

Abstract
Therapies capable of decelerating, or perhaps even halting, neurodegeneration in Parkinson’s disease (PD) remain elusive. Clinical trials of PD gene therapy testing the delivery of neurotrophic factors, such as the glial cell-line derived neurotrophic factor (GDNF), have been largely ineffective due to poor vector distribution throughout the diseased regions in the brain. In addition, current delivery strategies involve invasive procedures that obviate the inclusion of early stage patients who are most likely to benefit from GDNF-based gene therapy. Here, we introduce a two-pronged treatment strategy, composed of MR image-guided focused ultrasound (FUS) and brain-penetrating nanoparticles (BPN), that provides widespread but targeted GDNF transgene expression in the brain following systemic administration. MR image-guided FUS allows circulating gene vectors to partition into the brain tissue by noninvasive and transient opening of the blood–brain barrier (BBB) within the areas where FUS is applied. Once beyond the BBB, BPN provide widespread and uniform GDNF expression throughout the targeted brain tissue. After only a single treatment, our strategy led to therapeutically relevant levels of GDNF protein content in the FUS-targeted regions in the striatum of the 6-OHDA-induced rat model of PD, which lasted at least up to 10 weeks. Importantly, our strategy restored both dopamine levels and dopaminergic neuron density and reversed behavioral indicators of PD-associated motor dysfunction with no evidence of local or systemic toxicity. Our combinatorial approach overcomes limitations of current delivery strategies, thereby potentially providing a novel means to treat PD.
Keywords: Focused ultrasound, nonviral gene delivery, Parkinson’s disease, blood–brain barrier
Graphical Abstract
Parkinson’s disease (PD) is a largely idiopathic neurodegenerative disorder affecting approximately 2% of the population over 65.1 It is estimated that PD costs more than 14 billion dollars each year in the US alone,2 and its incidence is expected to double as early as 2040.3 One of the primary hallmarks of PD is the degeneration of dopaminergic neurons with cell bodies in the substantia nigra pars compacta (SNpc) and axon projections extending into the striatum. The resulting dopamine deficiency leads to progressive and debilitating motor control deficits including bradykinesia, rigidity, and resting tremor. While pharmacological dopamine replacement or surgical therapies like deep brain stimulation can ameliorate symptoms at early stages in PD, they are not neuroprotective, and the continued neuronal degeneration ultimately leads to a recurrence of symptoms.4,5 Furthermore, late-stage patients often develop motor symptoms that are refractory to dopamine replacement therapies or complications stemming from long-term dopamine-replacing drug use.6 Therapies that can slow or stop the neurodegenerative process have remained elusive.7
Gene therapy approaches aimed at slowing or reversing neurodegeneration in PD have been developed and tested in both preclinical and clinical settings for many years. Neurotrophic factors, like the glial cell-line derived neurotrophic factor (GDNF), are attractive candidates for gene therapy due to their ability to protect neurons from continued degeneration, induce neuronal regeneration and sprouting, and enhance dopamine generation from the remaining neuronal population.8,9 Numerous gene therapy clinical trials for PD have been completed using genes that encode for neurotrophic factors like GDNF or its close structural and functional relative, neurturin (NTRN). While these clinical trials showed safety, therapeutic outcomes were disappointing. Going forward, it has been hypothesized that therapeutic outcomes may be improved by (a) enhancing delivery efficiency, transfection volume, and reproducibility of delivery within the target structures,10 (b) treating earlier stage (or even prodromal) patients prior to the onset of extensive irreversible neurodegeneration,11,12 and (c) further adjusting dosing parameters to ensure appropriate levels of neurotrophic factor expression throughout the target volume. While advances in direct injection strategies, including convection enhanced delivery (CED), may improve outcomes, concerns over their invasive and risky nature have obviated the inclusion of early stage PD patients in clinical trials. More effective and minimally invasive approaches that can be used for patients regardless of their disease stages are required.
To date, clinical gene therapy studies have relied upon direct administration methods that are invasive and may yield poor transgene distribution. Systemic administration has not been considered because the blood–brain barrier (BBB) prevents nearly 100% of molecules larger than ~400 Da in size from entering the brain. Indeed, the BBB remains one of the most significant impediments to therapeutic delivery to the brain following systemic administration.13 To achieve efficacy, both viral vectors and nanoparticles with BBB-targeting ligands often require very high systemic doses, which can lead to peripheral adverse side effects.14 Other strategies for circumventing the BBB, such as intranasal administration or intra-arterial infusion of the osmotic agent mannitol, have been proposed and are being tested. However, they also have weaknesses that may hinder translation to the clinic, including invasiveness, inconsistency, and/or poor targeting and tissue distribution.15,16
MR image-guided focused ultrasound (FUS) is currently the only modality capable of noninvasively opening the BBB for spatially targeted therapeutic delivery into the brain.17 Through the activation of ultrasound contrast agent microbubbles (MBs) in stable cavitation, FUS permits the targeted delivery of nanoparticles as large as 100 nm across the BBB.17,18 Activated MBs exert mechanical forces on the vessel wall, temporarily disrupting tight junctional complexes19 and inducing active transport processes.20 Barrier function is typically fully restored within 4–6 h,21,22 and safety has been demonstrated in several large animal models.23,24 Advances in transducer technology now permit submillimeter precision, and with guidance from magnetic resonance (MR) imaging, it is possible to apply FUS across the human skull in an extremely localized manner, limiting the potential for unwanted side effects.25,26 Furthermore, MR imaging allows semireal time intraoperative treatment feedback and postoperative confirmation of success. Indeed, MR image-guided FUS is now FDA approved for use in humans with Essential Tremor,26 and clinical trials for other CNS disorders including tremor-dominant PD are underway.27
Once across the BBB, vectors must traverse a dense, nanoporous, and negatively charged extracellular matrix (ECM) that impedes the dispersion of traditional nanoparticles28,29 and viruses30 through both adhesive interactions and steric obstruction. Importantly, it has recently been shown that sub-114 nm nanoparticles densely coated with hydrophilic and neutrally charged polyethylene glycol (PEG) are able to overcome this barrier and diffuse rapidly within the brain parenchyma.28,31–33 These “brain-penetrating nanoparticles” (BPN) can be complexed into nanosized and colloidally stable gene vectors.34,35 We have previously demonstrated that FUS can target BPN delivery to rat brain,18 which can provide robust and long-term reporter gene expression in the FUS-targeted region when the BPN is loaded with plasmid DNA.36 In the current study, we used a BPN formulation to deliver a GDNF gene-bearing plasmid (GDNF-BPN) to the striatum of PD rats whose BBB was transiently opened in a targeted manner with MR image-guided FUS. We demonstrate a clinically relevant strategy to restore dopaminergic neuronal function without the need for invasive surgical procedures.
Results
Characterization of GDNF-BPN
The GDNF plasmid was previously engineered for long-term expression in the brain.37 By using a long-lasting polyubiquitin C promoter, an optimized Kozak consensus sequence to improve translation initiation, and a CpG depleted plasmid backbone to reduce DNA silencing,38 this gene cassette was found to have peak expression after approximately 7 days and lasting at least 6 months after direct injection into mouse striatum.37 We condensed GDNF plasmids or negative control reporter gene bearing plasmids (i.e., pBAL) with a blend of polyethylenimine (PEI) and PEI–PEG conjugates to yield GDNF-BPN or pBAL-BPN, respectively. We have shown that the large amount of PEG in these systems greatly reduces, if not eliminates, potential in vivo toxicity induced by the highly cationic nature of PEI.31,36,39 The GDNF-BPN and pBAL-BPN formulations both possessed a very small hydrodynamic diameter (50 ± 3 nm), a near neutral surface charge (ζ-potential of 1.5 ± 0.2 mV), and a tight size distribution (polydispersity index of 0.2); we have shown that nanoparticles that possess this combination of physicochemical properties rapidly penetrate within brain tissue31 and provide uniform and efficient reporter gene expression in the rat striatum after delivery with FUS.36 GDNF-BPN and pBAL-BPN were colloidally stable without any significant change in size and morphology for at least 6 h when incubated in artificial cerebrospinal fluid (aCSF) at 37 °C (Figure S1).
MR Imaging Assessment of Blood–Brain Barrier Opening
FUS treatments were guided by peri-operative MRI (Figure 1). Pre-FUS T1-weighted and T2-weighted (Figure 1A) MR images were acquired and used to plan 4 equally spaced treatment sites within the ipsilateral (6-OHDA treated) striatum. We targeted only the striatum because previous studies suggest that this approach leads to a greater functional recovery than treatment of both the striatum and SNpc.40 FUS parameters (1.15 MHz, 0.6 MPa nonderated peak-negative pressure), MB dose (3 × 105 MB/g), and GDNF-BPN dose (0.5 μg/g body weight) were chosen based on previous BPN delivery and safety studies from our group.18,36 To confirm BBB disruption after FUS application, Gd-DPTA was injected intravenously, and contrast-enhanced T1-weighted images were acquired (Figure 1C) and compared to pre-FUS T1-weighted images. Enhancement in signal intensity in the FUS-treated striatum was found in all (n = 32) animals treated with FUS. In addition, T2*-weighted imaging, which detects extensive red blood cell extravasation, was performed pre- and post-FUS (Figure 1B,D). Red blood cell extravasation, which would have been indicated by hypointense regions on T2* MRI, was never detected at the FUS-treated sites (n = 128) in this study.
Figure 1.

MR imaging for guidance, confirmation, and safety evaluation of FUS treatments. (A) The treatment site (i.e., 6-OHDA lesioned striatum) was targeted with T2 pre-FUS images. (C) BBB opening in the striatum was confirmed with post-FUS contrast-enhanced T1 imaging. (B, D) Treatment safety was assessed by comparing pre- and post-FUS T2* images. Hypointensities in T2* images, indicating red-blood cell accumulation, were never observed in FUS-treated animals (n = 32).
Localized Delivery of GDNF-BPN with FUS Elicits Robust GDNF Protein Expression in the Striatum
Two weeks after unilateral partial lesioning with 6-OHDA, GDNF-BPN were injected intravenously and delivered to the 6-OHDA lesioned striatum with MRI-guided FUS. Delivery of GDNF-BPN with FUS to the striatum led to an 11-fold increase in ipsilateral striatal GDNF protein when compared to 6-OHDA only control animals (Figure 2). GDNF protein levels remained elevated through week 12, reaching 12.3 ± 3.5 ng/mg. The application of 6-OHDA alone, in conjunction with GDNF-BPN without FUS, or in conjunction with FUS-mediated delivery of pBAL-BPN, had no significant effect on GDNF protein levels. GDNF levels were not elevated above baseline levels in the contralateral striatum or the SNpc (Figure S2A) of either hemisphere in any group, indicating that FUS application yielded target-site specific GDNF expression. Importantly, GDNF expression in animals receiving GDNF-BPN was not significantly elevated in off-target major organs (Figure S2B) when compared to 6-OHDA only animals.
Figure 2.

FUS-mediated delivery of GDNF-BPN to the striatum of PD rats leads to a significant increase in GDNF protein levels in the striatum. Bar graphs show GDNF protein levels in the ipsilateral (black) and contralateral (gray) striatum. n = 5 (6-OHDA + FUS + GDNF-BPN), n = 4 or n = 3 (6-OHDA + GDNF-BPN) in each group at each time point. *Significantly different from all groups at the same time point (p < 0.05).
Dopamine Levels are Elevated in 6-OHDA Lesioned Rats that Subsequently Received FUS-Targeted GDNF-BPN Delivery
Two weeks after the administration of 6-OHDA into the striatum of control animals, ipsilateral striatal dopamine (DA) levels dropped to ~10% of those measured in the contralateral striatum (Figure 3). Furthermore, DA levels remained low at weeks 6 and 12 in the 6-OHDA only control group. In agreement with the locomotor function data, animals in both the 6-OHDA + GDNF-BPN (no FUS) and 6-OHDA + FUS + pBAL-BPN control groups showed no improvement in ipsilateral DA levels compared to animals who received no treatment after 6-OHDA administration. Importantly, 6-OHDA lesioned animals treated with GDNF-BPN and FUS exhibited 2.8-fold and 2.2-fold increases in DA levels compared to 6-OHDA only control animals at weeks 6 and 12, respectively. Ipsilateral DA levels in these treated animals were not significantly different from contralateral striatal DA levels at week 12, suggesting that the treatment regimen normalized DA levels. In addition, in the ipsilateral striatum, the DA metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were elevated 3.8-fold and 4.3-fold, respectively, in 6-OHDA + FUS + GDNF-BPN animals at week 12 compared to 6-OHDA rats, indicative of DA turnover (Table S1).
Figure 3.

FUS-mediated delivery of GDNF-BPN improves dopamine levels in the ipsilateral hemisphere. Bar graph of dopamine levels in the striatum in the ipsilateral (black) and contralateral (gray) hemisphere at weeks 2, 6, or 12 after 6-OHDA administration. n = 5 (6-OHDA + FUS + GDNF-BPN), n = 4 (6-OHDA only), or n = 3 in each group at each time point. *p < 0.05 vs all groups at the same time point and same hemisphere. **p < 0.05. +p < 0.05 vs contralateral at same time point.
Dopaminergic Neuron Density Is Restored in 6-OHDA Lesioned Rats that Subsequently Received FUS-Targeted GDNF-BPN Delivery
Both dopaminergic (TH+) neuron cell number in the SNpc and nigrostriatal neuronal projection density in the striatum were assessed through quantitative morphology at weeks 2, 6, and 12 after 6-OHDA administration (Figure 4). In control animals, 6-OHDA administration led to a 74% reduction in dopaminergic cell number in the SNpc and an 87% reduction in dopaminergic staining density in the striatum at 2 weeks. Control animals receiving 6-OHDA only, 6-OHDA + GDNF-BPN, or 6-OHDA + FUS + pBAL-BPN showed no improvement in dopaminergic neuron number in the SNpc nor in dopaminergic staining density in the striatum. In contrast, 6-OHDA rats treated with GDNF-BPN and FUS showed, respectively, 3.2-fold and 5.0-fold increases in dopaminergic cell number in the SNpc and dopaminergic density in the striatum at week 12. Qualitatively, 6-OHDA + FUS + GDNF-BPN treatment also appeared to enhance TH+ fiber density in the substantia nigra pars reticulata (SNpr), which lies ventral to the SNpc, when compared to 6-OHDA controls.
Figure 4.

Delivery of GDNF-BPN to the striatum with FUS increases dopaminergic neuron density in the striatum and SNpc. Representative images of TH-stained coronal sections through the striatum (A) or SNpc (B) at either week 6 (top row) or week 12 (bottom row) after 6-OHDA administration. Bar graphs show inverse TH staining intensity (C) or TH+ cell number (D) normalized to contralateral. n = 5 (6-OHDA + FUS + GDNF-BPN) or n = 4 in each group and time point. *p, 0.05 vs all groups at same time point.
Locomotor Function Is Markedly Improved in 6-OHDA Lesioned Rats that Subsequently Received FUS-Targeted GDNF-BPN Delivery
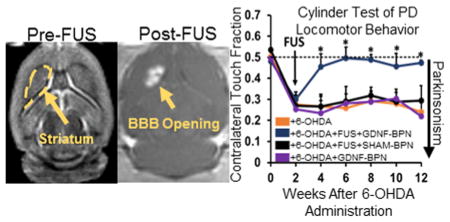
Unilateral striatal 6-OHDA lesioning without treatment led to extensive locomotor deficits, including apomorphine-induced rotational bias and forepaw use bias after 2 weeks (Figure 5). Control animals that received no treatment after 6-OHDA administration, intravenous GDNF-BPN only, or pBAL-BPN and FUS maintained both drug induced and voluntary locomotor bias throughout the entire 12-week course of the study, indicating extensive neurodegeneration (Figure 5). In contrast, 6-OHDA lesioned animals that received the FUS + GDNF-BPN treatment demonstrated markedly improved locomotor function. At week 2, treated animals exhibited a 54% reduction in apomorphine-induced rotational bias when compared to 6-OHDA only control animals. At week 4, forepaw use bias was restored to normal levels. In the 6-OHDA + FUS + GDNF-BPN treatment group, these improvements in locomotor function, as assessed by both tests, were fully retained through the 12 week duration of the experiment.
Figure 5.

Delivery of GDNF-BPN with FUS restores locomotor function in PD rats. (A) Line graph of average contralateral rotations per minute after apomorphine administration. (B) Line graph of contralateral touch fraction in the forepaw use bias test. n > 14 in each group at weeks 0 through 6; n > 7 in each group at weeks 8 through 12. *Significantly different than all other groups at the same time point (p < 0.01).
Safety Analysis
Animals in the 6-OHDA + FUS + GDNF-BPN treatment group did not show changes in weight gain compared to controls (Figure S3A). Brain tissue sections were stained with H&E and scored for signs of toxicity by a board certified neuropathologist (JWM) blinded to the treatment conditions. No evidence of neural damage, ischemic injury, or gliosis was found beyond that caused by the 6-OHDA probe injection tract in any slide throughout the course of the 12-week study (Figure S3B).
Discussion
Clinical strategies capable of slowing or reversing neurodegeneration in Parkinson’s disease remain elusive. It has been hypothesized that clinical gene therapy trials using viral vectors to deliver neurotrophic factors, including GDNF, have failed to meet primary efficacy outcomes due to both incomplete delivery to the target structures and invasive surgical strategies that obviate the inclusion of early stage PD patients.10,11 Using a clinically relevant treatment paradigm, wherein treatment was applied when the nigrostriatal motor neuron pathway had experienced an ~80% reduction in dopaminergic neuron density, we demonstrated that the MR image-guided delivery of brain-penetrating GDNF gene nanovectors with FUS is capable of generating marked improvements in locomotor function, nigrostriatal dopaminergic neuron density, and DA levels, with some metrics showing complete restoration. Given the minimally invasive nature of this gene delivery strategy, we postulate that it may eventually be translated to the clinic for treatment of early stage or prodromal PD patients.
Gene Delivery to the CNS with FUS
It is now well-established that MR image-guided FUS may be used to noninvasively and temporarily open the BBB, allowing localized delivery of systemically circulating agents as large as 100 nm in size to the CNS.17,18 We have previously demonstrated the ability of FUS to target the delivery of reporter gene-bearing BPN to the brains of healthy rats, leading to efficient, targeted, and localized transgene expression without signs of toxicity or astrocyte activation.36 Importantly, we found that ~30% of cells, including an equal ratio of neurons and astrocytes, were transfected at a similar dose as the current study.36 We note that FUS has recently been shown to facilitate the delivery of systemically administered GDNF-loaded liposomes ~200 nm in diameter to the brain, leading to neuroprotection in a mouse model of PD.41 While a modest but significant increase in GDNF protein content was observed (<2-fold increase over control animals), a total of 12 FUS + GDNF plasmid treatments, at doses of 1 μg/g, were required to achieve this level of GDNF expression. Moreover, transgene expression was detected in nontargeted brain and some peripheral organs, as determined by in vivo imaging. In contrast, our GDNF-BPN, possessing smaller diameters of ~50 nm, elicited an 11-fold increase in GDNF protein content that was achieved with a single treatment only (dose = 0.5 μg/g) and precisely localized to the FUS-treated striatum. Thus, we postulate that GDNF plasmid delivered via this unique combination of FUS-targeted BBB opening and BPN-mediated brain distribution offers a single treatment approach with improved spatial specificity, greater GDNF expression, and enhanced functional therapeutic outcomes.
FUS has also been shown, in studies by other investigators, to be capable of delivering naked plasmids42,43 and adeno-associated viruses (~20 nm size)44,45 to the CNS after systemic administration. The appeal of viral-based gene vectors is reduced by neutralizing immune responses to the virus, which are often observed following administration or prior exposure, that reduce efficiency of transgene delivery.46 Other limitations of viral vector approaches include low packaging capacity, high production costs, and/or safety risks.47 The nonviral BPN vectors used in the current study are versatile, highly tunable, and devoid of the intrinsic limitations associated with viral vectors.
Comparison to Previous Preclinical Studies with GDNF
Our treatment strategy yielded long-term and spatially localized GDNF expression at levels that did not elicit local toxicity. GDNF protein content remained elevated (~10-fold over controls) in the FUS + GDNF-BPN treated striatum for at least 10 weeks post-treatment, suggesting long-term GDNF expression at functional levels at least on the order seen in other gene delivery studies with the same plasmid.48 Elevated GDNF levels were not found in the ipsilateral SNpc, the contralateral striatum, or the contralateral SNpc, highlighting the ability of FUS to spatially localize the delivery of GDNF-BPN. While the mechanisms of restoration of dopaminergic cell number in the SNpc due to enhanced GDNF expression are not yet clear, it has been postulated49 that this could be elicited by GDNF-induced signaling at axon terminals rather than retrograde GDNF transport. This is consistent with our finding that dopaminergic neurons were restored in the SNpc despite GDNF levels remaining at normal levels. There appears to be a fairly large therapeutic window for GDNF in the brain, as previous studies have shown that, while small (3–4 fold) increases in GDNF protein levels in the striatum were sufficient to protect most neurons from 6-OHDA lesioning, much higher levels (~100–300-fold increases) of GDNF expression can lead to adverse side effects, including lower tissue DA and TH levels after 6–12 weeks.50,51 However, appropriate targeting within the brain is critical, as off-target GDNF overexpression in the hypothalamus52,53 or infusion into the ventricles54 can lead to weight loss. Studies using systemically administered recombinant GDNF protein have not reported adverse events associated with off-target GDNF uptake,55 which is consistent with the observation that animal weight gain was normal in our study.
Previous studies have used direct administration of viral vectors to overexpress GDNF in both toxin-based and α-synuclein-based preclinical models of PD. While studies in toxin-based models have demonstrated restoration of motor function and/or dopaminergic density after GDNF gene therapy, these studies have utilized less severe models of PD, wherein animals only exhibited ~30–50% dopaminergic neuron loss at the time of treatment.49,56,57 In contrast, animals in the current study were treated when ~70–80% of dopaminergic neurons had been lost, thereby replicating the level of neurodegeneration seen in early stage human PD patients.58,59 While the 6-OHDA model recapitulates many aspects of human PD, going forward, it may be useful to determine whether our approach is effcacious in other PD models, such as an α-synuclein overexpression model. Nonetheless, it is important to note that α-synuclein-based PD models have previously demonstrated a lack of efficacy of GDNF gene therapies,60 presumably due to GDNF receptor (Ret) downregulation;61 therefore, alternative models may be more appropriate for this application.
Relevance to Clinical Trials
Despite the success of gene therapies for neurotrophic factors in preclinical animal studies,49,50,56,62 clinical PD trials have generally failed to meet primary outcomes. While early gene therapy trials with AAV2-NTRN administered intracranially by convection-enhanced delivery did produce preliminary evidence for clinical improvement, results were inconsistent.63,64 Following this clinical trial, post-mortem results demonstrated that only ~20% of the targeted putamen expressed detectable levels of NTRN,65 suggesting that increasing putaminal coverage could improve outcomes. A follow-up phase 2b trial used an ~3-fold higher dose of AAV2-NTRN and increased the CED-injected volume ~4-fold.66 Unfortunately, putaminal coverage was increased to only ~30%, and these trials again failed to meet primary outcomes. Ongoing clinical trials using AAV2-GDNF (http://www.clinicaltrials.gov, NCT01621581) are aimed at improving volume of distribution using highly invasive intracranial infusion.
Additional data argue for treatments at earlier stage or even prodromal PD. PD symptoms typically appear after loss of ~70–80% of dopaminergic neurons,58,59 and degeneration is nearly complete within 10 years after diagnosis, rendering the disease irreversible.67 Consistent with this paradigm, a recent posthoc analysis of earlier AAV2-NTRN clinical trials demonstrated that patients treated less than 5 years after PD diagnosis had a significantly (~2.5-fold) greater improvement in unified PD rating scale (UPDRS) scores when compared to patients treated more than 10 years after diagnosis. Importantly, PD patients treated with AAV2-NTRN more than 10 years after diagnosis (constituting more than half of the treated patients) exhibited no improvement when compared to sham controls.66 The FDA has so far been unwilling to treat PD patients at earlier stages owing to the invasive nature and risks associated with intracranial injection/infusion strategies.11
In contrast, FUS has the advantage of being a noninvasive treatment strategy that can even be performed on awake patients, obviating risks associated with surgical procedures and general anesthetics.68 Moreover, the high capillary density in the brain provides a large surface area for delivery from the systemic circulation after FUS application. Coupling of FUS with highly diffusive BPN that can access all of the cells between opened capillaries allows exceptionally homogeneous and efficient DNA delivery over a large volume within the FUS-targeted region. FUS mediated delivery of GDNF-BPN, therefore, represents an enticing alternative capable of overcoming many of the hurdles associated with conventional treatment strategies.
Materials and Methods
Animals
Female Sprague–Dawley rats were purchased from Envigo, housed on a 12/12 h light/dark cycle, and given food ad libitum. Animal experiments were approved by the Animal Care and Use Committee at the University of Virginia and conformed to the National Institutes of Health guidelines for the use of animals in research.
6-OHDA Lesions
Partial striatal lesioning with 6-OHDA was performed as previously described.49,56 Female Sprague–Dawley rats (~160–180 g) were anesthetized and maintained with 2–2.5% isofluorane in oxygen. The top of the animals’ heads were shaved and depilated before being secured prone in a stereotaxic frame (Stoelting, Wood Dale, IL). Buprenorphine (0.03 mg/kg, Reckitt Benckiser Healthcare, Hull, UK) was administered to achieve local anesthesia. A midline scalp incision was made, and the skull was exposed. A total of 12 μg of 6-OHDA (Sigma-Aldrich, St. Louis, MO) dissolved in 2 μL of 0.02% ascorbic acid/0.9% saline was injected in two sites in the right striatum at the coordinates: AP: +0.5, ML: +2.1, DV: −5 and AP: −0.5, ML: +3.8, DV: −5 mm at 0.5 μL/min.
Plasmid Design
The GDNF plasmid (~4 kB) was constructed as previously described37,48 using standard molecular biology techniques and kindly donated to us by Dr. Mark Cooper (Copernicus Therapeutics) and Dr. David Yurek for use in this study. In brief, human GDNF was placed downstream of the ubiquitously active and long-lasting polyubiquitin C promoter and first exon and intron sequences. GDNF was codon-optimized, and prokaryotic elements were CpG depleted to reduce potential methylation sites known to increase likelihood of gene silencing. The pBAL plasmid was produced by Copernicus Therapeutics Inc. (Cleveland, OH) and is described elsewhere.36 Briefly, this plasmid contains the luciferase reporter gene driven by the ubiquitously active β-actin promoter.
DNA-BPN Fabrication and Characterization
DNA-BPN were fabricated and characterized as previously described.31,36 Briefly, PEG5k-PEI copolymer was synthesized by conjugating molar excess of 5 kDa methoxy-PEG-N-hydrosuccinimide (mPEG-NHS, 5 kDa, Sigma-Aldrich, St. Louis, MO) to primary amine groups of 25 kDa branched PEI (Sigma-Aldrich, St. Louis, MO). The resulting PEI–PEG conjugate possessed a high degree of PEG conjugation (average of 41 PEG chains per PEI molecule). The final product was extensively dialyzed against ultrapure water, and the conjugation was confirmed by nuclear magnetic resonance. DNA-BPN were formulated by dropwise addition of 10 volumes of plasmid DNA to 1 volume of polymer mixture solution of PEI and PEG5k-PEI. DNA-BPN were purified with 3 volumes of ultrapure water to eliminate free polymers and concentrated to 1 mg/mL using Amicon Ultra Centrifugal Filters (100 000 MWCO; Millipore Corp., Billerica, MA). The DNA concentration of DNA-BPN was confirmed using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA). The hydrodynamic diameter, ζ-potential, and polydispersity index of DNA-BPN were measured using Zetasizer Nano ZS (Malvern Instruments, Southborough, MA). The colloidal stability of DNA-BPN was observed with transmission electron microscopy (TEM, Hitachi H7600, Japan) after incubating DNA-BPN in aCSF (Harvard Apparatus, Holliston, MA) at 37 °C for 6 h.
Microbubble Fabrication and Characterization
MBs used in the current study have a formulation similar to Optison (GE Healthcare, Little Chalfont, Buckinghamshire, UK). A 1% solution of serum albumin in saline was sonicated (20 kHz, 30 s) with an ultrasound disintegrator (XL2020; Misonix, Farmingdale, NY) with an extended 1/2-in. titanium probe. The flask containing the solution had its headspace filled with octofluoropropane gas prior to sonication. MBs were sized and counted using a Coulter counter (Multisizer 3, Beckman Coulter, Fullterton, CA).
FUS-Mediated DNA-BPN Delivery
Female Sprague–Dawley rats were anesthetized with an intraperitoneal injection of ketamine (40 mg/kg, Zoetis, Fort Dodge, IA) and dexdomitor (0.2 mg/kg, Pfizer, New York, NY) in sterilized 0.9% saline. A tail vein catheter was inserted to allow intravenous injections of DNA-BPN and microbubbles. In a previous study by our group,18 BPN with a similar size and surface chemistry were found to be circulating 24 h after i.v. administration into rats. Rat heads were shaved and depilated before being secured supine in a degassed water bath coupled to the FUS system (RK-100, FUS Instruments, Toronto), and the entire system was placed in the 3T MR scanner (Magnetom Trio, Siemens Medical Solutions, Malvern, PA). A home-built 2-in. cylindrical transmit-receive RF coil was placed around the rat’s head to maximize imaging SNR. Baseline T2-weighted and T2*-weighted images were acquired using turbo spin–echo and spoiled gradient-echo pulse sequences, respectively. T2-weighted imaging parameters included: TR/TE = 3600/46 ms, flip angle = 90°/150°, readout bandwidth = 219 Hz/pixel, 9 slices, thickness = 1 mm, field of view = 50 mm, matrix = 192 × 192, turbo factor = 18, 100% phase oversampling, 4 averages, total time = 5:22. T2*-weighted imaging parameters included: TR/TE = 101/9 ms, fiip angle = 50°, readout bandwidth = 320 Hz/pixel, 7 slices, thickness = 2 mm, field of view = 64 mm, matrix = 192 × 192, 100% phase oversampling, 4 averages, total time = 2:37. T2-weighted images were used to select 4 evenly spaced target locations within the right striatum and 5 mm from the top of the skull.
Rats received a coinjection of GDNF-BPN (100 μg/200g body weight) and MBs (3 × 105 MBs/g body weight) followed by 0.3 mL of 2% heparinized saline to clear the catheter. Sonication began immediately after clearance of the catheter. Sonications were performed at 0.6 MPa (not accounting for skull/tissue attenuation) using a 1.15 MHz single element focused transducer (FUS Instruments, Toronto, Canada) operating in 10 ms bursts, 0.5 Hz pulse repetition frequency and 2 min total duration. Immediately following BBB opening, post-treatment T2*-weighted images were acquired followed by intravenous administration of MRI contrast agent (0.5 ul/g body weight, Magnevist, Bayer Health Care, PA), and T1-weighted contrast-enhanced images were acquired to verify BBB opening. T1-weighted imaging parameters were identical to those used for the T2*-weighted images, except: TE = 3.1 ms, 2 averages, total time = 1:19. Animals were removed from the MRI table and placed on a warm pad for 30 min prior to reversal of the anesthetic with antisedan (2 mg/kg, Orion Pharma, Espoo, Finland).
Behavioral Testing
The assessment of behavioral function was performed 1–3 days before and 2, 4, 6, 8, 10, and 12 weeks after injection of 6-OHDA (Figure S4).
1. Cylinder Test of Spontaneous Forelimb Use
Cylinder tests were performed similar to those previously described.69 In a dark testing room, rats were placed a 20 cm diameter glass cylinder and their activity was recorded from above. Left and right weight-bearing forepaw contacts with the wall were quantified by a trained and blinded observer. A minimum of 20 contacts were required to complete the test. Data are presented as percentage of contacts with the contralateral (left) forepaw.
2. Apomorphine-Induced Rotational Activity
Rotational tests were performed similar to those previously described.56 Rotational analysis was performed with automated bowls (Med Associates, St.Albans, VT) and the RotoRat software. Rats received a subcutaneous injection of apomorphine (0.4 mg/kg, Sigma-Aldrich, St. Louis, MO). Left (contralateral) and right (ipsilateral) rotations were recorded over 40 min. Data are expressed as net full body rotations per minute, with contralateral turns assigned a positive value.
Histological Processing
Immediately following euthanasia, animals were perfused via the left and right carotid arteries with 20 mL of 2% heparinized 0.9% saline followed by 10 mL of 4% formalin. Brains were suffusion-fixed in 4% formalin for 72h at 4 °C. Brains were then placed in a brain matrix (Stoelting, Wood Dale, IL). The first coronal slice containing the striatum was prepared by cutting 3 and 6 mm anterior to the front of the brain. The second coronal slide containing the SNpc was prepared by cutting 9 and 12 mm anterior to the front of the brain. Each slice was embedded in paraffin and serially cut in 8 μm coronal sections. Slices were cut in either 6 sections, 240 μm apart, or 8 sections, 120 μm apart, for the striatum-containing and SNpc-containing slices, respectively. Sections were immunolabeled against tyrosine hydroxylase.
Immunohistochemistry
Mounted sections were deparaffinized in xylene and rehydrated in decreasing concentrations of ethanol (100%–70%). Slides were heated to ~95 °C for 20 min in 10 mM sodium citrate buffer (pH 6.0) to unmask antigens. Endogenous peroxidase activity was blocked using 3% hydrogen peroxide. Sections were washed in PBS and blocked with blocking solution (Vector Laboratories, Burlingame, CA). Next, sections were incubated overnight at 4 °C with rabbit anti-TH primary antibody (1:250, Millipore, Temecula, CA). Sections were rinsed in PBS and incubated with goat anti-rabbit HRP conjugated secondary antibody (1:250, Abcam, Cambridge, MA) for 1 h at room temperature. After washing in PBS, sections were incubated with DAB-peroxidase substrate solution (IHC-Tek, Ellicott City, MD) and mounted with permanent mounting medium (Vector Laboratories, Burlingame, CA). Sections were imaged on a macroscope (Wild, Heerbrugg, Switzerland) equipped with a CCD camera (Olympus, Center Valley, NJ) at either 16X (striatal sections) or 35X (SNpc sections).
Quantitative Morphology
Images of striatal and SNpc sections were analyzed with in-house written software using MATLAB (Mathworks, Natick, MA). To assess inverse staining intensity in the striatum, images were inverted, and the averaged background was subtracted. Striatal regions were manually selected from the image, and the pixel intensity was averaged throughout the entire striatum. The average pixel intensity was then averaged across all six striatal sections. Data are presented as a ratio of average pixel intensity on the ipsilateral side compared to the contralateral side. To assess TH+ cell bodies in the SNpc, the averaged background was subtracted using a Gaussian averaging function. SNpc regions were manually selected from the image and converted to binary images using Otsu’s method. Images were then used to create linear structuring elements, and individual cell bodies were counted automatically. Cell body counts were averaged across all SNpc sections. Data are presented as a ratio of the average cell body number on the ipsilateral side compared to the contralateral side.
Sample Preparation for Biochemical Analysis
Striatum, SNpc, liver, spleen, heart, kidneys, and lungs were rapidly collected on ice and stored at −80 °C until further processing. Organs were pulverized in liquid nitrogen and separated into equal fractions for further analysis.
Measurement of Striatal Catecholamines
Striatal samples were lysed with 50 μL of 0.1 M perchloric acid per milligram of tissue using a tissue homogenizer (Omni International, Kennesaw, GA) and centrifuged at 13 000g for 10 min at 4 °C. The supernatant was passed through a 0.22 PVDF filter (Merck-Millipore, Cork, Ireland), and analyzed by HPLC, a modification of conditions from previously published methods.70 Samples were separated on a C18 reverse-phase HPLC column (Thermo-Scientific, BDS Hypersil, 100 × 2.1 mm). Dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) were quantified by electrochemical detection (Antec, Decade) in a mobile phase consisting of 50 mM citrate/acetate, pH 4.5, 1 mM decyl sulfonic acid, 0.1 mM EDTA, and 7% acetonitrile modifying agent. The flow rate was 0.35 mL/min, and the detector potential was 0.7 mV relative to a Ag/AgCl reference electrode.
GDNF Protein Levels Measured by ELISA
Striatal, SNpc, liver, spleen, lung, heart, and kidney samples were lysed with 10 μL of lysis buffer (137 mM NaCl, 20 mM Tris pH 8.0, 1% NP-40, 10% glycerol) supplemented with 1 mM PMSF, 0.5 mM sodium orthovanadate, and 10 μL of protease inhibitor cocktail (Sigma-Aldrich, cat. nr.: 8340) per milliliter of lysis buffer with a tissue homogenizer (Onmi International, Kennesaw, GA). The GDNF ELISA (enzyme-linked immunosorbent assay) was performed according to the instructions provided by the manufacturer (Promega, cat. nr., G7621). The total protein was measured using a BCA protein assay (Thermo Scientific, Rockford, IL), and GDNF protein was compared to the total amount of protein in the sample.
Statistical Analysis
Sample sizes for all groups were determined empirically. Animals which exhibited severe (>9 rotations/min) or inadequate (<5 rotations/min) apomorphine-induced rotational behavior 2 weeks following 6-OHDA administration were excluded. All statistics were conducted using SigmaPlot software (Version 13.0), which automatically suggests parametric or nonparametric tests depending on the results of normality testing. Group comparisons were performed using either three-way ANOVA (locomotor bias tests, body weight comparisons) or two-way ANOVA followed by Mann–Whitney rank sum tests or Holm Sidak methods. Repeated measure models were used to estimate the effects in the ipsilateral and contralateral sides of the brain. The statistical significance was set at p < 0.05. All values are presented as mean ± SEM.
Data Availability
The data supporting the findings in the current study are available upon request from the corresponding authors.
Supplementary Material
Acknowledgments
This work was supported by the Focused Ultrasound Foundation, NIH R01 CA164789, R01 CA197111, R01 EB020147, and R03 EB016784. B.P.M. was supported by the NHLBI-sponsored Basic Cardiovascular Research Training Grant (5 T32 HL007284) and the Wagner Fellowship. J.H. is supported by NIH R01GM084128. We are grateful to Dr. David Yurek and Dr. Mark Cooper for donating the GDNF plasmid used in this study.
Footnotes
Author Contributions
B.P.M., J.S.S., J.H., and R.J.P. conceived of the experiments. B.P.M. conducted animal surgeries and behavioral experiments and ex vivo analysis. G.W.M. designed MR imaging sequences and, with B.P.M., performed MR image guided FUS procedures. B.P.M. and D.H. performed immunohistochemistry and quantitative morphology. N.K. and P.M. fabricated and characterized B.P.N.. A.L.K. designed and fabricated micro-bubbles. J.W.M. assessed H&E stained sections for safety analysis. J.H. designed HPLC parameters. All authors contributed to writing the manuscript.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nano-lett.7b00616.
Figure S1: GDNF-BPN are colloidally stable when incubated in water or artificial cerebrospinal fluid. Figure S2: FUS-mediated delivery of GDNF-BPN to the striatum of PD rats does not change GDNF protein levels in the SNpc or other major organs. Figure S3: FUS-mediated delivery of GDNF-BPN to the striatum of PD rats does not lead to systemic or local toxicity. Figure S4: Time schedule of study. Table S1: Dopamine metabolites in the ipsilateral striatum (PDF)
References
- 1.Pringsheim T, Jette N, Frolkis A, Steeves T. Mov Disord. 2014;29:1583–1590. doi: 10.1002/mds.25945. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharjee S, Sambamoorthi U. Park Relat Disord. 2013;19:746–750. doi: 10.1016/j.parkreldis.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hegarty SV, O’Keeffe GW, Sullivan AM. Neural Regener Res. 2014;9:1708–1711. doi: 10.4103/1673-5374.143410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jankovic J, Poewe W. Curr Opin Neurol. 2012;25:433–447. doi: 10.1097/WCO.0b013e3283542fc2. [DOI] [PubMed] [Google Scholar]
- 5.Toulouse A, Sullivan AM. Prog Neurobiol. 2008;85:376–392. doi: 10.1016/j.pneurobio.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Kalia LV, Lang AE. Lancet. 2015;386:896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 7.Garbayo E, Ansorena E, Blanco-Prieto MJ. Maturitas. 2013;76:272–278. doi: 10.1016/j.maturitas.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 8.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 9.Kotzbauer PT, Lampe P, Heuckeroth RO, Golden JP, Creedon DJ, Johnson EM, Milbrandt J. Nature. 1996;384:467–470. doi: 10.1038/384467a0. [DOI] [PubMed] [Google Scholar]
- 10.Bartus RT, Johnson EM. Neurobiol Dis. 2017;97:156–168. doi: 10.1016/j.nbd.2016.03.027. [DOI] [PubMed] [Google Scholar]
- 11.Bartus RT, Johnson EM. Neurobiol Dis. 2017;97:169–178. doi: 10.1016/j.nbd.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 12.Kordower JH, Bjorklund A. Mov Disord. 2013;28:96–109. doi: 10.1002/mds.25344. [DOI] [PubMed] [Google Scholar]
- 13.Pardridge WM. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deleu D, Northway MG, Hanssens Y. Clin Pharmacokinet. 2002;41:261–309. doi: 10.2165/00003088-200241040-00003. [DOI] [PubMed] [Google Scholar]
- 15.Kroll RA, Neuwelt EA. Neurosurgery. 1998;42:1083–1099. doi: 10.1097/00006123-199805000-00082. [DOI] [PubMed] [Google Scholar]
- 16.Pardridge WM. Drug Discovery Today. 2007;12:54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Timbie K, Mead BP, Price RJ. J Controlled Release. 2015;219:61–75. doi: 10.1016/j.jconrel.2015.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nance E, Timbie K, Miller GW, Song J, Louttit C, Klibanov AL, Shih TY, Swaminathan G, Tamargo RJ, Woodworth GF, Hanes J, Price RJ. J Controlled Release. 2014;189:123–132. doi: 10.1016/j.jconrel.2014.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheikov N, McDannold NJ, Vykhodtseva NI, Jolesz FA, Hynynen K. Ultrasound Med Biol. 2004;30:979–989. doi: 10.1016/j.ultrasmedbio.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 20.Shang X, Wang P, Liu Y, Zhang Z. J Mol Neurosci. 2011;43:364–369. doi: 10.1007/s12031-010-9451-9. [DOI] [PubMed] [Google Scholar]
- 21.Hynynen K, McDannold NJ, Vykhodtseva N, Jolesz FA. Radiology. 2001;220:640–646. doi: 10.1148/radiol.2202001804. [DOI] [PubMed] [Google Scholar]
- 22.Vlachos F, Tung YS, Konofagou EE. Phys Med Biol. 2010;55:5451–5466. doi: 10.1088/0031-9155/55/18/012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDannold NJ, Arvanitis CDD, Vykhodtseva NI, Livingstone MSS. Cancer Res. 2012;72:3652–3663. doi: 10.1158/0008-5472.CAN-12-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Downs ME, Buch A, Sierra C, Karakatsani ME, Chen S, Konofagou EE, Ferrera VP. PLoS One. 2015;10:1–26. [Google Scholar]
- 25.Ghanouni P, Pauly KB, Elias WJ, Henderson J, Sheehan J, Monteith S, Wintermark M. AJR, Am J Roentgenol. 2015;205:150–159. doi: 10.2214/AJR.14.13632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elias WJ, Lipsman N, Ondo WG, Ghanouni P, Kim YG, Lee W, Schwartz M, Hynynen K, Lozano AM, Shah BB, et al. N Engl J Med. 2016;375:730–739. doi: 10.1056/NEJMoa1600159. [DOI] [PubMed] [Google Scholar]
- 27.Schlesinger I, Eran A, Sinai A, Erikh I, Nassar M, Goldsher D, Zaaroor M. Mov Disord. 2014;29:1–4. [Google Scholar]
- 28.Nance EA, Woodworth GF, Sailor KA, Shih TY, Xu Q, Swaminathan G, Xiang D, Eberhart C, Hanes J. Sci Transl Med. 2012;4:1–8. doi: 10.1126/scitranslmed.3003594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kenny GD, Bienemann AS, Tagalakis AD, Pugh JA, Welser K, Campbell F, Tabor AB, Hailes HC, Gill SS, Lythgoe MF, et al. Biomaterials. 2013;34:9190–9200. doi: 10.1016/j.biomaterials.2013.07.081. [DOI] [PubMed] [Google Scholar]
- 30.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Proc Natl Acad Sci U S A. 1994;91:2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mastorakos P, Zhang C, Berry S, Oh Y, Lee S, Eberhart CG, Woodworth GF, Suk JS, Hanes J. Adv Healthcare Mater. 2015;4:1023–1033. doi: 10.1002/adhm.201400800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. Adv Drug Delivery Rev. 2016;99:28–51. doi: 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nance E, Zhang C, Shih T, Xu Q, Schuster B, Hanes J. ACS Nano. 2014;8:10655–10664. doi: 10.1021/nn504210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mastorakos P, da Silva AL, Chisholm J, Song E, Choi WK, Boyle MP, Morales MM, Hanes J, Suk JS. Proc Natl Acad Sci U S A. 2015;112:8720–8725. doi: 10.1073/pnas.1502281112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suk JS, Kim AJ, Trehan K, Schneider CS, Cebotaru L, Woodward OM, Boylan NJ, Boyle MP, Lai SK, Guggino WB, Hanes J. J Controlled Release. 2014;178:8–17. doi: 10.1016/j.jconrel.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mead BP, Mastorakos P, Suk JS, Klibanov AL, Hanes J, Price RJ. J Controlled Release. 2016;223:109–117. doi: 10.1016/j.jconrel.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fletcher AM, Kowalczyk TH, Padegimas L, Cooper MJ, Yurek DM. Neuroscience. 2011;194:220–226. doi: 10.1016/j.neuroscience.2011.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yurek DM, Fletcher AM, Mcshane M, Kowalczyk TH, Weatherspoon MR, Kaytor MD, Cooper MJ, Assem G. Mol Imaging. 2011;10:327–339. doi: 10.2310/7290.2010.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burke CW, Suk JS, Kim AJ, Hsiang YHJ, Klibanov AL, Hanes J, Price RJ. J Controlled Release. 2012;162:414–421. doi: 10.1016/j.jconrel.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Björklund A, Kirik D, Rosenblad C, Georgievska B, Lundberg C, Mandel RJ. Brain Res. 2000;886:82–98. doi: 10.1016/s0006-8993(00)02915-2. [DOI] [PubMed] [Google Scholar]
- 41.Lin CY, Hsieh HY, Chen CM, Wu SR, Tsai CH, Huang CY, Hua MY, Wei KC, Yeh CK, Liu HL. J Controlled Release. 2016;235:72–81. doi: 10.1016/j.jconrel.2016.05.052. [DOI] [PubMed] [Google Scholar]
- 42.Huang Q, Deng J, Xie Z, Wang F, Chen S, Lei B, Liao P, Huang N, Wang Z, Wang Z, Cheng Y. Ultrasound Med Biol. 2012;38:1234–1243. doi: 10.1016/j.ultrasmedbio.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 43.Huang Q, Deng J, Wang F, Chen S, Liu Y, Wang ZZ, Wang ZZ, Cheng Y. Exp Neurol. 2012;233:350–356. doi: 10.1016/j.expneurol.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 44.Wang S, Olumolade OO, Sun T, Samiotaki G, Konofagou EE. Gene Ther. 2015;22:104–110. doi: 10.1038/gt.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thevenot E, Jordão JF, O’Reilly MA, Markham K, Weng YQ, Foust KD, Kaspar BK, Hynynen K, Aubert I. Hum Gene Ther. 2012;23:1144–1155. doi: 10.1089/hum.2012.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calcedo R, Wilson JM. Front Immunol. 2013;4:1–7. doi: 10.3389/fimmu.2013.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergen JM, Park IK, Horner PJ, Pun SH. Pharm Res. 2008;25:983–998. doi: 10.1007/s11095-007-9439-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yurek DM, Fletcher AM, Smith GM, Seroogy KB, Ziady AG, Molter J, Kowalczyk TH, Padegimas L, Cooper M. Mol Ther. 2009;17:641–650. doi: 10.1038/mt.2009.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drinkut A, Tereshchenko Y, Schulz JB, Bahr M, Kügler S. Mol Ther. 2012;20:534–543. doi: 10.1038/mt.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eslamboli A, Georgievska B, Ridley RM, Baker HF, Muzyczka N, Burger C, Mandel RJ, Annett L, Kirik D. J Neurosci. 2005;25:769–777. doi: 10.1523/JNEUROSCI.4421-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Georgievska B, Kirik D, Bjo A. J Neurosci. 2004;24:6437–6445. doi: 10.1523/JNEUROSCI.1122-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tumer N, Scarpace PJ, Dogan MD, Broxson CS, Matheny M, Yurek DM, Peden CS, Burger C, Muzyczka N, Mandel R. Neurobiol Aging. 2006;27:459–470. doi: 10.1016/j.neurobiolaging.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 53.Manfredsson FP, Tumer N, Erdos B, Landa T, Broxson CS, Sullivan LF, Rising AC, Foust KD, Zhang Y, Muzyczka N, et al. Mol Ther. 2009;17:980–991. doi: 10.1038/mt.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Isacson O, Kordower JH. Ann Neurol. 2008;64:122–138. doi: 10.1002/ana.21473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pardridge WM, Boado RJ. Pharm Res. 2009;26:2227–2236. doi: 10.1007/s11095-009-9939-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tereshchenko J, Maddalena A, Bahr M, Kügler S. Neurobiol Dis. 2014;65:35–42. doi: 10.1016/j.nbd.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 57.Garbayo E, Montero-Menei CN, Ansorena E, Lanciego JL, Aymerich MS, Blanco-Prieto MJ. J Controlled Release. 2009;135:119–126. doi: 10.1016/j.jconrel.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 58.Sarkar S, Raymick J, Imam S. Int J Mol Sci. 2016;17:904–935. doi: 10.3390/ijms17060904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bohnen NI, Albm RL, Koeppe RA, Wernette KA, Kilbourn MR, Minoshima S, Frey KA. J Cereb Blood Flow Metab. 2006;26:1198–1212. doi: 10.1038/sj.jcbfm.9600276. [DOI] [PubMed] [Google Scholar]
- 60.Decressac M, Ulusoy A, Mattsson B, Georgievska B, Romero-Ramos M, Kirik D, Björklund A. Brain. 2011;134:2302–2311. doi: 10.1093/brain/awr149. [DOI] [PubMed] [Google Scholar]
- 61.Decressac M, Kadkhodaei B, Mattsson B, Laguna A, Perlmann T, Björklund A, Bjorklund A. Sci Transl Med. 2012;4:1–15. doi: 10.1126/scitranslmed.3004676. [DOI] [PubMed] [Google Scholar]
- 62.Kirik D, Rosenblad C, Bjo A, Mandel RJ. J Neurosci. 2000;20:4686–4700. doi: 10.1523/JNEUROSCI.20-12-04686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marks WJ, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N, Vitek J, Stacy M, Turner D, Verhagen L, et al. Lancet Neurol. 2010;9:1164–1172. doi: 10.1016/S1474-4422(10)70254-4. [DOI] [PubMed] [Google Scholar]
- 64.Marks WJ, Ostrem JL, Verhagen L, Starr PA, Larson PS, Bakay RA, Taylor R, Cahn-Weiner DA, Stoessl AJ, Olanow CW, Bartus RT. Lancet Neurol. 2008;7:400–408. doi: 10.1016/S1474-4422(08)70065-6. [DOI] [PubMed] [Google Scholar]
- 65.Bartus RT, Kordower JH, Johnson EM, Brown L, Kruegel BR, Chu Y, Baumann TL, Lang AE, Olanow CW, Herzog CD. Neurobiol Dis. 2015;78:162–171. doi: 10.1016/j.nbd.2015.03.023. [DOI] [PubMed] [Google Scholar]
- 66.Olanow WC, Bartus RT, Baumann TL, Factor S, Boulis N, Stacy M, Turner D, Marks W, Larson P, Starr P, et al. Ann Neurol. 2015;78:162–171. doi: 10.1002/ana.24436. [DOI] [PubMed] [Google Scholar]
- 67.Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carpentier A, Canney M, Vignot A, Reina V, Beccaria K, Horodyckid C, Karachi C, Leclercq D, Lafon C, Chapelon J, et al. Sci Transl Med. 2016;8:1–8. doi: 10.1126/scitranslmed.aaf6086. [DOI] [PubMed] [Google Scholar]
- 69.Gasmi M, Brandon EP, Herzog CD, Wilson A, Bishop KM, Hofer EK, Cunningham JJ, Printz Ma, Kordower JH, Bartus RT. Neurobiol Dis. 2007;27:67–76. doi: 10.1016/j.nbd.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 70.Hardie SL, Hirsh J. J Neurosci Methods. 2006;153:243–249. doi: 10.1016/j.jneumeth.2005.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings in the current study are available upon request from the corresponding authors.