

Abstract
Expression of the EVI1 proto-oncogene is deregulated by chromosomal translocations in some cases of acute myeloid leukemia (AML) and is associated with poor clinical outcome. Here, through transcriptomic and metabolomic profiling of hematopoietic cells, we reveal that EVI1 overexpression alters cellular metabolism. A pooled shRNA screen identified the ATP-buffering, mitochondrial creatine kinase CKMT1 as a metabolic dependency in EVI1-positive AML. EVI1 promotes CKMT1 expression by repressing the myeloid differentiation regulator RUNX1. Suppression of arginine-creatine metabolism by CKMT1-directed shRNAs or by the small molecule cyclocreatine selectively decreased the viability, promoted cell cycle arrest and apoptosis of human EVI1-positive AML cells, and prolonged survival in human orthotopic and mouse primary AML models. CKMT1 inhibition alters mitochondrial respiration and ATP production, an effect that is abrogated by phospho-creatine-mediated reactivation of the arginine-creatine pathway. Targeting CKMT1 is a promising therapeutic strategy for this EVI1-driven AML subtype that is highly resistant to current treatment regimens.
Keywords: AML, RUNX1, CKMT1, cyclocreatine, arginine metabolism
INTRODUCTION
Recent investigations into the relationship between cancer and cellular metabolism have revealed the strong dependency of different cancers on a diverse array of metabolic pathways. Therefore, identifying mechanisms by which these metabolic pathways become deregulated in the first place en route to, or as a result of, the development of cancer has emerged as a major area of therapeutic and biological interest.
Deregulation of metabolic pathways can occur through multiple mechanisms, including alterations in the cellular landscape brought about by mutations in metabolic enzymes (e.g., IDH1/IDH2 and SDH)1–4. IDH1 and IDH2 mutations, which are initiating events in high-grade gliomas and frequently occur in acute myeloid leukemia (AML), interfere with normal IDH1/2 function to produce the pro-oncogenic metabolite R(−)-2-hydroxyglutarate which acts as an epigenetic modulator5–8. Deregulation of metabolic pathways can also occur through aberrant expression of transcription factors, such as the proto-oncogene MYC. Transcriptional changes resulting from aberrantly activated MYC increase glucose uptake and glycolysis in cancer cells and stimulate glutaminolysis, serine/glycine metabolism, and lipid biosynthesis9. Although MYC and other transcription factors are typically considered pharmacologically challenging targets, the diverse metabolic alterations induced by these transcription factors may constitute another source of unique cancer dependencies and provide multiple downstream opportunities for therapeutic intervention. It is thus necessary not only to identify the metabolic changes which occur with cancer development, but to link these programs to initiating events and oncogenic drivers; the latter will enable identification of patient populations that may benefit from specific metabolic interventions.
Transcriptional modulators are deregulated in AML either by translocation (e.g., MLL-AF9, AML1-ETO, or PML-RARA) or aberrant expression, as is true of EVI110,11. Abnormal expression of this transcription factor through chromosome 3q26 rearrangements (e.g., t(3;3)(q21;q26) or inv3(q21;q26)) promotes the development or progression of high-risk acute myeloid leukemia (AML), for which current treatment regimens remain inadequate12–15. Here we integrate genomic and metabolic screening to identify a new druggable metabolic dependency in mouse and human EVI1-positive AML, including cell lines and primary patient samples.
RESULTS
EVI1 modulates metabolism in AML cells
To characterize the transcriptional consequences of EVI1 overexpression in AML, we generated an EVI1-dependent gene signature from murine bone marrow LinLow cells infected with a retroviral construct encoding Evi1 (GSE 3472916). We then used this signature to conduct an open-ended enrichment analysis of 4725 curated gene sets from the c2 collection assembled from various sources such as online pathway databases, publications in PubMed, and knowledge of domain experts available from MSigDB (Figure 1A). We confirmed the findings of other investigators that gene sets related to cell proliferation and differentiation were enriched in genes whose expression is suppressed by enforced Evi1 overexpression16–18. Multiple cell metabolism-related gene sets were also significantly enriched in an unbiased search of the complete c2 collection. Evi1 overexpression modulated genes from cellular energy-related pathways, including glycolysis, the TCA cycle, and oxidative phosphorylation, purine and pyrimidine synthesis, and the metabolism of various amino acids.(Figure 1B). Using an unbiased metabolomic profiling approach, we established that of 292 small metabolites detected by mass spectrometry, steady state levels of 82 were significantly (−log10 (p value) ≤ 1) changed by Evi1-expression (Supplementary Figure S1). The most highly enriched metabolic pathways were related to purine and pyrimidine metabolism, amino acid metabolism (alanine, aspartate, glutamate, arginine, and proline), the pentose phosphate pathway and glycolysis (Figure 1C).
Figure 1. EVI1 overexpression imparts new metabolic dependencies on AML cells.
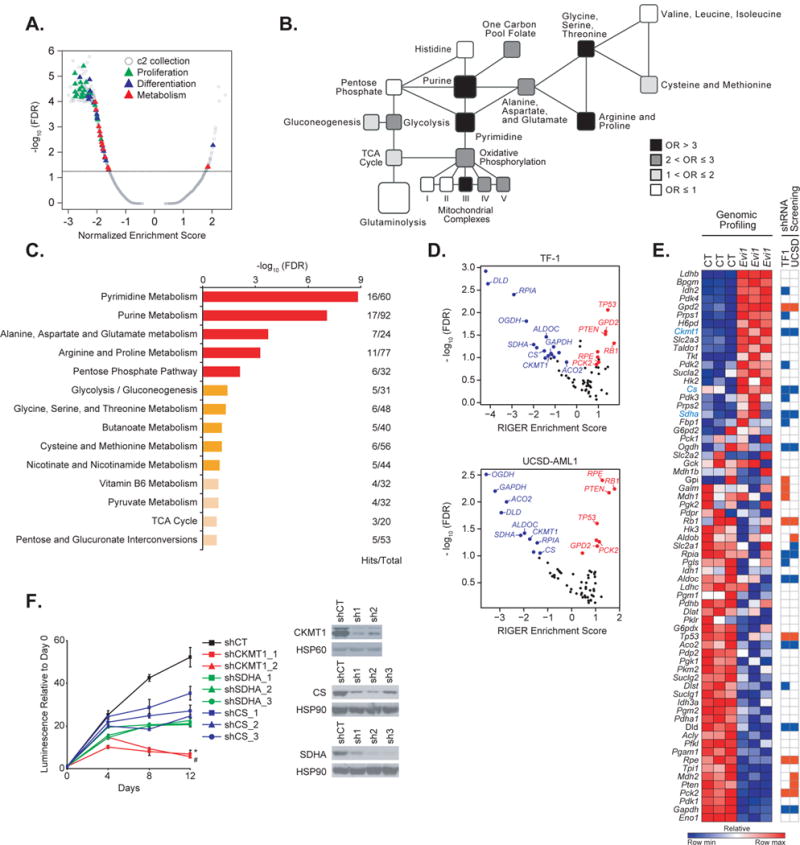
(A) Quantitative comparison by GSEA of the c2 collection of curated gene sets available from MSigDB v.5.0 for murine LinLow Evi1-overexpressing versus control bone marrow samples (GSE 34729). Data are presented as a volcano plot of −log10 (FDR) versus the normalized enrichment score for each evaluated gene set. Triangles indicate sets related to proliferation (green), cell differentiation (blue), and metabolism (red), and gray dots indicate all other c2 gene sets.
(B) Metabolic network showing gene sets altered in the Evi1 overexpression signature generated from GSE 34729. Black, dark grey, light grey, and white colors indicate an enrichment odds ratio (OR) above 3, between 2 and 3, between 1 and 2, and below 1, respectively.
(C) Pathway analysis integrating enrichment and pathway topology analyses (MetaboAnalyst) of the steady-state metabolite profile from control versus Evi1-overexpressing LinLow murine bone marrow cells. Top 15 out of a total of 45 metabolic pathways are represented based on a FDR ≤ 0.1 and p value ≤ 0.05. “Hits” represents the number of metabolites that scored in the steady-state profile and “Total” represents the number of metabolites present in the given metabolic pathway. Metabolic pathways with a - log10 (FDR) ≥ 1.5 are depicted in red, 1 ≤ − log10 (FDR) < 1.5 are in orange, and − log10 (FDR) < 1 in beige.
(D) Scatterplot of hits significantly depleted (in blue) or enriched (in red) from an shRNA library targeting 67 metabolism-related genes in the human TF-1 (top) and UCSD-AML1 (bottom) cell lines. Hits which did not score are depicted in black. Data are presented as volcano plots of −log10 (FDR) versus the RIGER enrichment score for each gene.
(E) Heatmap of the metabolism genes differentially expressed by genomic profiling in GSE 34729 upon Evi1 overexpression in LinLow bone marrow cells (left panel), and heatmap of genes either depleted (in blue) or enriched (in red) in the shRNA screen in TF-1 and UCSD-AML1 cells (right panel). Shown in blue text are genes both upregulated upon Evi1 overexpression and depleted in the shRNA screen. CT, control. Each column for each condition represents a technical replicate (n=3 per condition).
(F) Growth of TF-1 cells infected with hairpins directed against either CKMT1, SDHA, or CS (left panel). Immunoblot confirming shRNA target knockdown (right panel). shCT, control shRNA. Error bars represent mean ± SD. * and # p value ≤ 0.05 was calculated on the latest time point using a nonparametric Kruskall-Wallis test and Dunn’s multiple comparisons test. Data are representative of two independent experiments.
To assess whether upregulation of EVI1 in human AML cell lines might promote oncogenic dependency on metabolic enzymes, we performed an shRNA screen against 67 genes encoding enzymes from glycolysis, the pentose phosphate pathway, the TCA cycle, and related metabolic pathways in the AML cell lines TF-1 and UCSD-AML1, both of which express EVI1. These cells were infected with the shRNA library and expanded until day 36 at which point genomic DNA was harvested and hairpin representation assessed by sequencing and compared to the representation of the input genomic DNA at day 0. We identified hairpin sequence that was depleted (hairpins target leukemia-promoting genes) or enriched (hairpins targeting leukemia-suppressing genes) at 36 days compared to the input genomic DNA. These experiments identified 9 depleted and 6 enriched genes overlapping in both lines, tested with at least two hairpins (Figure 1D and Supplementary Table S1). As expected, three positive control tumor suppressor genes, PTEN, TP53 and RB1, scored among the top enriched hits. Three genes of interest – Ckmt1, Cs, and Sdha genes (murine homologs of human CKMT1, Citrate Synthase (CS) and Succinate Dehydrogenase Complex, Subunit A (SDHA), respectively) – were both upregulated by genomic profiling in Evi1-transduced cells and depleted by multiple shRNAs in the TF-1 and UCSD-AML1 AML cell lines (Figure 1E). We then infected TF-1 AML cells with multiple hairpins that markedly reduced CKMT1, SDHA, and CS protein levels (Figure 1F, right panel). While all hairpins significantly decreased cell growth over a 12-day time course, CKMT1 depletion caused the most pronounced growth inhibition (Figure 1F).
CKMT1 is a target in EVI1-positive leukemia
To address the potential relationship between EVI1 expression and dependence on CKMT1, we expanded our investigation to a larger panel of human AML cell lines with and without elevated EVI1 expression. All four EVI1-expressing AML cell lines exhibited elevated CKMT1 protein expression and activity (Figure 2A and 2B). The viability of these four cell lines was more strongly decreased compared to all the other lines tested in response to a treatment with cyclocreatine, an analog of the CKMT1 substrate creatine and inhibitor of the creatine biosynthesis pathway (Figure 2C). Furthermore, three AML cell lines with elevated EVI1 expression – UT-7, TF-1, and UCSD-AML1 – exhibited enhanced sensitivity to a reduction in CKMT1 expression mediated by doxycycline-inducible shRNAs in comparison with the other cell lines tested (Figure 2D). The human Kasumi-3 cell line was not tested in this experiment due to its poor transduction efficiency. Similar growth defects were observed with non-inducible CKMT1-targeting shRNAs (Supplementary Figure S2A and S2B). These AML cell lines are grown in GM-CSF, and withdrawal of GM-CSF neither affected CKMT1 expression nor sensitivity of EVI1-expressing cells to cyclocreatine (Supplementary Figures S2C and S2D). Conversely, GM-CSF supplementation did not promote CKMT1 expression in the two AML cell lines tested to represent EVI1-null, CKMT1-null cells: HL-60 and U937 (Supplementary Figures S2E).
Figure 2. EVI1-positive cells highly express and are dependent on CKMT1.
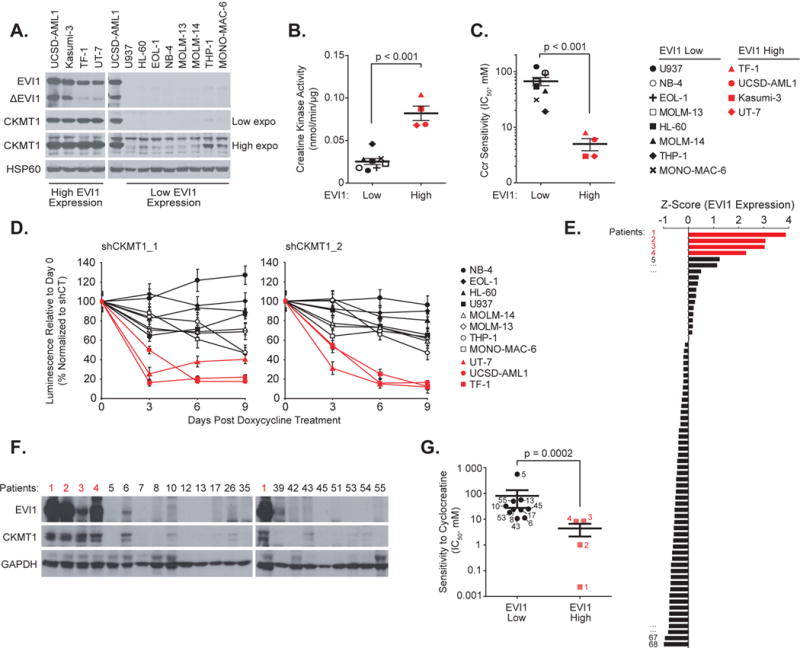
(A) Immunoblot for EVI1, CKMT1, and HSP60 (loading control) from a panel of human AML cell lines with high or low EVI1 expression.
(B-C) Creatine kinase activity (B) and distribution of IC50 (C) for human EVI1 high versus low cell lines treated with cyclocreatine (Ccr). p value calculated using nonparametric Mann-Whitney test. Error bars represent mean ± SD of 8 low EVI1-expressing and four high EVI1-expressing human cell lines.
(D) Eleven human AML cell lines were infected with two CKMT1-directed miR30-shRNAs. Growth after treatment with 0.5 μg/ml doxycycline is normalized to the control shRNA and shown relative to day 0 (time of seeding), with error bars representing the mean ± SD of seven technical replicates. Each experiment in A–D was performed at least two independent times.
(E) Z Score normalization of EVI1 expression across a panel of CD13/33-gated bone marrow cells from 68 patient AML samples. Red bars indicate the four patients with the highest EVI1 expression level.
(F) Immunoblot for EVI1, CKMT1, and GAPDH (loading control) from bone marrow samples sub-selected from panel (E).
(G) Distribution of IC50 for EVI1 high versus low patient samples in response to treatment with cyclocreatine (Ccr). Red squares represent the four patient samples from panel (F) highlighted in red. p value calculated using nonparametric Mann-Whitney test. Error bars represent mean ± SD of 10 low EVI1-expressing and four high EVI1-expressing human primary samples.
Finally, we assayed EVI1 expression by flow cytometry in a panel of 68 unselected primary AML patient specimens (Figure 2E and Supplementary Table S2). Samples with at least 5×106 blast cells were subsequently analyzed by flow cytometry and immunoblot. The four leukemias with the highest levels of EVI1 expression showed higher CKMT1 protein levels and enhanced sensitivity to cyclocreatine treatment compared to the leukemias with low EVI1 expression (Figures 2E–2G). Together, these studies indicate that elevated CKMT1 expression and biochemical dependence on this metabolic pathway are correlated with high levels of EVI1 expression in both human AML cell lines and primary patient specimens.
EVI1 promotes CKMT1 expression through RUNX1 repression
To clarify how EVI1 regulates cellular CKMT1 expression, we first infected mouse Linlow, c-kit+ bone marrow cells with either an empty MSCV vector or with a construct encoding EVI1. Consistent with our analysis of human AMLs, enforced Evi1 expression increased Ckmt1 protein and mRNA levels (Figures 3A and 3B). Moreover, doxycycline induction of three independent shRNA molecules targeting EVI1 reduced CKMT1 expression in TF-1 and UCSD-AML1 cells (Figure 3C). Then, we used a luciferase reporter system in which the CKMT1 gene promoter was cloned upstream of a luciferase cassette, and levels of CKMT1 transactivation were assessed using bioluminescence (Figure 3D). Co-expression of EVI1 and the CKMT1-Luc promoter in HEK293E cells activated CKMT1 transcription. Deleting a promoter region between −1169 and −943 bp upstream of the transcriptional start site increased basal CKMT1 transactivation at a similar fold-increase to that observed upon EVI1 overexpression. This effect was recapitulated by deletion of a RUNX1 consensus binding motif ACCACA (ΔACCACA) from the −1169/−943 CKMT1 promoter sequence (Figure 3E), suggesting that RUNX1 might repress CKMT1 expression through direct binding to its promoter. In accordance with this idea, RUNX1 overexpression impeded the capacity of EVI1 to transactivate CKMT1 (Figure 3F). EVI1 knockdown promoted both an increase in RUNX1 and a decrease in CKMT1 expression. Furthermore, RUNX1-directed shRNAs, which abrogated RUNX1 upregulation, prevented a decrease of CKMT1 expression upon EVI1 knockdown (Figure 3G).
Figure 3. EVI1-mediated RUNX1 downregulation promotes CKMT1 expression.
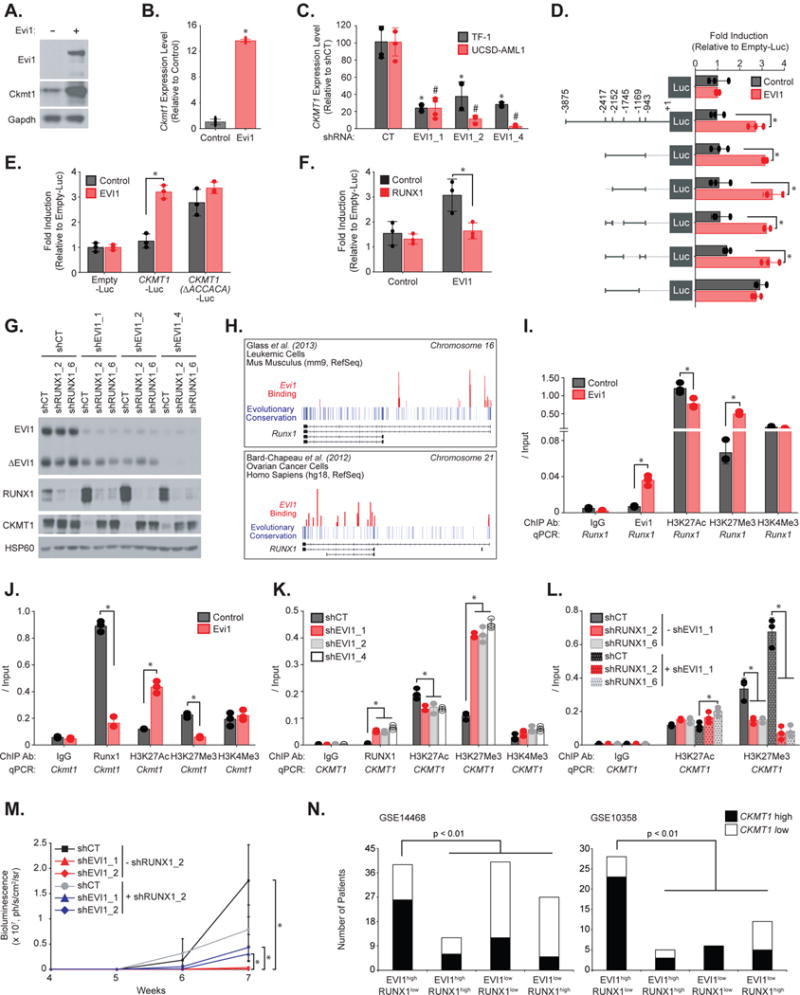
(A–B) Immunoblot for Evi1, Ckmt1 and Gapdh (A) and qRT-PCR for Ckmt1 expression (B) from LinLow, c-Kit+ murine bone marrow cells infected with an empty or Evi1-encoding construct. Error bars represent the mean ± SD of four technical replicates. * p value ≤ 0.05 in comparison with empty control vector calculated using a Mann-Whitney test.
(C) qRT-PCR for CKMT1 expression level in human TF-1 and UCSD-AML1 cells infected with shControl (shCT) or three EVI1-targeting shRNAs. Error bars represent mean ± SD of four technical replicates. * and # p value ≤ 0.05 in comparison with shCT calculated using a Mann-Whitney test.
(D) Luciferase reporter assay from human 293E cells co-expressing a vector encoding EVI1, and wild-type or several truncated forms of the human CKMT1 promoter flanking a luciferase cassette.
(E) Luciferase reporter assay from human 293E cells co-expressing a vector encoding EVI1 and a luciferase-flanked human CKMT1 promoter either wild-type or deleted for the RUNX1-binding site.
(F) Luciferase reporter assay from human 293E cells co-expressing vectors encoding EVI1 and RUNX1.
(G) Immunoblot for EVI1, RUNX1, CKMT1, and HSP60 (loading control) from human TF-1 cells infected with the indicated combinations of EVI1- and RUNX1-directed shRNAs.
(H) Representation of EVI1 binding peaks on RUNX1 gene tracks available from two ChIP-sequencing experiments performed by Bard-Chapeau et al.19 and Glass et al.20.
(I-L) LinLow, c-Kit+ murine bone marrow cells overexpressing a vector encoding Evi1 (I–J), or human UCSD-AML1 cells infected with shControl (shCT) and three EVI1-directed shRNAs (K), or human UCSD-AML1 cells infected with combinations of EVI1- and RUNX1-directed shRNAs (L) following ChIP with the indicated ChIP antibodies (ChIP Ab) and qPCR on the indicated promoter regions. Results are represented as fold enrichment over input.
(D–L) Error bars represent mean ± SD of three technical replicates.* p value ≤ 0.05 calculated using a Welch’s t-test. Each experiment in A–L was performed at least two independent times.
(M) TF-1-Luc cells engineered to overexpress CSF2 for efficient engraftment were infected with a combination of two EVI1- and one RUNX1-directed shRNAs before tail vein injection. Bioluminescence was quantified weekly as a measure of disease burden. Data are represented as mean ± SEM of 5 mice per cohort. * p value ≤ 0.05 was calculated on the latest time point using nonparametric Mann-Whitney test.
(N) Bar graph showing the number of primary patient AML samples with either EVI1high/RUNX1low, EVI1high/RUNX1high, EVI1low/RUNX1low, or EVI1low/RUNX1high expression and displaying CKMT1 high versus low expression level (absolute cut-off of a z-score ≥ 0.58 is high expression and a z-score ≤ − 0.58 is low expression) in two cohorts: GSE14468 and GSE10358. p value calculated using a Fisher exact test.
We then used a ChIP-qPCR approach to dissect the sequential events involved in EVI1-induced CKMT1 upregulation and the possible role of RUNX1 as an intermediate in this process (Figure 3H–3L). Multiple red peaks corresponding to EVI1-binding regions appeared on both murine and human RUNX1 gene tracks from two ChIP-sequencing experiments19,20 (Figure 3H). After immunoprecipitation of Evi1 from Evi1-overexpressing primary bone marrow murine cells and Runx1 promoter amplification by qPCR, we found a significant increase in Evi1 binding at the Runx1 promoter, accompanied by an increased amount of histone repressive mark H3K27Me3 at the same promoter region (Figure 3I). This was associated with a decrease in the expression of RUNX1 mRNA and protein levels (Supplementary Figure S3A and S3B). At basal conditions, Runx1 is bound to Ckmt1 promoter in normal bone marrow cells. Evi1 overexpression, however, decreased Runx1 binding to the Ckmt1 promoter, as well as the amount of bound repressive mark H3K27me3, and increased the binding signal of the transcriptional activation histone mark H3K27Ac (Figure 3J). This was associated with increased expression of Ckmt1 mRNA and protein levels (Supplementary Figures S3A and S3B). In contrast, EVI1 knockdown in UCSD-AML1 cells enhanced RUNX1 expression and binding at the CKMT1 promoter (Figure 3K, and Supplementary Figure S3C and 3D). This led to an increased H3K27Me3 binding signal and a reduced amount of bound H3K27Ac at the CKMT1 promoter and was associated with a decreased expression of CKMT1 (Figure 3K). In contrast, concomitant EVI1 and RUNX1 knockdown decreased repressive H3K27Me3 histone mark level at the CKMT1 promoter (Figure 3L). Taken together, these results show that EVI1 represses RUNX1 expression by direct binding to its promoter. This, in turn, eliminates repressive RUNX1 binding at the CKMT1 promoter and thereby promotes CKMT1 expression. Moreover, we also observed that RUNX1-directed shRNAs attenuated by 17-fold (based on the average bioluminescence across the two hairpins) the reduced disease burden caused by EVI1 depletion (Figure 3M). Finally, we explored the relationship between EVI1 and RUNX1 expression with CKMT1 mRNA levels in two AML transcriptional datasets (GSE14468 and GSE10358). We divided these cohorts into four subgroups: EVI1high/RUNX1low, EVI1high/RUNX1high, EVI1low/RUNX1low, and EVI1low/RUNX1high. Consistent with our mechanistic analysis, AML samples with high CKMT1 levels were significantly more represented within the EVI1high/RUNX1low subgroup than in the three other subgroups (Figure 3N).
Creatine kinase activity drives mitochondrial function and ATP production
CKMT1 promotes the metabolism of arginine to creatinine. To determine the effect of CKMT1 suppression on this pathway, we measured the metabolic flux of stable-isotope labeled L-arginine 13C6 through creatine synthesis, ornithine / spermine and ornithine / urea cycle, and the glutamate pathway using mass spectrometry (Figure 4A). As expected, both CKMT1 knockdown and cyclocreatine treatment in UCSD-AML1 and TF-1 cells diminished incorporation of L-Arginine 13C6 into creatinine. Creatinine is the end metabolite product of the creatine synthesis pathway and is spontaneously synthesized from phospho-creatine (Figure 4A–C and Supplementary Figure S4A). We also observed a significant decrease in arginine 13C6 incorporation into the precursor metabolite of the creatine synthesis pathway, guanidinoacetate, suggesting an inhibitory feedback loop of end metabolite products on the guanidinoacetate-producing enzyme glycine amidinotransferase, GATM. Of note, cyclocreatine treatment had a broader effect than shRNA-mediated CKMT1 depletion on arginine metabolism because it also impaired production of glutamate, α-ketoglutarate, and L-arginino-succinate (Figure 4C). Co-treatment of cells with exogenous phospho-creatine activated production of guanidinoacetate 13C and creatinine 13C from arginine 13C6 and thereby rescued cyclocreatine-induced defects on the creatine synthesis pathway alone, without affecting production of glutamate, α-ketoglutarate, and L-arginino-succinate (Figure 4C and Supplementary Figure S4B). Activation of the arginine-creatine pathway by phospho-creatine prevented the loss of viability of human EVI1-positive AML cells induced by cyclocreatine or CKMT1-directed shRNAs, and also maintained the serial replating activity of Evi1-transformed bone marrow cells (Figure 4D–4F).
Figure 4. Blockade of the arginine-creatine pathway upon CKMT1 inhibition impairs both mitochondrial respiration and ATP production in EVI1-positive AML cells.
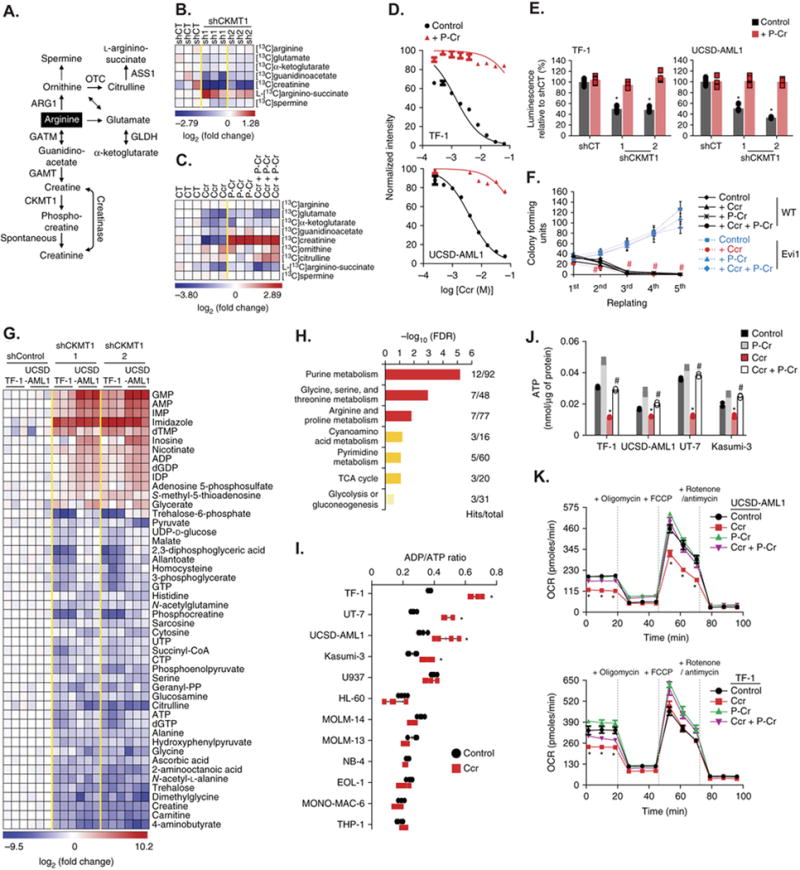
(A) Schematic of arginine metabolism and enzymes involved in this process.
(B–C) Heatmaps of metabolite products of arginine metabolism that are differentially regulated in human UCSD-AML1 cells infected with either control (shCT) or CKMT1-directed shRNAs (B) or in human TF-1 cells treated with 3 mM cyclocreatine (Ccr) for 12 hours in combination with 1 mM phospho-creatine (P-Cr) (C) before a 30-min pulse labeling with L-Arginine 13C6. Metabolites scoring with a p value ≤ 0.05 are shown as log2 fold change normalized to the average control condition. Each column for each condition represents a technical replicate (n=3 per condition).
(D–E) Growth inhibition of human TF-1 and UCSD-AML1 cells treated with increasing concentrations of cyclocreatine (Ccr) (D) or infected with control (shCT) or CKMT1-directed shRNAs (E) in combination with 1 mM phospho-creatine (P-Cr). Error bars represent mean ± SD of five technical replicates.* p value ≤ 0.05 in comparison with shControl calculated using a Mann-Whitney test.
(F) Colony-forming assay for 5 rounds of serial replating from control or Evi1-overexpressing murine LinLow, c-Kit+ bone marrow cells treated with 3 mM cyclocreatine (Ccr) in combination with 1 mM phospho-creatine (P-Cr). # p value ≤ 0.05 calculated using Welch’s t-test in comparison with the control condition. Error bars represent mean ± SEM of three technical replicates.
(G) Heatmap of the top upregulated or downregulated metabolites (p value ≤ 0.05 and absolute log2 fold change ≥ 1.5 in at least one of the two CKMT1-directed shRNAs condition versus average shControl condition) identified by steady-state metabolism profiling in human TF-1 and UCSD-AML1 cells infected with either control (shCT) or two CKMT1-directed shRNAs. Each column for each condition represents a technical replicate (n=3 per condition in two individual human EVI1-positive cell lines).
(H) Pathway analysis integrating enrichment and pathway topology analyses (MetaboAnalyst) of the list of top metabolites from panel (G). Top enriched metabolic pathways are represented based on a FDR ≤ 0.25 and p value ≤ 0.05. “Hits” represents the number of metabolites that scored in the steady-state profile and “Total” represents the number of metabolites present in the given metabolic pathway. Metabolic pathways with a − log10 (FDR) ≥ 1.5 are depicted in red, 1 ≤ − log10 (FDR) < 1.5 are in orange, and − log10 (FDR) < 1 in beige.
(I) ADP / ATP ratio in a panel of 12 human AML cell lines treated with 5 mM cyclocreatine (Ccr) for 12 hours. * p value ≤ 0.05 in comparison with the control condition calculated using a Mann-Whitney test. Error bars represent mean ± SD of four technical replicates.
(J) Intracellular ATP level in the indicated human EVI1-positive AML cell lines treated with 5 mM cyclocreatine (Ccr) in combination with 1 mM phospho-creatine (P-Cr) for 12 hours. * and # p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with the control condition and Ccr-treated condition respectively. Error bars represent mean ± SD of four technical replicates.
(K) Mitochondrial Oxygen Consumption Rate (OCR) of human UCSD-AML1 and TF-1 cells treated with 5 mM cyclocreatine (Ccr) in combination with 1 mM phospho-creatine (P-Cr) for 12 hours prior to measurement under basal conditions, following sequential injections of 1 μM ATP synthase inhibitor oligomycin, 1 μM of the uncoupling agent FCCP, and 1 μM of the complex I and III inhibitors rotenone and antimycin A, respectively. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with the control condition. Error bars represent mean ± SD of 4 technical replicates.
Each experiment was performed at least two independent times except in G and H in which two different EVI1-positive human cell lines transduced with two different CKMT1-directed shRNAs were profiled using metabolomics.
We next used a metabolism profiling approach to characterize the effect of CK pathway alteration on global cell metabolism. Steady state levels of 48 metabolites highly enriched in pathways related to purine metabolism, amino acid metabolism (glycine, serine, threonine, arginine, and proline), and energy metabolism (TCA cycle and glycolysis), were significantly changed by CKMT1 suppression in TF-1 and UCSD-AML1 cells (Figure 4G and 4H). This analysis revealed a significant (p value ≤ 0.05) decrease in intracellular ATP levels accompanied by an increase in ADP and AMP levels (Figure 4G). This increase in the ADP/ATP ratio induced by CK pathway inhibition was only observed in the four AML cell lines expressing high level of CKMT1 – TF-1, UT-7, UCSD-AML1, and Kasumi-3 – but not in those expressing low levels of CKMT1 (Figure 4I). Finally, activation of the creatine pathway by phospho-creatine rescued cyclocreatine-induced decrease in intracellular ATP level, suggesting that the creatine pathway may regulate ATP production in CKMT1-expressing cells (Figure 4J).
To investigate this hypothesis, we evaluated the impact of creatine pathway inhibition on the respiration capacity of the mitochondria, the main organelles involved in ATP production. Measurement of mitochondrial oxygen consumption rate (OCR) reflects the capacity to produce ATP. Cyclocreatine treatment decreased basal mitochondrial respiration capacity in both TF-1 and UCSD-AML1 cells as well as the maximal mitochondrial respiration capacity visualized after FCCP supplementation in UCSD-AML1 cells (Figure 4K). Co-treatment with phospho-creatine counteracted the effect of cyclocreatine and restored normal mitochondrial respiration capacity, suggesting that an active creatine pathway is required for promoting mitochondrial activity.
Finally, we overexpressed a wild-type (WT) or a metabolically inactive mutant form of CKMT1 (C316G21) in two AML cell lines, NB-4 and U937, that do not express endogenous CKMT1 (Supplementary Figure S5A). Wild-type, but not the C316G mutant form of CKMT1, conferred cyclocreatine sensitivity to these two cell types (Supplementary Figure S5B), and cyclocreatine treatment significantly reduced ATP production and basal mitochondrial respiration in cells expressing wild-type CKMT1 (Supplementary Figures S5C and S5D). All together, these data demonstrate that expression of CKMT1 renders mitochondria dependent on creatine pathway activity for ATP production.
Blockade of CK activity diminishes EVI1-expressing cell viability
To determine the transcriptional consequences of CK pathway inhibition in EVI1-expressing cells, we profiled three EVI1-expressing cell lines, TF-1, UT-7 and UCSD-AML1, treated with cyclocreatine for 24 hours using RNA sequencing (RNAseq). We identified 54 and 53 genes significantly 1.5-fold upregulated and downregulated, respectively, compared to control treatment (Figure 5A). An open-ended enrichment analysis was next conducted using a relaxed cyclocreatine signature (defined based on the cut-offs SNR permutation p value ≤ 0.05, Benjamini-Hochberg false discovery rate (FDR) ≤ 0.05 and absolute fold change for log2 (FPKM) scores ≥ 0.5.) on the entire set of signatures from the c2 (MsigDB) and DMAP collections (Supplementary Tables S3 and S4). Gene sets related to cell cycle, energy metabolism and cell stemness were enriched in genes whose expression was suppressed by cyclocreatine and those related to cell differentiation were more enriched in genes whose downregulation was induced by both pro-differentiating agents and cyclocreatine. Finally, amino acid metabolism gene sets were more enriched in genes upregulated by cyclocreatine (Figure 5B, 5C, and Supplementary Table S5). At the intersection of the three enriched functional groups – cell cycle, cell stemness, and cell differentiation – a leading edge analysis isolated a core of ten highly significant genes downregulated by cyclocreatine treatment in the three lines profiled (Supplementary Figure S6A). We validated the transcriptional dowregulation of MYC and CCND1 in all of the EVI1-positive cell lines treated with cyclocreatine by real-time PCR (Supplementary Figure S6B). Cyclocreatine or CKMT1-directed shRNAs increased cell size and cell vacuolization (Figures 5D–5F). This was accompanied by a G0/G1 cell cycle arrest in UCSD-AML1 and Kasumi-3 cell lines while TF-1 and UT-7 underwent apoptosis (Figures 5G and 5H). Finally, expression of the immature cell surface marker CD117 decreased in response to cyclocreatine or CKMT1-direct shRNAs, and this was associated with inhibition of the serial replating activity of UCSD-AML1, TF-1, and UT-7 cells (Figures 5I and 5J).
Figure 5. Inhibition of the creatine kinase pathway alters the viability of EVI-1-positive AML cells via cell cycle blockade and apoptosis induction.
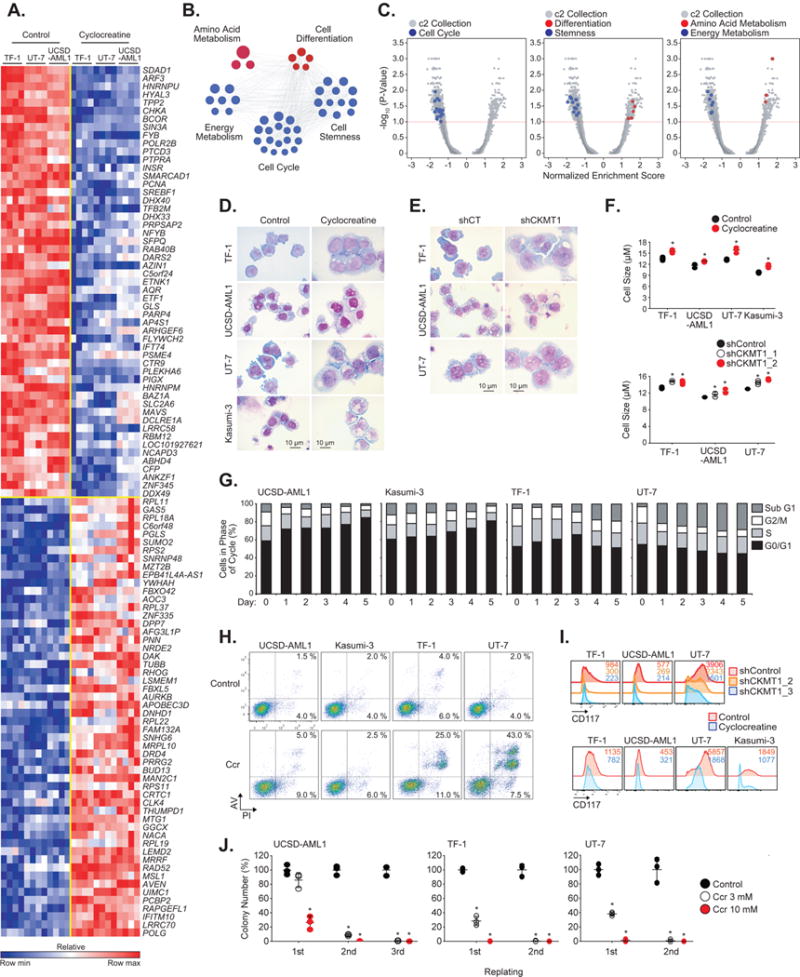
(A) Heatmap of the top common up- and down-regulated genes in human TF-1, UT-7, and UCSD-AML1 cell lines treated with cyclocreatine for 24 hours. p value ≤ 0.05, FDR ≤ 0.05, and absolute fold change for log2 (FPKM) scores ≥ 1.5. Each column for each condition represents an independent biological replicate (n=3 per condition in three individual human EVI1-positive cell lines).
(B–C) A cyclocreatine signature identified by RNAseq was interrogated by GSEA across the MsigDB and DMAP database for vehicle versus cyclocreatine-treated AML cells. (B) Top enriched upregulated (in red) and downregulated (in blue) gene sets upon cyclocreatine treatment are depicted in a functional network. (C) Quantitative comparison of these gene sets of interest (in blue or red) versus all other available gene sets (in gray). Data are presented as a volcano plot of -log10 (p value) versus Normalized Enrichment Score (NES) for each evaluated gene set.
(D–E) May-Grünwald-Giemsa staining of indicated human AML cell lines treated with 8 mM cyclocreatine for 9 days (D), or infected with a control (shCT) or a CKMT1-directed shRNA (E).
(F) Cell size measurement of indicated human AML cell lines treated with 8 mM cyclocreatine for 9 days (top panel) or infected with CKMT1-directed shRNAs (bottom panel). * p value ≤ 0.05 calculated using Welch’s t-test in comparison with the control condition. Error bars represent mean ± SD of three technical replicates.
(G) Cell cycle analysis at the indicated time points of human EVI1-positive cell lines treated with cyclocreatine.
(H) FACS plots showing Annexin V (AV) / propidium iodide (PI) profile of the indicated human AML cell lines treated with 8 mM cyclocreatine (Ccr) for 5 days.
(I) FACS analysis of the expression of CD117 cell surface marker in the indicated human AML cell lines treated with 8 mM cyclocreatine for 9 days (bottom panel) or infected with CKMT1-directed shRNAs (top panel). A representative FACS plot from each cell line is shown.
(J) Colony formation assay for 2 to 3 rounds of serial replating from indicated human AML cell lines treated with either 3 mM or 10 mM cyclocreatine (Ccr). Results represent the average of triplicate assays. * p value ≤ 0.05 calculated using a Welch’s t-test in comparison to the control condition. Error bars represent mean ± SEM.
Each experiment in D–J was performed at least two independent times.
Progression of EVI1-expressing AML depends on CKMT1
We next sought to investigate whether CKMT1 was required for EVI1-dependent AML disease development in vivo. Li et al. generated transplantable primary AMLs by performing retroviral insertional mutagenesis in Mx1-Cre; NrasG12D/+ mice22,23. We compared two Nras mutant AMLs from this screen: NrasG12D + Evi1 contains a clonal Evi1 integration and expresses high transcript levels while the control NrasG12D AML does not. Immunoblot analysis revealed markedly elevated levels of Evi1 and Ckmt1 in the NrasG12D + Evi1 AML (Figure 6A), and this leukemia was also more sensitive to inhibition by cyclocreatine treatment than WT or NrasG12D bone marrow cells (Supplementary Figure S7A). To examine the oncogenic Evi1-dependency on Ckmt1 expression, we transduced NrasG12D and NrasG12D + Evi1 AML cells with a doxycycline-inducible non-targeting control (CT) or two Ckmt1-directed shRNAs. Venus-positive transduced cells were then sorted and injected into irradiated recipient mice. Once disease onset was detected, mice were treated with doxycycline to induce hairpin expression in tandem with an RFP reporter cassette, and knockdown was confirmed by immunoblot (Figure 6B). Mice transplanted with NrasG12D AML cells transduced with either control or Ckmt1-directed shRNAs, or NrasG12D + Evi1 AML cells expressing shControl cells succumbed to aggressive AML with a median latency of approximately 20 days. By contrast, recipients of NRasG12D + Evi1 AML cells expressing either shCkmt1_2 or shCkmt1_3 exhibited attenuated disease and survived for approximately 40 days (Figure 6C). Splenomegaly and leukocytosis, which reflect the AML disease burden, were also markedly reduced in mice transplanted with NRasG12D + Evi1 AML cells expressing either shCkmt1_2 or shCkmt1_3 (Figure 6D), and the splenic proportion of Gr-1+/Mac-1+ granulocytes was substantially reduced compared to NRasG12D + Evi1 shControl mice (Figure 6E).
Figure 6. CKMT1 knockdown preferentially impairs development of EVI1-positive human and murine myeloid leukemias, without affecting normal progenitor cell viability.
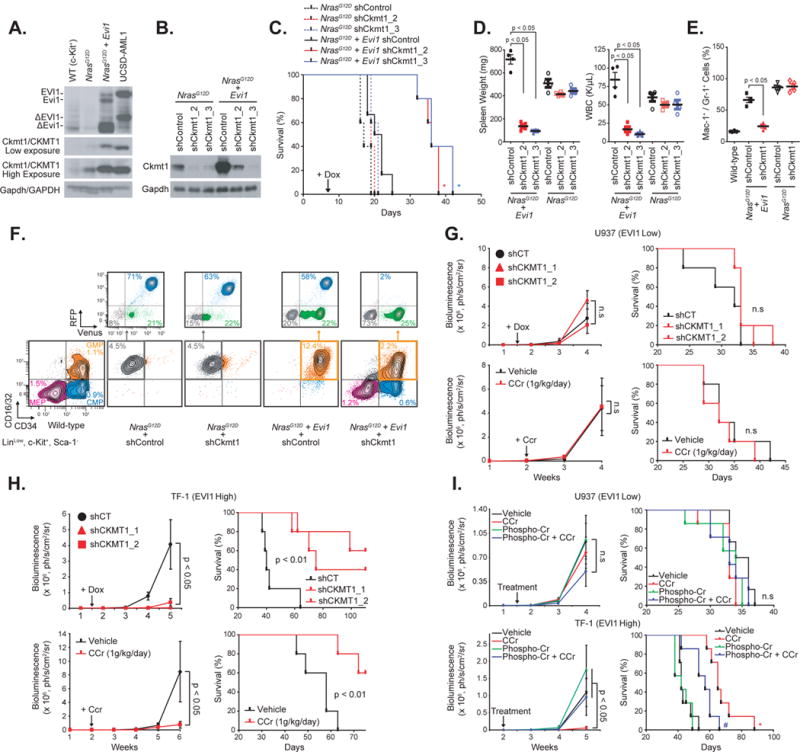
(A) Western immunoblot for Evi1/EVI1, Ckmt1/CKMT1, and Gapdh/GAPDH (loading control) in murine bone marrow cells from wild-type (WT), N-RasG12D, or N-RasG12D + Evi1 mouse. Human UCSD-AML1 cell line is used as positive control for EVI1 and CKMT1 expression.
(B) Immunoblot indicating the level of Ckmt1 knockdown in murine RFP-sorted bone marrow cells seven days post-doxycycline induction.
(C) Kaplan-Meier curves showing overall survival of mice (n = 5 for each group except for N-RasG12D + Evi1 shCT + dox group, for which n = 6) transplanted with cells expressing each combination of indicated constructs. Statistical significance determined by log-rank (Mantel-Cox) test. * p value ≤ 0.05 by comparison with shControl (shCT) within the N-RasG12D or the N-RasG12D + Evi1 group.
(D) Spleen weight and white blood cell (WBC) count of four mice per group when shControl mice became moribund. P value calculated using a Mann-Whitney test. Error bars represent mean ± SEM.
(E) Proportion of Mac-1+ / Gr-1+ cells in spleens from four mice per group. p value calculated using a Mann-Whitney test. Error bars represent mean ± SEM.
(F) Proportion of CMP (CD16/32−/CD34+), GMP (CD16/32+/CD34+), and MEP (CD16/32−/CD34−) cell populations on gated LinLow/Sca-1−/c-Kit+ myeloid progenitors when shControl became moribund. RFP and Venus expression were evaluated on every leukemic cell population. One representative moribund mouse from each group is shown.
(G–H) Low U937-Luc (G) and high TF-1 CSF2+-Luc (H) EVI1-expressing human cell lines were either infected with two doxycycline-inducible CKMT1-directed miR30-shRNAs (top panel) before tail vein injection, or treated with 1g/kg/day cyclocreatine (Ccr) 10 days post-injection (bottom panel). TF-1-Luc cells were engineered to overexpress CSF2 for efficient engraftment in mice. Bioluminescence was quantified weekly as a measure of disease burden. Arrows indicates beginning of doxycycline (Dox) or cyclocreatine (Ccr) treatments. Data are represented as mean ± SEM of 5 mice per cohort. p value was calculated on the latest time point using nonparametric Mann-Whitney test. Kaplan-Meier curves showing overall survival of each group of mice is shown in the right panel. Statistical significance determined by log-rank (Mantel-Cox) test. n.s, not significant.
(I) Low U937-Luc (top panel) and high TF-1 CSF2+-Luc (bottom panel) EVI1-expressing human cell lines were treated with 1g/kg/day cyclocreatine (Ccr) in combination with phospho-creatine (P-Cr) at the indicated time point. Bioluminescence was quantified weekly as a measure of disease burden. Data are represented as mean ± SEM of seven mice per cohort. p value was calculated on the latest time point using a nonparametric Kruskall-Wallis test and Dunn’s multiple comparisons test. Kaplan-Meier curves showing overall survival (right panel). Statistical significance determined by log-rank (Mantel-Cox) test. * and # p value ≤ 0.05 by comparison with vehicle- and Ccr-treated groups respectively. n.s, not significant.
While all NrasG12D + Evi1 mice showed a recurrent expansion within the LinLow/Sca-1−/c-Kit+/CD16/32+/CD34+ GMP compartment in comparison to wild-type mice, NrasG12D mice exhibited dominant growth of a less immature LinLow/Sca-1−/c-Kit+/CD16/32+/CD34− population (representative examples in Figure 6F). In NrasG12D mice, Ckmt1 knockdown did not alter the growth of the leukemic population compared to shControl mice. In recipients transplanted with NrasG12D + Evi1 AML cells, Ckmt1 suppression suppressed the expansion of the RFP+/Venus+ leukemic population from 58% in shControl mice to 2% in shCkmt1 group, thus reducing the overall growth advantage of the GMP fraction over MEP and CMP populations. To assess the effects of Ckmt1 deletion on normal hematopoiesis, CD45.2 bone marrow cells infected with shCT were transplanted into lethally irradiated recipient mice with an equal number of CD45.1 competitor bone marrow cells infected with Ckmt1-directed shRNAs (Supplementary Figure S7B). Chimerism was confirmed based on evaluation of the ratio of CD45.1 to CD45.2 in bone marrow of recipient mice. Reducing Ckmt1 expression did not alter the repopulating potential on normal hematopoietic cells in competitive transplantation assays, suggesting no toxic effect of Ckmt1 targeting in normal bone marrow cells (Supplementary Figure S7C).
Finally, we investigated the effects of either doxycycline-inducible CKMT1-directed shRNAs or cyclocreatine treatment on leukemia development in xenografts of EVI1-low U937-Luc and EVI1-high TF-1 CSF2+-Luc human AML cell lines. Leukemia burden was monitored in recipients treated therapeutically with doxycycline or cyclocreatine after detectable disease burden was increasing at day 8 in U937 cells and day 10 in TF-1 cells (Supplementary Figure S7D and S7E). Consistent with our in vivo analysis of primary murine Nras mutant AMLs, pharmacologic or genetic inhibition of the CKMT1-dependent pathway blocked disease progression and prolonged the survival of mice injected with TF-1 cells but not with U937 cells (Figures 6G and 6H). Co-treatment with phospho-creatine significantly rescued the effects of cyclocreatine in mice injected with TF-1 but had no effect in the U937 cell leukemia model (Figure 6I).
DISCUSSION
Here we found that overexpression of the zinc finger transcription factor EVI1 remodeled the metabolic landscape of immature hematopoietic cells, particularly by impacting cell energy-related pathways, arginine metabolism, and de novo synthesis of purines and pyrimidines. The broad transcriptional effects of EVI1 overexpression on these different metabolic pathways are reminiscent of a recent study which identified glutaminolysis as a critical pathway for T-cell acute lymphoblastic leukemia growth downstream of the hyperactivated, mutant form of the transcription factor NOTCH1 and as a key determinant of the response to anti-NOTCH1 therapies24. Other results suggest that glycolysis is important for MLL-AF9-driven AML maintenance and progression and that those leukemic cells are more sensitive to the inhibition of aerobic glycolysis than normal hematopoietic cells25. This supports the idea that the pro-leukemic effects of EVI1 expression in AML may be mediated, at least in part, by perturbations of cell metabolism and that targeting these metabolic dependencies may be promising therapeutic avenues in EVI1-positive leukemias.
Our results indicate a higher dependency of EVI1-positive leukemias, compared to other subgroups of AML, on the creatine kinase pathway. Creatine is one of the end products of the arginine pathway, which is altered upon EVI1 overexpression. In healthy cells, arginine can be produced by the ornithine-citrulline-arginine cycle. However, recent investigations identified that the majority of blasts from patients with AML are deficient in enzymes specialized in promoting this cycle and are therefore strongly reliant on extracellular arginine availability26,27. This study suggests that a constitutive blockade of the ornithine-citrulline-arginine cycle would preferentially redirect transported arginine through other arginine-dependent metabolic pathways such as the creatine pathway. Interestingly, a mycoplasma-derived enzyme, ADI-PEG 20, that degrades extracellular arginine and thus prevents intracellular import of arginine showed good in vitro anti-leukemic efficacy in 19 samples from a cohort of 38 primary AML patient samples28. From the entire cohort, only one AML sample harbored a translocation involving EVI1, and this sample was sensitive to ADI-PEG 20. This finding would be consistent with the dependency of EVI-positive AML on the creatine kinase pathway and suggests that EVI1-positive patients may also be good responders to a therapy targeting the import of arginine.
CKMT1 has two main functions in energy metabolism: it buffers the cellular ATP pool by maintaining cytosolic concentrations of phospho-creatine for high cellular energy demand to regenerate ATP, and it maintains an energy shuttle between sites of energy supply and sites of energy demand using the easily diffusible creatine29. Our results showed that CKMT1 directly impacts mitochondrial respiration and ATP production. This is reminiscent of the role of hexokinases, another class of mitochondria-coupled enzymes that utilize ATP produced by mitochondria to catalyze conversion of glucose into glucose-6-phosphate, the first rate-limiting step of glycolysis30. In turn, the ADP formed from this reaction stimulates mitochondrial respiration31,32. Similarly, the increase of the creatine/phospho-creatine interconversion rate subsequent to CKMT1 overexpression seems to regulate mitochondrial respiration and then influence mitochondrial ATP production. The expression of CKMT1 alone is sufficient to reprogram mitochondrial activity, suggesting that CKMT1 upregulation itself may be one of the main contributors to metabolic perturbations that are necessary to the pro-leukemic effects of EVI1 in AML.
Although CKMT1 is ubiquitously expressed, several studies have shown that some cancer types, such as mesotheliomas, small cell lung and brain tumors, expressed higher CK protein levels which predict higher sensitivity to cyclocreatine33. Moreover, high CKMT1 expression is a marker of poor prognosis for patients with breast cancer34. We established that EVI1 promotes CKMT1 expression by direct repression of the transcription factor RUNX1. A transcriptomic analysis of 269 patients with AML revealed that those who harbor t(8;21) or t(11q23) alterations exhibit a wild-type RUNX1 signature, while patients bearing del(7q)/7− or inv3/t(3;3) – responsible for EVI1 overexpression – express a RUNX1 mutation-derived gene expression signature35. This corroborates our observation that CKMT1, whose expression is repressed by RUNX1, is predominantly upregulated in the EVI1-overexpressing leukemia subgroup.
The direct targeting of aberrant transcription factors as a therapeutic strategy remains a significant challenge in cancer therapy. An alternative approach is to target the unique liabilities engendered by the altered cellular circuitry present in these transcription-factor driven malignancies. To this end, we have integrated “omic” approaches to identify CKMT1 as a druggable liability in EVI-positive AML. In considering the clinical translation of this finding, it is notable that cyclocreatine can be found in marketed nutritional and sports supplements. In addition, while not specifically for the mechanism of CKMT1 inhibition, but rather for use as a cell permeable analogue of creatine, cyclocreatine was granted orphan designation by the European Commission to Pharma Gateway AB, Sweden, in 2016 for the treatment of creatine deficiency syndromes. Despite enthusiasm about the likely safety of this molecule for multiple disease indications, clinical trials testing the safety and efficacy of cyclocreatine, however, will still need to be conducted. Another path forward for targeting CKMT1 might be the development of new selective agents targeting this enzyme. In summary, this study supports a potential therapeutic avenue for targeting the CK pathway in EVI1-positive AML, which remains one of the worst outcome subtypes of this disease.
ONLINE METHODS
Plasmids and shRNA Constructs
Empty and Evi GCDN vectors were a kind gift from Drs. Tomohiko Sato and Mineo Kurokawa (The University of Tokyo, Tōkyō, Japan). Empty and HA-tagged pHAGE-CMV-CKMT1 and pHAGE-CMV-RUNX1 vectors were generated by enzyme-mediated site-specific recombination between pENTR223-CKMT1 (Harvard PlasmID database, HsCD00372650) and pENTR223-RUNX1 (Harvard PlasmID database, HsCD00378663) donor vectors and pHAGE-CMV-HA destination vector (gift from Dr. J. Wade Harper, Harvard Medical School, Boston, MA, USA).
shRNA constructs targeting human CKMT1 (shCKMT1_1 and shCKMT1_2), EVI1 (shEVI1_1, shEVI1_2, shEVI1_4), CS (shCS_1, shCS_2, shCS3), SDHA (shSDHA_1, shSDHA_2, shSDHA_3), or RUNX1 (shRUNX1_2, shRUNX1_6) – sequences listed below – were designed by adapting BIOPREDsi small interfering RNA predictions. shRNAs were cloned into the TRMPVIR retroviral vector36, containing a miR30 expression cassette, as 116-nt XhoI–EcoRI fragments, which were generated by amplifying 97-mer oligonucleotides (Invitrogen) using 5′miR30-XhoI (TACAATACTCGAGAAGGTATATTGCTGTTGACAGTGAGCG) and 3′miR30-EcoRI (ACTTAGAAGAATTCCGAGGCAGTAGGCA) primers and the Vent polymerase kit (Invitrogen) with the following conditions: 50 μl reaction containing 0.05 ng oligonucleotide template, 1× Vent buffer, 0.3 mM of each dNTP, 0.8 μM of each primer, and 1.25 U Vent polymerase; cycling: 94 °C for 3 min; 35 cycles of 94 °C for 30 s, 54 °C for 30 s, and 75 °C for 20 s; 75 °C for 5 min.
| Designation | Species | Accession Number | Target Sequence |
|
| |||
| shCKMT1_1 | Homo Sapiens | NM 020990.3 | ATCACCCGTGTATGATCCTCC |
| shCKMT1_2 | Homo Sapiens | NM_020990.3 | TTTACTGGCATCTAGATCCGT |
| shCS_1 | Homo Sapiens | NM 004077.2 | ATCGCGGATCAGTCTTCCTTA |
| shCS_2 | Homo Sapiens | NM 004077.2 | AAGAACTGATGTTTCATAGAC |
| shCS_3 | Homo Sapiens | NM_004077.2 | TGTAACAGCTGCACTGAGCTG |
| shEVI1_1 | Homo Sapiens | NM 001105077.31 NM 005241.3|NM 001105078.3|NM 004991.3|NM 001163999.1|NM 001164000.1 | TTTCTGTGAAGTAAGCATCTT |
| shEVI1_2 | Homo Sapiens | NM 001105077.31 NM 005241.3|NM 001105078.3|NM 004991.3|NM 001163999.1|NM 001164000.1 | TTGCACTTGATTTGGGTTCTG |
| shEVI1_4 | Homo Sapiens | NM_001105077.31 NM_005241.3 | NM_001105078.3 | NM_004991.31 NM_001163999.1| NM_001164000.1 | TTCCAGTT CAGAATGAGGCGA |
| shRUNX1_2 | Homo Sapiens | NM 001001890.2| NM 001754.4|NM 001122607.1 | TGATCATCTAGTTTCTGCCGAT |
| shRUNX1_6 | Homo Sapiens | NM 001001890.2| NM 001754.4|NM 001122607.1 | TCCACTGTGATTTTGATGGCTC |
| shSDHA_1 | Homo Sapiens | NM_004168.2 | CAGATGCCCTCTTGTTCCCAT |
| shSDHA_2 | Homo Sapiens | NM_004168.2 | ATGACTCTTCGATGCTCAGGG |
| shSDHA_3 | Homo Sapiens | NM_004168.2 | AACAGTGTTCTTTGCTCTTAT |
| shControl | AGGAATTATAATGCTTATCTA | ||
|
| |||
shRNA constructs targeting murine Ckmt1 (shCkmt1_2 and shCkmt1_3) were purchased from GE Healthcare and were subcloned from the GIPZ vector into the TRIPZ-Venus lentiviral vector. The TRIPZ-Venus vector was designed by substitution in the TRIPZ vector (GE Healthcare), of the puromycin resistance cassette for the sequence encoding the yellow-green fluorescent protein Venus reporter isolated from the TRMPVIR vector. Non-inducible human CKMT1-directed shRNAs (CKMT1_3 and CKMT1_4) and murine Ckmt1-directed shRNAs used for the in vivo chimerism assay were purchased from TransOMIC Technology Inc.
For virus production, 12 μg of the above plasmids and 6 μg ψ-eco (for retroviral infection of murine cells), 6 μg pCMV-GAG/POL and pCMV-VSVG (for retroviral infection of human cells), or 6 μg pCMV8.9 or psPAX2 and pCMV-VSVG (for lentiviral infection of human and murine cells) packaging vectors were transfected into the 293T packaging cell line using X-tremeGENE 9 (Roche), and the resulting viral supernatants were harvested as previously described37. The lentiviral viruses were then concentrated using PEG-it™ Virus Precipitation Solution (SBI System Biosciences).
Pooled shRNA Screening
A custom library of 361 hairpins directed against 67 human genes involved in glycolysis, pentose phosphate pathway, TCA cycle, and related metabolic pathways with at least 3 hairpins per gene, was cloned into a retroviral pMLP2.1 vector and was purchased from TransOMIC Technology Inc. To preserve library complexity, a minimum of 1000-fold coverage of the shRNA library was maintained at each step of the screen. Two series of quadruplicates, from TF-1 and UCSD-AML1 cells, were spin-infected using plates coated with 10 μg/mL retronectin (Takara) per the manufacturer’s instructions. Fresh viral supernatant was added onto each retronectin-coated well before spinning for two hours at 2000g at 32°C to allow virus binding to the retronectin. 7.5×106 cells in 6 mL transplant media containing 5 μg/mL polybrene (Sigma) and 7.5 mM HEPES buffer (Sigma) were then seeded into each of these wells and centrifuged for 1 hour at 1800g to promote cell transduction. The final transduction rate estimated was between 5% and 10% of GFP-positive cells. Cells were expanded for 48 hours before selection with 1 μg/ml puromycin. At 72 hours post-puromycin selection, half of the volume of each quadruplicate was pelleted and frozen down to constitute the day 0 time point, and cells were maintained in culture and split every two days to keep at least 6×106 cells per replicate until day 36. Genomic DNA was isolated by proteinase K digestion and isopropanol precipitation from each pellet at day 0 and day 36 time-points. The antisense strand of shRNA was amplified from genomic DNA using primers that include 1-basepair mutations to barcode individual samples. Hairpins were amplified in multiple 50 μl reactions using HotStar Taq (Qiagen). After PCR amplification, samples were pooled and prepared for sequencing with Illumina’s genomic adaptor kit. At least 41 bases of the PCR product were sequenced with an Illumina HiSeq 2000 machine. shRNAs with less than 100 reads in the input sample were excluded from further analysis, and read numbers for each shRNA were normalized to the total read numbers per sample to allow for cross-comparison between samples. We then compared using an EdgeR/RIGER method the hairpin representation between day 0 and day 36 and thus identified genes whose depletion blocked or enhanced cell proliferation. EdgeR (R/Bioconductor v.3.3 package38) provided the statistical analysis of count data from the day 0 versus day 36 time-points per cell lines. Next, the RNAi gene enrichment ranking (RIGER) score was given by examination of the position of each hairpin targeting each gene in the full ranked list of the 361 shRNAs. The enrichment scores and the gene ranking were finally computed based on the RIGER v.0.1 module39 available from the GENE-E platform (http://www.broadinstitute.org/cancer/software/GENE-E/). The GENE-E platform is now replaced by the Morpheus platform which is available at the same URL as GENE-E.
Metabolite Profiling for Steady-state and Targeted Flux Analyses
To determine the relative levels of intracellular metabolites, extracts were prepared and analyzed by LC/MS/MS. Twenty-four hours after sorting and 16 hours prior to metabolite extraction, 7.5×106 cells were plated in triplicate in an incubation media: DMEM media with no glucose, no glutamine, and no sodium pyruvate (Gibco) supplemented with 1 g/L D-glucose (Sigma), 1.5 mM L-glutamine (Sigma), 5 mM sodium pyruvate (ThermoFisher Scientific), 4 g/L BSA (Sigma), 1 μg/ml insulin (#91077, Sigma), 30 μg/mL transferrin (#T0665, Sigma), 20 ng/ml IL-3 (#213-13, Peprotech), 20 ng/ml IL-6 (#216-16, Peprotech), 100 ng/ml FLT3-Ligand (#250-31L, Peprotech), and 100 ng/ml SCF (#250-03, Peprotech). For L-Arginine 13C6 flux studies, cells were washed twice with PBS and then incubated in DMEM no glutamine, no lysine, and no arginine media supplemented with 466 μM L-Arginine 13C6, 1 mM Lysine, 1.5 mM L-glutamine, 5 mM sodium pyruvate, and 10% dialyzed FBS. Metabolites were extracted on dry ice with 4 mL of 80% methanol (−80°C), as described previously40. Insoluble material was pelleted by centrifugation at 3000g for 5 min, followed by two subsequent extractions of the insoluble pellet with 0.5 ml of 80% methanol, with centrifugation at 16000g for 5 min. The 5 ml metabolite extract from the pooled supernatants was dried down under nitrogen gas using an N-EVAP (Organomation Associates, Inc).
Dried pellets were re-suspended using 20 μL HPLC grade water for mass spectrometry. 10 μl were injected and analyzed using a 5500 QTRAP triple quadrupole mass spectrometer (AB/SCIEX, BIDMC Mass Spec Core, Boston, MA, USA) coupled to a Prominence UFLC HPLC system (Shimadzu) via selected reaction monitoring (SRM) of a total of 292 endogenous water soluble metabolites for steady-state analyses of samples. Some metabolites were targeted in both positive and negative ion mode for a total of 391 SRM transitions using pos/neg polarity switching. ESI voltage was +4900V in positive ion mode and –4500V in negative ion mode. The dwell time was 3 ms per SRM transition and the total cycle time was 1.55 seconds. Approximately 10–14 data points were acquired per detected metabolite. Samples were delivered to the MS via normal phase chromatography using a 4.6 mm i.d × 10 cm Amide Xbridge HILIC column (Waters Corp.) at 350 μL/min. Gradients were run starting from 85% buffer B (HPLC grade acetonitrile) to 42% B from 0–5 minutes; 42% B to 0% B from 5–16 minutes; 0% B was held from 16–24 minutes; 0% B to 85% B from 24–25 minutes; 85% B was held for 7 minutes to re-equilibrate the column. Buffer A was comprised of 20 mM ammonium hydroxide/20 mM ammonium acetate (pH=9.0) in 95:5 water:acetonitrile. Peak areas from the total ion current for each metabolite SRM transition were integrated using MultiQuant v2.0 software (AB/SCIEX). For stable isotope labeling experiments, custom SRMs were created for expected 13C incorporation in various forms for targeted LC/MS/MS. Peak areas of metabolites detected by mass spectrometry were normalized to median and then normalized to protein concentrations. A Student’s t-test was performed to assess significance of all pairwise comparisons. All data are log2-transformed and normalized against the average control condition. No or small variations in metabolite production compared to average control condition appears closest to the white color on the heatmap. Decreased or increased metabolite production compared to the average control condition is represented by a gradient of color from blue to red respectively. The MetaboAnalyst software (www.metaboanalyst.ca) was used to perform pathway enrichment analysis on the set of top metabolites hits identified by steady-state profiling. No specific manual settings were needed to analyze the list of metabolites.
ChIP-sequencing and ChIP-qPCR
A Runx1 gene track generated by ChIP-sequencing for the analysis of murine Evi1 binding peaks was obtained from Dr. Archibald S. Perkins (University of Rochester Medical Center, New-York, NY, USA). The human RUNX1 gene track was obtained from publically available ChIP-sequencing data generated by Bard-Chapeau et al.19. The conservation score between species was computed based on phasCons data for hg18 and mm941. Gene tracks and EVI1-binding peaks were aligned using IGV software42.
ChIP-qPCR was performed as described by Bernt et al.43. Briefly, crosslinking was performed in cell culture medium containing 1% formaldehyde with gentle rotation for 5 minutes at room temperature. Fixation was stopped by the addition of glycine (125 mM final concentration). Fixed cells were washed twice in ice-cold PBS and then resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, supplemented with Complete-mini-protease inhibitor cocktail (Roche)). Chromatin was sheared to approximately 200bp fragments by Covaris ultra-sonication and immunoprecipitated. Precipitates were washed sequentially with ice cold low salt wash (0.1% SDS, 1% Triton-X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), high salt wash (0.1% SDS, 1% Triton-X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), LiCl wash (0.25 M LiCl, 1% IGEPAL CA-630, 1% deoxycholic acid, 1 mM EDTA, 10 mM Tris-HCl, pH 8.1) and TE wash (1 mM EDTA, 10 mM Tris-HCl, pH 8.1) and eluted in elution buffer (1% SDS, 0.1M NaHCO3). Eluted DNA fragments were analyzed by qPCR. Primers used for ChIP-qPCR are listed below.
| Targets | Species | Forward | Reverse |
|
| |||
| Runxl | Mus Musculus | CTCCCTCCCTCTCTCCTCTATTAAA | GAGTTCTTGTCTTTCTGCGTCATG |
| Ckmtl | Mus Musculus | GTCTGAGTATGCACTTCGCAAAA | GGTCATCAGGGAGTCTCTGTCTTC |
| CKMT1 | Homo Sapiens | AGACCTGTGCCTGCCTGTTC | GGAGGAGAAGGTAAAGTGAGGTGAA |
|
| |||
Western Immunoblotting
Western immunoblotting was performed as described previously44 using cell lysates normalized for total protein content. Lysates were boiled in Laemmli sample buffer and run on SDS–polyacrylamide gel electrophoresis (PAGE) prior to transfer to nitrocellulose membranes, and immunoblotting. A list of antibodies used for western immunoblotting is listed below.
Flow Cytometry
Cell lines were washed in PBS-0.1% BSA-2 mM EDTA before a 45 minute incubation with an anti-human CD117 antibody. Cells were then washed twice with PBS containing 0.1% BSA and 2 mM EDTA before analysis with BC FACScanto II (BD Biosciences).
Primary patient AML cells were washed in PBS containing 0.1% BSA and 2 mM EDTA before a 45-minute incubation with a combination of anti-human PE-Cy7 CD13 and CD33 (used at 1/50) or PE-Cy7 isotype control antibodies. Cells were then washed twice with PBS-0.1% BSA-2 mM EDTA before a 20-minute fixation with BD Cytofix/CytoPerm Fixation and Permeabilization Solution (BD Biosciences). Cells were then washed three times with BD Perm/Wash Buffer and incubated for 45 minutes at 4°C with a non-conjugated anti-human EVI1 antibody (used at 1/50), or the corresponding isotype control antibody. Cells were washed three times with Perm/Wash Buffer and were incubated for 35 minutes with an Alexa Fluor 488 goat anti-rabbit IgG antibody used at 1/500 (Invitrogen). Cells were washed three times in Perm/Wash Buffer and analyzed using a BD FACSCanto II analyzer. To account for any background fluorescence, the EVI1 mean fluorescence level was normalized to the mean fluorescence level of its corresponding isotype control. This normalized value was then scaled for each patient using the Z-Score calculation across all patient samples.
Murine bone marrow and spleen were collected from each organ, washed with PBS containing 0.1% BSA and 2 mM EDTA and lysed for 10 minutes with red blood cell lysis buffer (Sigma). After two washing steps with PBS containing 0.1% BSA and 2 mM EDTA, bone marrow cells were stained for 30 minutes at 4°C with a combination of anti-lineage cocktail, Sca-1, c-KIT, CD16/32 and CD34 antibodies and spleen cells were incubated for 5 minutes at 4°C with mouse BD Fc Block™ (BD Biosciences) before staining with the combination of Mac-1/Gr-1 antibodies. Stained cell subpopulations were analyzed with BD LSR-II analyzer (BD Biosciences).
A list of antibodies used for western immunoblotting and flow cytometry is listed below.
| Targets | Conjugate | References | Manufacturer |
|
| |||
| ACTIN | Non | MS-1295-P | Neomarkers |
| CD5 | APC | 17-0051 | eBioscience |
| CD13 | PE-Cy7 | 25-0138-42 | eBioscience |
| CD16/32 | PerCP-Cy5.5 | 45-0161-82 | eBioscience |
| CD33 | PE-Cy7 | 25-0338-42 | eBioscience |
| CD34 | eFluor 450 | 48-0341-82 | eBioscience |
| CD45.1 | PE-Cy7 | 25-0453-82 | eBioscience |
| CD45.2 | APC | 17-0454-82 | eBioscience |
| CD45R | APC | 17-0452 | eBioscience |
| CD117 | PE-Cy7 | 313212 | BioLegend |
| CD127 | APC | 17-1271 | eBioscience |
| c-KIT | PE-Cy5 | 105810 | BioLegend |
| CKMT1 | Non | H00001159M16 | Abnova |
| CKMT1 | Non | H00001159-D01 | Abnova |
| CS | Non | 14309S | Cell Signaling Technology |
| EVI1 | Non | 2593 | Cell Signaling Technology |
| Evi1 | Non | Custom | Dr. Archibald Perkins |
| GR-1 | APC | 17-5931-81 | eBioscience |
| GAPDH | Non | sc-25778 | Santa-Cruz Biotechnology |
| H3K4Me3 | Non | Ab8580 | Abcam |
| H3K27Ac | Non | Ab4729 | Abcam |
| H3K27Me3 | Non | Ab6002 | Abcam |
| HSP60 | Non | sc-1722 | Santa-Cruz Biotechnology |
| HSP90 | Non | sc-13119 | Santa-Cruz Biotechnology |
| Lineage | APC | BDB558074 | BD Biosciences |
| Mac-1 | PE-Cy7 | 101216 | BioLegend |
| RUNX1 | Non | Ab23980 | Abcam |
| RUNX1 | Non | 8529 | Cell Signaling Technology |
| Sca-1 | PE-Cy7 | 25-5981-82 | eBioscience |
| SDHA | Non | 11998P | Cell Signaling Technology |
| VINCULIN | Non | Ab18058 | Abcam |
|
| |||
Genome-wide Expression Analysis
Genome Profiling of Evi1-overexpressing Murine Progenitor Cells
eGFP-positive control and Evi1-overexpressing progenitor cells were sorted and total RNA was extracted and profiled using the GeneChip Mouse Gene 1.0 ST Array from Affymetrix (GSE3910316).
Comparative Marker and GSEA Analyses
The 6 samples in the GSE39103 data available from the Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) were separated into 2 groups: control versus Evi1-overexpressing samples. The Comparative Marker Selection module from GenePattern v3.9.345 was used to identify individual genes differentially expressed between the two groups. A permutation p value ≤ 0.05, a Benjamini–Hochberg false discovery rate (FDR) ≤ 0.05 accounting for multiple hypothesis testing, and an absolute fold change ≥ 2 served as cutoff for significant genes. The Gene Set Enrichment Analysis (GSEA) v.2.1.0 software46,47 was used to identify pathways, or groups of functionally related genes, deregulated by Evi1 overexpression. The goal of GSEA is to identify groups of genes sharing common biologic function (gene sets), which are distributed at the top or at the bottom of the ranked list of differentially expressed genes. The significance of the enrichment score is estimated on the basis of a permutation p value and adjusted for multiple hypotheses testing through FDR. The set of differentially expressed genes accounting for the enrichment signal is called the leading edge. GSEA was run on the c2 collection of 4725 curated gene sets from MSigDB v.5.0 (http://software.broadinstitute.org/gsea/msigdb/). Gene sets with less than 15 genes or more than 500 genes were excluded from the analysis. Gene sets with a nominal p value ≤ 0.05 and FDR ≤ 0.25 were considered significant. The gene ranking metric in the weighted enrichment score was the 2-sided SNR, and the p values were calculated using 1,000 permutations of the phenotype.
Metabolic Network
Each metabolic gene deregulated upon Evi1 overexpression was assigned to a functional group based on the c2 collection from MSigDB (http://www.genomespace.org). The enrichment results were visualized with the Enrichment Map module,48,49 which organizes the significant gene sets into a network called an “enrichment map.” In the enrichment map, the nodes correspond to gene sets and the edges reflect significant overlap between the nodes according to a two-tailed Fisher exact test. The hubs correspond to collections of gene sets with a unifying class label according to gene ontology (GO) biologic processes. The size of the nodes is correlated with the number of genes in the gene set.
Gene Expression Profiling of Cyclocreatine Treatment
TF-1, UT-7 and UCSD-AML1 cells were treated in quadruplicate with either vehicle or 3 mM cyclocreatine for 24 hours. Total RNA was extracted and profiled by RNA sequencing (HiSeq, Illumina) at the BioMicroCenter, Massachusetts Institute of Technology (Cambridge, MA, USA). The total number of reads for individual samples ranged from 44 to 48 Mb. Quality control tests for the unmapped reads were performed using the FASTQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The reads were aligned to the GRCh37 (hg19) GENCODE v17 annotated human reference genome by using TopHat250. FPKM scores for genes were computed based on the RSEM R software package51. Expression data was evaluated as log2 (FPKM). The significance of the differential expression between the control and cyclocreatine treatment phenotypes was estimated using the EBSeq method implemented in R (Bioconductor v.3.352), based on the significance cutoff 0.05 for the posterior probability. The aligned RNASeq data is available at the GEO repository (GSE86151).
Comparative Marker Analysis
The 24 samples available in the RNAseq data were separated into two groups: 12 =vehicle-treated and 12 =cyclocreatine treated. The Comparative Marker Selection module from GenePattern v.3.9.345 was employed to identify individual genes that were differentially expressed between the two conditions. The analysis was performed by applying a 2-sided Signal-to-Noise Ratio (SNR) test followed by 1000 permutations of phenotype labels. The settings for the SNR parameters were log-transformed-data:no, complete:no, balanced:no, smooth p values: yes.
A “core” cyclocreatine signature consisting of 54 genes upregulated and of 53 genes downregulated between the vehicle and cyclocreatine-treated samples was defined based on the cut-offs SNR permutation p value ≤ 0.05, Benjamini-Hochberg false discovery rate (FDR) ≤ 0.05, and absolute fold change for log2 (FPKM) scores ≥ 1.5. In addition, a “relaxed” cyclocreatine signature consisting of 366 upregulated and 474 downregulated genes between the two conditions was defined based on the cut-offs SNR permutation p value ≤ 0.05, Benjamini-Hochberg false discovery rate (FDR) ≤ 0.05 and absolute fold change for log2 (FPKM) scores ≥ 0.5. The “core” cyclocreatine signature is presented in Supplementary Tables S3 and S4.
GSEA analysis and Scatter Plot
The Gene Set Enrichment Analysis (GSEA v2.1.0) software46,47 was used to identify functional associations of the molecular phenotypes induced by cyclocreatine with a compendia of gene signatures including the MSigDB v5.047,53 collections c2 of 4,725 curated gene sets and c6 of 189 oncogenic signatures, the DMAP collection of hematopoietic lineage differentiation signatures53, and with four AML differentiation gene signatures available from Gene Expression Omnibus (GEO) database: Vitamin D- and PMA-induced differentiation signatures (accession no. GSE99554); JQ1-induced differentiation signature (accession no. GSE2979955), and DOT1L inhibitor-induced differentiation signature (accession no. GSE2982856). Gene sets with less than 15 genes or more than 500 genes were excluded from the analysis. Gene sets with an FDR ≤ 0.25 and a nominal p value ≤ 0.05 were considered significant hits. The results were plotted on volcano plots and visualized on a functional network called an “enrichment map”. In the enrichment map, the nodes correspond to gene sets and the edges reflect significant overlap between the nodes according to a two-tailed Fisher exact test. The size of the nodes is correlated with the gene set enrichment significance (−log10 (p value)). The hubs correspond to collections of gene sets with a unifying functional category label.
In Silico Tests for CKMT1 High Versus CKMT1 Low Expression in Primary AML Samples
The in silico test used to determine the proportion of EVI1high/RUNX1low, EVI1high/RUNX1high, EVI1low/RUNX1low, and EVI1low/RUNX1high patients exhibiting high CKMT1 versus low CKMT1 expression levels was carried out on the Affymetrix U133 Plus2 gene expression data GSE14468857 (526 AML samples) and GSE1035858 (279 AML samples). Data were downloaded from the InSilico DB Genomic Datasets Hub (http://www.genomespace.org). EVI1, RUNX1, and CKMT1 expression levels were computed for each sample of these two cohorts and were further transformed into a z-score by subtracting the average of the EVI1, RUNX1, and CKMT1 expression levels assigned to all other samples and by dividing the result to their standard deviation. For each patient, high versus low EVI1, RUNX1, and CKMT1 expression levels were evaluated based on the absolute cut-off of a z-score ≥ 0.58 is high expression and a z-score ≤ −0.58 is low expression. The significance of the differences between the proportions of patients with CKMT1 high versus low expression within patient subgroups with high and low EVI1 and RUNX1 expressions was estimated by applying the two-tailed Fisher’s Exact Test implemented in the function fisher.test (library stats, R 3.2, http://cran.r-project.org/).
Supplementary Material
Figure S1. Effect of Evi1 overexpression on metabolism of hematopoietic progenitor cells.
Heatmap of the top up- or down-regulated metabolites identified by steady-state metabolism profiling in control versus Evi1-overexpressing LinLow murine bone marrow cells. Results are presented as an absolute log2 Fold Change of Evi1 overexpression normalized to the average empty condition. Each column for each condition represents a technical replicate (n=3 per condition).
Figure S2. Effect of doxycycline and GM-CSF supplementation on CKMT1 expression and sensitivity to CKMT1 depletion.
(A–B) Immunoblot (A) and relative growth (B) of TF-1 and UCSD-AML1 human cell lines transduced with either a control (CT) or two CKMT1-directed shRNAs. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with the shControl condition. Error bars represent the mean ± SD of seven technical replicates. (C–D) Effect of GM-CSF withdrawal on CKMT1 expression by immunoblot and cyclocreatine (Ccr) sensitivity in the indicated human AML cell lines. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with + GM-CSF condition. Error bars represent mean ± SD of four different EVI1 high human cell lines. n.s, not significant. (E) Immunoblot analysis of the effect of a 10 day GM-CSF supplementation on the expression of CKMT1 in two AML cell lines, HL-60 and U937.
Each experiment was performed at least two independent times.
Figure S3. Effect of the modulation of EVI1 expression on RUNX1 and CKMT1 mRNA and protein levels.
(A–B) qRT-PCR for Evi1, Runx1, and Ckmt1 expression (A) and immunoblot for Evi1, Runx1, Ckmt1 and Gapdh (loading control) (B) from LinLow, c-Kit+ murine bone marrow cells infected with an empty or Evi1-encoding construct. (C–D) qRT-PCR for EVI1, RUNX1, and CKMT1 expression (C) and immunoblot for EVI1, RUNX1, CKMT1, and HSP60 (loading control) from human UCSD-AML1 cells infected with either a control shRNA (shCT) or three EVI1-directed shRNAs (D). (A and C) * p value ≤ 0.05 calculated using Mann-Whitney test in comparison with the control (either empty for panel A or shCT for panel C) condition. Error bars represent mean ± SD of four technical replicates. Data are representative of at least two independent experiments.
Figure S4. Effect of CK pathway inhibition on levels of creatinine, guanidinoacetate, and L-arginino-succinate.
(A–B) Normalized peak areas of 13C-labeled metabolites in human UCSD-AML1 cells infected with either shControl or two CKMT1-directed shRNAs (A), or in TF-1 human cells treated with 3 mM cyclocreatine (Ccr) for 15h in combination with phospho-creatine (P-Cr) (B). * p value ≤ 0.05 calculated using Welch’s t-test in comparison with the control condition. ns = not significant. Error bars represent mean ± SD of three technical replicates.
Figure S5. Effect of CKMT1 overexpression on mitochondria function.
(A) Immunoblot for CKMT1 and VINCULIN (loading control) from human NB-4 and U937 cells overexpressing a construct encoding a wild-type (WT) or a mutant (C316G) form of CKMT1.
(B) Growth inhibition of human NB-4 and U937 cells expressing a construct encoding either a wild-type (WT) or a mutant (C316G) form of CKMT1 and treated with increasing concentrations of cyclocreatine (Ccr). Error bars represent mean ± SD of seven technical replicates per dose.
(C) Intracellular ATP levels in human NB-4 and U937 AML cells expressing a construct encoding either a WT or a mutant (C316G) form of CKMT1 and treated with 12 mM cyclocreatine (Ccr) for 12 hours. Results represent the average of triplicate assays. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with the control condition. Error bars represent mean ± SD of four technical replicates per condition.
(D) OCR of either eGFP- or wild-type (WT) CKMT1-overexpressing human U937 cells treated with 12 mM cyclocreatine (Ccr) for 12 hours prior to measurement under basal conditions, following sequential injections of 1 μM ATP synthase inhibitor oligomycin, 1 μM uncoupler agent FCCP, and 1 μM complex I and III inhibitors rotenone and antimycin A. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with the CKMT1 WT control condition. Error bars represent mean ± SD of four technical replicates.
Each experiment was performed at least two independent times.
Figure S6. Top scoring genes identified by Leading Edge Analysis (A) Heatmaps of the core cyclocreatine (Ccr)-downregulated genes that were identified by Leading Edge Analysis of the three functional gene set categories “Cell Stemness”, “Cell Cycle” and “Cell Differentiation” enriched by GSEA. In red are selected genes of interest validated by qRT-PCR. Each column for each condition represents an independent biological replicate (n=3 per condition in three individual human EVI1-positive cell lines).
(B) qRT-PCR for MYC and CCND1 expression level in indicated AML cells treated with cyclocreatine (Ccr) for 24 hours. Error bars represent mean ± SD of four technical replicates. * p value ≤ 0.05 in comparison with the control condition calculated using a Mann-Whitney test. Data are representative of at least two independent experiments.
Figure S7. Characterization of the in vivo mouse models used to study the effect of CKMT1 depletion on disease development.
(A) Growth inhibition of wild-type (WT), N-RasG12D, or N-RasG12D + Evi1 cells treated with increasing concentrations of cyclocreatine (Ccr). Error bars represent mean ± SD of seven technical replicates per dose.
(B) Model of the competitive bone marrow transplantation assay developed to characterize the toxicity of Ckmt1 depletion on normal murine progenitors using two Ckmt1-directed shRNAs versus a control shRNA.
(C) Chimerism of CD45.1+ versus CD45.2+ murine cell populations (top panel) and shCkmt1-expressing CD45.1+ versus shControl-expressing CD45.2+ cell populations (bottom panel) were evaluated by FACS on the whole bone marrow at day 10 post-transplantation (n=4 mice per group). Data are representative of at least two independent time points.
(D–E) Bioluminescence of human U937-Luc (D) and TF-1 CSF2+-Luc (E) cells injected into nude mice was quantified at each indicated time point post-injection to assess emergence of the disease. Data are represented as mean ± SEM of five mice per group. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with no luciferin (no Lucif.) condition. Error bars represent mean ± SEM. At day 8 and day 10 post-injection, luciferase signal from U937 and TF-1 cells respectively, started to be significantly detected.
Table S1. List of top depleted and enriched genes identified in the in vitro shRNA screening on UCSD-AML1 and TF-1 cells.
Genes are categorized based on Normalized Enrichment Score (NES) and −log10 (p value). Core depletion / enrichment means that genes scored in both cell lines; TF-1_ or UCSD-AML1_ depletion / enrichment means that genes scored only in the indicated cell line; 0 means no significant depletion or enrichment in any cell lines.
Table S2. Cytogenetics and clinical features of patients samples used in this study. n/a: limited patient data available, unknown: test was not done and thus data not available.
Table S3. Top genes significantly downregulated by cyclocreatine treatment in TF-1, UT-7, and UCSD-AML1 cell lines.
Significance cut-offs: Fold Change (Log2 FPKM) = −0.5, permutation p value = 0.05, False Discovery Rate (Benjamini-Hochberg) = 0.05. Top scoring genes identified from Leading Edge Analysis are underlined.
Table S4. Top genes significantly upregulated by cyclocreatine treatment in TF-1, UT-7, and UCSD-AML1 cell lines.
Significance cut-offs: Fold Change (Log2 FPKM) = 0.5, permutation p value = 0.05, False Discovery Rate (Benjamini-Hochberg) = 0.05.
Table S5. Table of selective gene sets enriched by GSEA in genes whose expression was downregulated (DOWN) or upregulated (UP) by cyclocreatine (Ccr) treatment in TF-1, UT-7, and UCSD-AML1 cell lines.
NES = Normalized Enrichment Score. Significant p value cut-off: 0.05.
Acknowledgments
We thank Drs. Tomohiko Sato and Mineo Kurokawa (The University of Tokyo, Tōkyō, Japan) for providing plasmid constructs and detailed procedures for Evi1 overexpression in murine hematopoietic cells. We also thank Dr. Joseph F. Clark (University of Cincinnati, Cincinnati, Ohio, USA) for advice on the use of cyclocreatine in vivo. This research was supported with grants from the National Cancer Institute (NIH 1R35 CA210030-01); Stand-up-to-Cancer; the Bridge Project, a collaboration between The Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center (DF/HCC) (KS and MH) and support from the Cubans Curing Children’s Cancers (4C’s Fund) (KS) and from Koch Institute Cancer Center Support (grant P30-CA14051, NCI) (MH). AP is a recipient of support from the ATIP/AVENIR French research program, the EHA research grant for Non-Clinical Advanced fellow, and supported by the St Louis Association for leukemia research, and KS is an LLS Scholar. AP, NF, and IBS were awarded the “Prix Jeune Chercheur” from the Bettencourt Foundation and the Franco-American Exchange Prize from Philippe Foundation Inc.
Footnotes
AUTHOR CONTRIBUTIONS
NF and CFB contributed equally to the manuscript as co-first authors. NF and CFB developed the study, established conditions for in vivo and in vitro experiments, acquired and analyzed the data, and wrote the manuscript. IB-S designed and performed metabolic-related experiments. LB, AR, YP, ASC, and FL designed, performed, and analyzed in vivo experiments. QL, MRB. and KSh revised the manuscript and provided NrasG12D and NrasG12D + Evi1 mouse models and methodology for in vivo functional analyses. GA revised the manuscript and performed statistical analysis, biostatistics, and computational analysis of the RNA sequencing, the publicly available patient sample cohorts, and the shRNA screen experiments. ASP and YZ provided reagents and ChIP-sequencing data for ChIP-qPCR experiments performed on endogenous murine Evi-1. IG, DJD, and RMS provided patient samples and revised the manuscript. PA revised the manuscript. MTH, AP and KS contributed equally to this work as co-senior authors. MTH, AP and KS supervised the study, wrote and revised the manuscript, designed the in vitro and in vivo experiments, analyzed the data, and provided funding for the study.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Baysal BE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 2.Janeway KA, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ricketts C, et al. Germline SDHB mutations and familial renal cell carcinoma. Journal of the National Cancer Institute. 2008;100:1260–1262. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 4.Kim S, Kim DH, Jung WH, Koo JS. Succinate dehydrogenase expression in breast cancer. SpringerPlus. 2013;2:299. doi: 10.1186/2193-1801-2-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3:730–741. doi: 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- 6.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–494. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 11.Patel JP, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glass C, Wilson M, Gonzalez R, Zhang Y, Perkins AS. The role of EVI1 in myeloid malignancies. Blood Cells Mol Dis. 2014;53:67–76. doi: 10.1016/j.bcmd.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Goyama S, Kurokawa M. Evi-1 as a critical regulator of leukemic cells. Int J Hematol. 2010;91:753–757. doi: 10.1007/s12185-010-0618-5. [DOI] [PubMed] [Google Scholar]
- 14.Groschel S, et al. High EVI1 expression predicts outcome in younger adult patients with acute myeloid leukemia and is associated with distinct cytogenetic abnormalities. J Clin Oncol. 2010;28:2101–2107. doi: 10.1200/JCO.2009.26.0646. [DOI] [PubMed] [Google Scholar]
- 15.Lugthart S, et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol. 2010;28:3890–3898. doi: 10.1200/JCO.2010.29.2771. [DOI] [PubMed] [Google Scholar]
- 16.Kustikova OS, et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia. 2013;27:1127–1138. doi: 10.1038/leu.2012.355. [DOI] [PubMed] [Google Scholar]
- 17.Goyama S, et al. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3:207–220. doi: 10.1016/j.stem.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Kataoka K, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. 2011;208:2403–2416. doi: 10.1084/jem.20110447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bard-Chapeau EA, et al. Ecotopic viral integration site 1 (EVI1) regulates multiple cellular processes important for cancer and is a synergistic partner for FOS protein in invasive tumors. Proc Natl Acad Sci U S A. 2012;109:2168–2173. doi: 10.1073/pnas.1119229109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glass C, et al. Global Identification of EVI1 Target Genes in Acute Myeloid Leukemia. PLoS One. 2013;8:e67134. doi: 10.1371/journal.pone.0067134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furter R, Furter-Graves EM, Wallimann T. Creatine kinase: the reactive cysteine is required for synergism but is nonessential for catalysis. Biochemistry. 1993;32:7022–7029. doi: 10.1021/bi00078a030. [DOI] [PubMed] [Google Scholar]
- 22.Burgess MR, et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood. 2014;124:3947–3955. doi: 10.1182/blood-2014-05-574582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Q, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117:2022–2032. doi: 10.1182/blood-2010-04-280750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herranz D, et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med. 2015;21:1182–1189. doi: 10.1038/nm.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang YH, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–1323. doi: 10.1016/j.cell.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mussai F, et al. Arginine dependence of acute myeloid leukemia blast proliferation: a novel therapeutic target. Blood. 2015;125:2386–2396. doi: 10.1182/blood-2014-09-600643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plunkett W. Arginine addiction in AML. Blood. 2015;125:3971–3972. doi: 10.1182/blood-2015-05-643411. [DOI] [PubMed] [Google Scholar]
- 28.Miraki-Moud F, et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood. 2015;125:4060–4068. doi: 10.1182/blood-2014-10-608133. [DOI] [PubMed] [Google Scholar]
- 29.Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281(Pt 1):21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell death and differentiation. 2015;22:248–257. doi: 10.1038/cdd.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arora KK, Pedersen PL. Functional significance of mitochondrial bound hexokinase in tumor cell metabolism. Evidence for preferential phosphorylation of glucose by intramitochondrially generated ATP. The Journal of biological chemistry. 1988;263:17422–17428. [PubMed] [Google Scholar]
- 32.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]
- 34.Cimino D, et al. Identification of new genes associated with breast cancer progression by gene expression analysis of predefined sets of neoplastic tissues. Int J Cancer. 2008;123:1327–1338. doi: 10.1002/ijc.23660. [DOI] [PubMed] [Google Scholar]
- 35.Gaidzik VI, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29:1364–1372. doi: 10.1200/JCO.2010.30.7926. [DOI] [PubMed] [Google Scholar]
Methods-only References
- 36.Zuber J, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol. 2011;29:79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerji V, et al. The intersection of genetic and chemical genomic screens identifies GSK-3alpha as a target in human acute myeloid leukemia. J Clin Invest. 2012;122:935–947. doi: 10.1172/JCI46465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo B, et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones SJ. Prediction of genomic functional elements. Annual review of genomics and human genetics. 2006;7:315–338. doi: 10.1146/annurev.genom.7.080505.115745. [DOI] [PubMed] [Google Scholar]
- 42.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puissant A, et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell. 2014;25:226–242. doi: 10.1016/j.ccr.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reich M, et al. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 46.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 47.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merico D, Isserlin R, Bader GD. Visualizing gene-set enrichment results using the Cytoscape plug-in enrichment map. Methods Mol Biol. 2011;781:257–277. doi: 10.1007/978-1-61779-276-2_12. [DOI] [PubMed] [Google Scholar]
- 49.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leng N, et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Novershtern N, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stegmaier K, et al. Gene expression-based high-throughput screening(GE-HTS) and application to leukemia differentiation. Nat Genet. 2004;36:257–263. doi: 10.1038/ng1305. [DOI] [PubMed] [Google Scholar]
- 55.Zuber J, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daigle SR, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wouters BJ, et al. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tomasson MH, et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood. 2008;111:4797–4808. doi: 10.1182/blood-2007-09-113027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of Evi1 overexpression on metabolism of hematopoietic progenitor cells.
Heatmap of the top up- or down-regulated metabolites identified by steady-state metabolism profiling in control versus Evi1-overexpressing LinLow murine bone marrow cells. Results are presented as an absolute log2 Fold Change of Evi1 overexpression normalized to the average empty condition. Each column for each condition represents a technical replicate (n=3 per condition).
Figure S2. Effect of doxycycline and GM-CSF supplementation on CKMT1 expression and sensitivity to CKMT1 depletion.
(A–B) Immunoblot (A) and relative growth (B) of TF-1 and UCSD-AML1 human cell lines transduced with either a control (CT) or two CKMT1-directed shRNAs. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with the shControl condition. Error bars represent the mean ± SD of seven technical replicates. (C–D) Effect of GM-CSF withdrawal on CKMT1 expression by immunoblot and cyclocreatine (Ccr) sensitivity in the indicated human AML cell lines. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with + GM-CSF condition. Error bars represent mean ± SD of four different EVI1 high human cell lines. n.s, not significant. (E) Immunoblot analysis of the effect of a 10 day GM-CSF supplementation on the expression of CKMT1 in two AML cell lines, HL-60 and U937.
Each experiment was performed at least two independent times.
Figure S3. Effect of the modulation of EVI1 expression on RUNX1 and CKMT1 mRNA and protein levels.
(A–B) qRT-PCR for Evi1, Runx1, and Ckmt1 expression (A) and immunoblot for Evi1, Runx1, Ckmt1 and Gapdh (loading control) (B) from LinLow, c-Kit+ murine bone marrow cells infected with an empty or Evi1-encoding construct. (C–D) qRT-PCR for EVI1, RUNX1, and CKMT1 expression (C) and immunoblot for EVI1, RUNX1, CKMT1, and HSP60 (loading control) from human UCSD-AML1 cells infected with either a control shRNA (shCT) or three EVI1-directed shRNAs (D). (A and C) * p value ≤ 0.05 calculated using Mann-Whitney test in comparison with the control (either empty for panel A or shCT for panel C) condition. Error bars represent mean ± SD of four technical replicates. Data are representative of at least two independent experiments.
Figure S4. Effect of CK pathway inhibition on levels of creatinine, guanidinoacetate, and L-arginino-succinate.
(A–B) Normalized peak areas of 13C-labeled metabolites in human UCSD-AML1 cells infected with either shControl or two CKMT1-directed shRNAs (A), or in TF-1 human cells treated with 3 mM cyclocreatine (Ccr) for 15h in combination with phospho-creatine (P-Cr) (B). * p value ≤ 0.05 calculated using Welch’s t-test in comparison with the control condition. ns = not significant. Error bars represent mean ± SD of three technical replicates.
Figure S5. Effect of CKMT1 overexpression on mitochondria function.
(A) Immunoblot for CKMT1 and VINCULIN (loading control) from human NB-4 and U937 cells overexpressing a construct encoding a wild-type (WT) or a mutant (C316G) form of CKMT1.
(B) Growth inhibition of human NB-4 and U937 cells expressing a construct encoding either a wild-type (WT) or a mutant (C316G) form of CKMT1 and treated with increasing concentrations of cyclocreatine (Ccr). Error bars represent mean ± SD of seven technical replicates per dose.
(C) Intracellular ATP levels in human NB-4 and U937 AML cells expressing a construct encoding either a WT or a mutant (C316G) form of CKMT1 and treated with 12 mM cyclocreatine (Ccr) for 12 hours. Results represent the average of triplicate assays. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with the control condition. Error bars represent mean ± SD of four technical replicates per condition.
(D) OCR of either eGFP- or wild-type (WT) CKMT1-overexpressing human U937 cells treated with 12 mM cyclocreatine (Ccr) for 12 hours prior to measurement under basal conditions, following sequential injections of 1 μM ATP synthase inhibitor oligomycin, 1 μM uncoupler agent FCCP, and 1 μM complex I and III inhibitors rotenone and antimycin A. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with the CKMT1 WT control condition. Error bars represent mean ± SD of four technical replicates.
Each experiment was performed at least two independent times.
Figure S6. Top scoring genes identified by Leading Edge Analysis (A) Heatmaps of the core cyclocreatine (Ccr)-downregulated genes that were identified by Leading Edge Analysis of the three functional gene set categories “Cell Stemness”, “Cell Cycle” and “Cell Differentiation” enriched by GSEA. In red are selected genes of interest validated by qRT-PCR. Each column for each condition represents an independent biological replicate (n=3 per condition in three individual human EVI1-positive cell lines).
(B) qRT-PCR for MYC and CCND1 expression level in indicated AML cells treated with cyclocreatine (Ccr) for 24 hours. Error bars represent mean ± SD of four technical replicates. * p value ≤ 0.05 in comparison with the control condition calculated using a Mann-Whitney test. Data are representative of at least two independent experiments.
Figure S7. Characterization of the in vivo mouse models used to study the effect of CKMT1 depletion on disease development.
(A) Growth inhibition of wild-type (WT), N-RasG12D, or N-RasG12D + Evi1 cells treated with increasing concentrations of cyclocreatine (Ccr). Error bars represent mean ± SD of seven technical replicates per dose.
(B) Model of the competitive bone marrow transplantation assay developed to characterize the toxicity of Ckmt1 depletion on normal murine progenitors using two Ckmt1-directed shRNAs versus a control shRNA.
(C) Chimerism of CD45.1+ versus CD45.2+ murine cell populations (top panel) and shCkmt1-expressing CD45.1+ versus shControl-expressing CD45.2+ cell populations (bottom panel) were evaluated by FACS on the whole bone marrow at day 10 post-transplantation (n=4 mice per group). Data are representative of at least two independent time points.
(D–E) Bioluminescence of human U937-Luc (D) and TF-1 CSF2+-Luc (E) cells injected into nude mice was quantified at each indicated time point post-injection to assess emergence of the disease. Data are represented as mean ± SEM of five mice per group. * p value ≤ 0.05 calculated using a Mann-Whitney test in comparison with no luciferin (no Lucif.) condition. Error bars represent mean ± SEM. At day 8 and day 10 post-injection, luciferase signal from U937 and TF-1 cells respectively, started to be significantly detected.
Table S1. List of top depleted and enriched genes identified in the in vitro shRNA screening on UCSD-AML1 and TF-1 cells.
Genes are categorized based on Normalized Enrichment Score (NES) and −log10 (p value). Core depletion / enrichment means that genes scored in both cell lines; TF-1_ or UCSD-AML1_ depletion / enrichment means that genes scored only in the indicated cell line; 0 means no significant depletion or enrichment in any cell lines.
Table S2. Cytogenetics and clinical features of patients samples used in this study. n/a: limited patient data available, unknown: test was not done and thus data not available.
Table S3. Top genes significantly downregulated by cyclocreatine treatment in TF-1, UT-7, and UCSD-AML1 cell lines.
Significance cut-offs: Fold Change (Log2 FPKM) = −0.5, permutation p value = 0.05, False Discovery Rate (Benjamini-Hochberg) = 0.05. Top scoring genes identified from Leading Edge Analysis are underlined.
Table S4. Top genes significantly upregulated by cyclocreatine treatment in TF-1, UT-7, and UCSD-AML1 cell lines.
Significance cut-offs: Fold Change (Log2 FPKM) = 0.5, permutation p value = 0.05, False Discovery Rate (Benjamini-Hochberg) = 0.05.
Table S5. Table of selective gene sets enriched by GSEA in genes whose expression was downregulated (DOWN) or upregulated (UP) by cyclocreatine (Ccr) treatment in TF-1, UT-7, and UCSD-AML1 cell lines.
NES = Normalized Enrichment Score. Significant p value cut-off: 0.05.