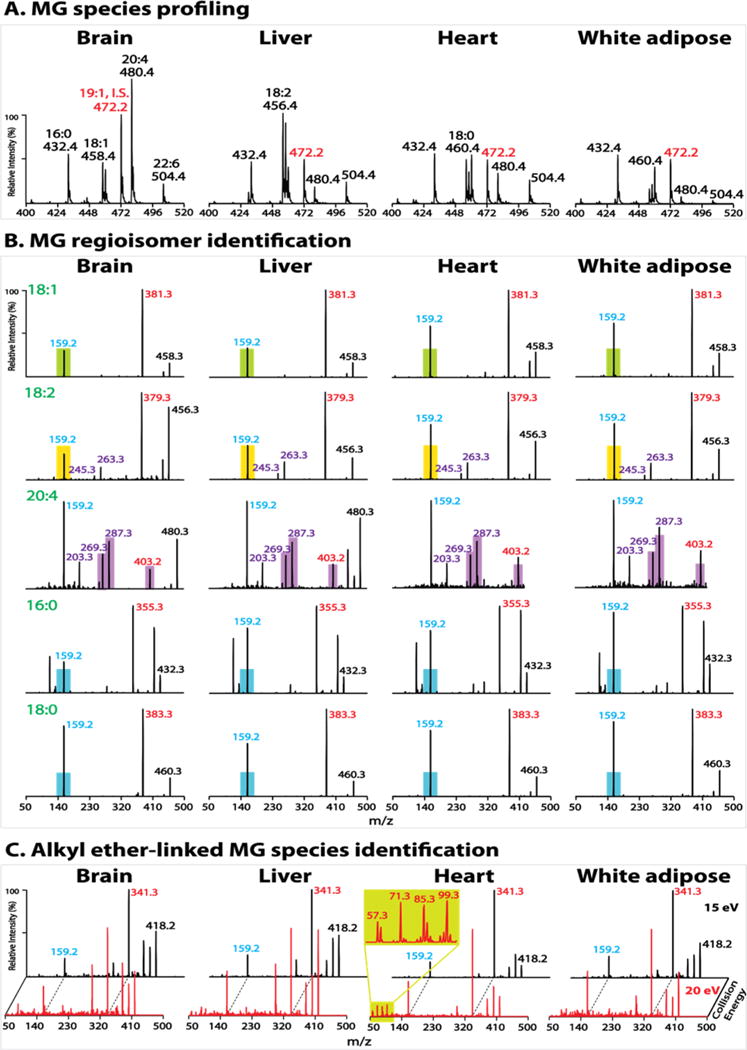
Tissue monoglyceride (MG) profiling and molecular species identification. Lipid extracts of mouse brain, liver, heart and white adipose tissues were prepared by a single-phase MTBE extraction without acid followed by acetonitrile enrichment, as described in Experimental Section. (A) Representative spectra of precursor ion scanning (PIS) of m/z 159 after normalization to the internal standard (I.S.) peak at m/z 472 (1–19:1 MG). Note that the amount of I.S. for brain tissue (20 pmol/mg tissue) is twice that in other tissues. (B) Product ion spectra of representative MG molecular species. The filled color bars (except purple bars) indicate the peak height of m/z 159 of the authentic sn-2 isomer (e.g., green bar for 2-OG, yellow bar for 2-LG, and blue bar for 2-PG and 2-18:0 MG). The m/z 159 peak height of an endogenous MG species close to or below the bar indicates its sn-2 identity while above the bar identifies the coexistence of sn-1 and sn-2 isomers whose composition can be quantified as described in the text. The filled purple bars indicate the peak heights of the two ketene fragment ions (m/z 287 and m/z 269), and of m/z 403 (from neutral loss of 77 amu) for authentic 2-AG. The peak heights of these fragment ions of an endogenous AG species close to or above the bars indicate its sn-2 identity while below the bars identify the coexistence of 1- and 2-AG which were not observed in the examined tissues. (C) Product ion spectra of molecular ion m/z 418 acquired at collision energies of 15 and 20 eV. The enlarged low mass range spectrum from heart extract demonstrates the fingerprint pattern for alkyl ether-linked species (i.e., 1-a16:0 MG).
In addition, the regiospecificity of each individual MG species was also remarkably diverse from tissue to tissue (Figure 5B). AG was nearly exclusively the sn-2 isomer in all examined tissues. LG and OG were also nearly exclusively the sn-2 isomer in brain and liver tissues but present in isomeric mixtures (i.e., coexisting sn-1 and sn-2 isomers) in heart and white adipose tissues. By contrast, saturated MG species (PG and 18:0 MG) were exclusively the sn-1 isomer in white adipose but present in isomeric mixtures in other examined tissues.
To quantify individual MG species including regioisomers by a MG internal standard (e.g., 1–19:1 MG) using PIS of m/z 159, correction factors are needed to adjust for the differential relative intensity of m/z 159 during fragmentation of the sn-1 or sn-2 isomer or their mixtures. To determine the correction factors, as an example, a series of LG solutions as used in Figures 3A and C were reanalyzed using PIS of m/z 159 in the presence of 1–19:1 MG at an equal molar concentration to LG (Figure S10A). The result demonstrated that the correction factors for LG were largely consistent with the linear relationship identified from their product ion spectra shown in Figure 3C (Figure S10B). Therefore, the correction factors for LG, PG or OG were then derived from the established linearity in Figures 3C, S3C or S3D, respectively, while the correction factors for other MG species containing acyl chains with 0–3 double bonds were derived from the linearity identified in Figure 3D (Table S1). PIS of m/z 159 of AG solutions in the presence of 1–19:1 MG demonstrated a much less altered response with respect to isomeric composition (Figure S10A; Table S1). Alternatively, stable isotope labeled 2-AG (e.g., d5-AG, or d8-AG) can be used as internal standard for AG quantitation without correction if endogenous 2-AG is of particular interest. The quantitation results of representative MG species including regioisomers in examined tissues are shown in Figure S11.
Furthermore, although naturally occurring 1-O-alkyl-sn-glycerols are precursors for potent biologically active compounds, they were found in nature mainly as the diacylglyceryl ether and phosphoether lipids.53 The strategy demonstrated in Figure 2B was utilized to examine the presence of alkyl ether-linked MG species in tissue samples. We observed the presence of 1-a16:0 MG (3.6 ± 0.3 pmol/mg tissue) in heart tissue, but not in other examined tissues (Figure 5C).
CONCLUSIONS
α-Hydroxy acyl migration has been a long-standing problem in lipidomics. Integrated extraction, enrichment and direct infusion-based mass spectrometric analysis were developed to identify and quantify low abundance monoglyceride regioisomers after a facile and robust diacetyl derivatization at low temperature. This strategy was employed to prevent acyl migration and to stabilize the regiospecificity of monoglycerides. Product ion analysis of these structurally preserved diacetyl monoglycerides identified distinct informative fragmentation patterns that could differentiate and quantify individual isomeric entities. Moreover, tandem mass spectrometric analysis of a characteristic diacetyl glycerol product ion (i.e., by precursor ion scanning of m/z 159) comprehensively profiled monoglyceride species in extracts of biologic tissues. Diverse profiles and differential regiospecificities of individual monoglyceride species were present from tissue to tissue. We note that isotope labeling-based approaches can be performed using stable isotope labeled derivatizing reagents (e.g., commercially available acetic anhydride-13C4, or -d6, or -1,1′-13C2, d6, or -13C4, d6) for relative quantitation or direct comparison of multiple samples. Furthermore, we observed efficient acetylation of other lipid classes, including hydroxyl-containing diglycerides, cholesterol, and N-acyl ethanolamine, and amine-containing ceramides. Further detailed investigations of their fragmentation patterns may facilitate identification and quantification of the hydroxyl- and amine-lipidome.
Supplementary Material
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grants RO1HL118639 and RO1DK100679 (R.W.G.) and P30DK056341 (Nutrition Obesity Research Center; K.Y.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b01862.
Experimental details and supporting figures and table (PDF).
Notes
The authors declare the following competing financial interest(s): R.W.G. has financial relationships with Lipo-Spectrum and Platomics.
References
- 1.Bilgin M, Bindila L, Graessler J, Shevchenko A. Anal Bioanal Chem. 2015;407:5125–5131. doi: 10.1007/s00216-015-8559-8. [DOI] [PubMed] [Google Scholar]
- 2.Keereetaweep J, Chapman KD. Neural Plast. 2016;2016:29. doi: 10.1155/2016/2426398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zoerner AA, Batkai S, Suchy MT, Gutzki FM, Engeli S, Jordan J, Tsikas D. J Chromatogr B: Anal Technol Biomed Life Sci. 2012;884:161–171. doi: 10.1016/j.jchromb.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 4.Wong A, Sagar DR, Ortori CA, Kendall DA, Chapman V, Barrett DA. J Lipid Res. 2014;55:1902–1913. doi: 10.1194/jlr.M048694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iannotti FA, Piscitelli F, Martella A, Mazzarella E, Allara M, Palmieri V, Parrella C, Capasso R, Di Marzo V. Prostaglandins, Leukotrienes Essent Fatty Acids. 2013;89:127–135. doi: 10.1016/j.plefa.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Viader A, Blankman JL, Zhong P, Liu X, Schlosburg JE, Joslyn CM, Liu QS, Tomarchio AJ, Lichtman AH, Selley DE, Sim-Selley LJ, Cravatt BF. Cell Rep. 2015;12:798–808. doi: 10.1016/j.celrep.2015.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams J, Wood J, Pandarinathan L, Karanian DA, Bahr BA, Vouros P, Makriyannis A. Anal Chem. 2007;79:5582–5593. doi: 10.1021/ac0624086. [DOI] [PubMed] [Google Scholar]
- 8.Bradshaw HB, Walker JM. Br J Pharmacol. 2005;144:459–465. doi: 10.1038/sj.bjp.0706093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood JT, Williams JS, Pandarinathan L, Courville A, Keplinger MR, Janero DR, Vouros P, Makriyannis A, Lammi-Keefe CJ. Clin Chem Lab Med. 2008;46:1289–1295. doi: 10.1515/CCLM.2008.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gouveia-Figueira S, Nording ML. Anal Chem. 2014;86:1186–1195. doi: 10.1021/ac403352e. [DOI] [PubMed] [Google Scholar]
- 11.Zoerner AA, Gutzki FM, Batkai S, May M, Rakers C, Engeli S, Jordan J, Tsikas D. Biochim Biophys Acta, Mol Cell Biol Lipids. 2011;1811:706–723. doi: 10.1016/j.bbalip.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Sergi M, Battista N, Montesano C, Curini R, Maccarrone M, Compagnone D. Anal Bioanal Chem. 2013;405:785–793. doi: 10.1007/s00216-012-6273-3. [DOI] [PubMed] [Google Scholar]
- 13.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardison S, Weintraub ST, Giuffrida A. Prostaglandins Other Lipid Mediators. 2006;81:106–112. doi: 10.1016/j.prostaglandins.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Destaillats F, Cruz-Hernandez C, Nagy K, Dionisi F. J Chromatogr A. 2010;1217:1543–1548. doi: 10.1016/j.chroma.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Schmid PC, Schwartz KD, Smith CN, Krebsbach RJ, Berdyshev EV, Schmid HH. Chem Phys Lipids. 2000;104:185–191. doi: 10.1016/s0009-3084(99)00124-3. [DOI] [PubMed] [Google Scholar]
- 17.Lee T, Hastilow C, Smith K. J Assoc Off Anal Chem. 1988;71:785–788. [PubMed] [Google Scholar]
- 18.Khaled MY, McNair HM, Hanson DJ. J Chromatogr Sci. 1993;31:375–379. [Google Scholar]
- 19.Thomas A, Hopfgartner G, Giroud C, Staub C. Rapid Commun Mass Spectrom. 2009;23:629–638. doi: 10.1002/rcm.3918. [DOI] [PubMed] [Google Scholar]
- 20.Richardson D, Ortori CA, Chapman V, Kendall DA, Barrett DA. Anal Biochem. 2007;360:216–226. doi: 10.1016/j.ab.2006.10.039. [DOI] [PubMed] [Google Scholar]
- 21.Vogeser M, Schelling G. Clin Chem Lab Med. 2007;45:1023–1025. doi: 10.1515/CCLM.2007.197. [DOI] [PubMed] [Google Scholar]
- 22.Fanelli F, Di Lallo VD, Belluomo I, De Iasio R, Baccini M, Casadio E, Gasparini DI, Colavita M, Gambineri A, Grossi G, Vicennati V, Pasquali R, Pagotto U. J Lipid Res. 2012;53:481–493. doi: 10.1194/jlr.M021378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Hayakawa J, Yang K, Han X. Anal Chem. 2014;86:2146–2155. doi: 10.1021/ac403798q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang K, Jenkins CM, Dilthey B, Gross RW. Anal Bioanal Chem. 2015;407:5199–5210. doi: 10.1007/s00216-015-8640-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogasawara D, Deng H, Viader A, Baggelaar MP, Breman A, den Dulk H, van den Nieuwendijk AM, Soethoudt M, van der Wel T, Zhou J, Overkleeft HS, Sanchez-Alavez M, Mo S, Nguyen W, Conti B, Liu X, Chen Y, Liu QS, Cravatt BF, van der Stelt M. Proc Natl Acad Sci U S A. 2016;113:26–33. doi: 10.1073/pnas.1522364112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tortoriello G, Rhodes BP, Takacs SM, Stuart JM, Basnet A, Raboune S, Widlanski TS, Doherty P, Harkany T, Bradshaw HB. PLoS One. 2013;8:e67865. doi: 10.1371/journal.pone.0067865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seyer A, Cantiello M, Bertrand-Michel J, Roques V, Nauze M, Bezirard V, Collet X, Touboul D, Brunelle A, Comera C. PLoS One. 2013;8:3. doi: 10.1371/journal.pone.0058224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Destaillats F, Angers P, Wolff RL, Arul J. Lipids. 2001;36:1247–1254. doi: 10.1007/s11745-001-0839-9. [DOI] [PubMed] [Google Scholar]
- 30.Petrosino T, Riccieri R, Blasi F, Brutti M, D’Arco G, Bosi A, Maurelli S, Cossignani L, Simonetti MS, Damiani P. J AOAC Int. 2007;90:1647–1654. [PubMed] [Google Scholar]
- 31.Leiker TJ, Barkley RM, Murphy RC. Int J Mass Spectrom. 2011;305:103–109. doi: 10.1016/j.ijms.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang X, Cheng H, Yang K, Gross RW, Han X. Anal Biochem. 2007;371:135–145. doi: 10.1016/j.ab.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han X, Yang K, Gross RW. Mass Spectrom Rev. 2012;31:134–178. doi: 10.1002/mas.20342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang K, Cheng H, Gross RW, Han X. Anal Chem. 2009;81:4356–4368. doi: 10.1021/ac900241u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kingsley PJ, Marnett LJ. Anal Biochem. 2003;314:8–15. doi: 10.1016/s0003-2697(02)00643-7. [DOI] [PubMed] [Google Scholar]
- 36.Balvers MG, Verhoeckx KC, Witkamp RF. J Chromatogr B: Anal Technol Biomed Life Sci. 2009;877:1583–1590. doi: 10.1016/j.jchromb.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Han X, Gross RW. Anal Biochem. 2001;295:88–100. doi: 10.1006/abio.2001.5178. [DOI] [PubMed] [Google Scholar]
- 38.Mancuso DJ, Sims HF, Yang K, Kiebish MA, Su X, Jenkins CM, Guan S, Moon SH, Pietka T, Nassir F, Schappe T, Moore K, Han X, Abumrad NA, Gross RW. J Biol Chem. 2010;285:36495–36510. doi: 10.1074/jbc.M110.115766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schweitzer GG, Chen Z, Gan C, McCommis KS, Soufi N, Chrast R, Mitra MS, Yang K, Gross RW, Finck BN. J Lipid Res. 2015;56:848–858. doi: 10.1194/jlr.M055962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marshall DL, Pham HT, Bhujel M, Chin JS, Yew JY, Mori K, Mitchell TW, Blanksby SJ. Anal Chem. 2016;88:2685–2692. doi: 10.1021/acs.analchem.5b04001. [DOI] [PubMed] [Google Scholar]
- 41.Han RH, Wang M, Fang X, Han X. J Lipid Res. 2013;54:1023–1032. doi: 10.1194/jlr.M033837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Evans JJ. Rapid Commun Mass Spectrom. 2005;19:2528–2538. doi: 10.1002/rcm.2087. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Collins EJ, Evans JJ. Rapid Commun Mass Spectrom. 2006;20:171–177. doi: 10.1002/rcm.2270. [DOI] [PubMed] [Google Scholar]
- 44.McAnoy AM, Wu CC, Murphy RC. J Am Soc Mass Spectrom. 2005;16:1498–1509. doi: 10.1016/j.jasms.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 45.Byrdwell WC, Neff WE. Rapid Commun Mass Spectrom. 2002;16:300–319. doi: 10.1002/rcm.581. [DOI] [PubMed] [Google Scholar]
- 46.Li M, Baughman E, Roth MR, Han X, Welti R, Wang X. Plant J. 2014;77:160–172. doi: 10.1111/tpj.12365. [DOI] [PubMed] [Google Scholar]
- 47.Gakwaya R, Li X, Wong YL, Chivukula S, Collins EJ, Evans JJ. Rapid Commun Mass Spectrom. 2007;21:3262–3268. doi: 10.1002/rcm.3208. [DOI] [PubMed] [Google Scholar]
- 48.Li M, Butka E, Wang X. Sci Rep. 2014;4:1–11. doi: 10.1038/srep06581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang MY, Gao Y, Btesh J, Kagan N, Kerns E, Samad TA, Chanda PK. J Mass Spectrom. 2010;45:167–177. doi: 10.1002/jms.1701. [DOI] [PubMed] [Google Scholar]
- 50.Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. J Lipid Res. 2008;49:1137–1146. doi: 10.1194/jlr.D700041-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wood PL, Medicherla S, Sheikh N, Terry B, Phillipps A, Kaye JA, Quinn JF, Woltjer RL. J Alzheimer’s Dis. 2015;48:537–546. doi: 10.3233/JAD-150336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halvarson H, Qvist O. J Am Oil Chem Soc. 1974;51:162–165. [Google Scholar]
- 53.Magnusson CD, Haraldsson GG. Chem Phys Lipids. 2011;164:315–340. doi: 10.1016/j.chemphyslip.2011.04.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.