
Figure 9.
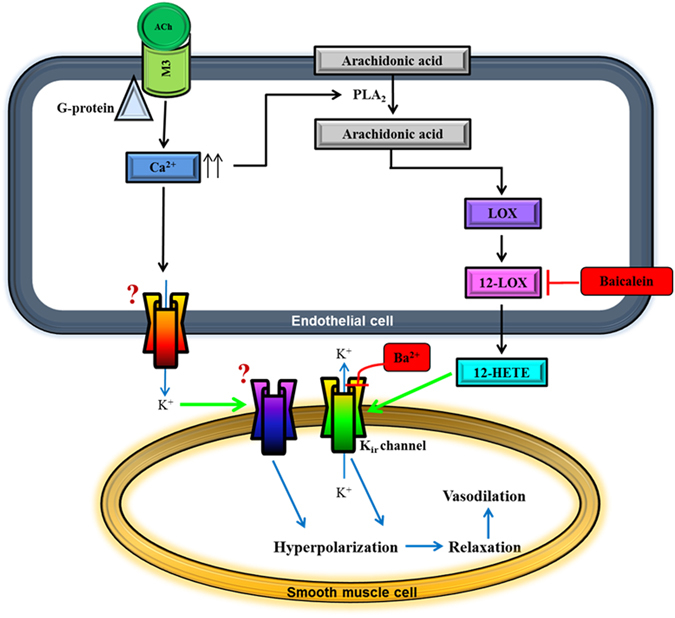
Proposed scheme for endothelium-dependent compensatory mechanisms responsible for mediating ACh-induced vasodilation in the ophthalmic artery of mice deficient in the eNOS gene. Endothelium-derived hyperpolarizing factor (EDHF)-mediated dilatory responses to ACh involve two mechanisms acting in parallel. In the first pathway, stimulation of the muscarinic ACh receptor (M3) coupled to G protein of class Gq on vascular endothelial cells mediates a transient increase in intracellular calcium, [Ca2+]i. Elevated endothelial [Ca2+]i activates phospholipase A2 (PLA2) to release arachidonic acid (AA) from cell membrane. Free AA is metabolized via the 12-LOX pathway to produce relaxing factors, namely 12-HETE, which activates Kir channels on the smooth muscle. In the second pathway, it is hypothesized that elevated [Ca2+]i also initiates activation of putative K+ channel(s) and release of K+ ions from the endothelial cell. These K+ ions diffuse to the adjacent vascular smooth muscle cell in concentrations sufficient to activate another putative K+ channel. Both pathways eventually cause hyperpolarization of the smooth muscle cell membrane and vasorelaxation. The precise identities of the putative channels hypothesized to be present in the ophthalmic artery of eNOS−/− mice that facilitate K+ efflux on the endothelial cell and are activated on the smooth muscle cell for downstream spread of hyperpolarization are unknown. Question marks represent unknown channels that remain to be identified. Inhibitor and blocker are indicated in red boxes. Green arrows indicate channel activation.12- HETE, hydroxyeicosatetraenoic acid; Kir, inward rectifying K+ channel.