

Abstract
d‐2‐Aminobutyric acid is an unnatural amino acid serving as an important intermediate in pharmaceutical production. Developing a synthetic method that uses cheaper starting materials and produces less by‐product is a pressing demand. A tri‐enzymatic catalytic system, which is composed of l‐threonine ammonia lyase (l‐TAL), d‐amino acid dehydrogenase (d‐AADH), and formate dehydrogenase (FDH), has thus been developed for the synthesis of d‐2‐aminobutyric acid with high optical purity. In this cascade reaction, the readily available l‐threonine serves as the starting material, carbon dioxide and water are the by‐products. d‐2‐Aminobutyric acid was obtained with >90 % yield and >99 % enantioselective excess, even without adding external ammonia, demonstrating that the ammonia from the first reaction can serve as the amino donor for the reductive amination step. This multi‐enzymatic system provides an attractive method with high atomic economy for the synthesis of d‐α‐amino acids from the corresponding l‐α‐amino acids, which are readily produced by fermentation.
Keywords: atom economy, d-2-aminobutyric acid, d-aminoacid dehydrogenase, enzymatic cascade, l-threonine ammonia lyase
1. Introduction
Although d‐α‐amino acids do not exist in the nature as widely as their l‐counterparts, naturally occurring d‐α‐amino acids play an important role in defence mechanisms (cell wall, venoms) of many microorganisms and plants.1 d‐α‐Amino acids have also been detected in a variety of peptides synthesized by animal cells2 and in human tissue, appearing as indicators of aging.3 Several enzymes producing or metabolizing d‐amino acids have been discovered.4 d‐2‐Aminobutyric acid, which is an unnatural amino acid, has been widely used in the synthesis of antibiotics,5 angiotensin‐converting enzyme 2 inhibitors,6 brain‐permeable polo‐like kinase‐2 (Plk‐2) inhibitors,7 matrix metalloproteinase inhibitors,8 and antiproliferatives.9 Generally speaking, resolution of the racemate of α‐amino acids is currently the main method to obtain the optically pure d‐amino acids. For d‐2‐aminobutyric acid, aminoacylase,1, 10 protease,11 or the formation of diastereomeric derivatives12 have been used to realize the separation of l/d‐amino acids. ω‐Transaminases have been used for the synthesis of d‐2‐aminobutyric acid through the amino‐transfer reaction, providing an alternative biocatalytic method.13 d‐2‐Aminobutyric acid has also been synthesized through asymmetric alkylation at low temperature,14 or de‐racemization of l/d‐amino acids using l‐amino acid oxidase from Proteus myxofaciens in combination with amine boranes.15
Compared to the previously reported methods, reductive amination of 2‐oxobutyric acid with ammonia by d‐amino acid dehydrogenase provides an attractive synthetic method for d‐2‐aminobutyric acid, owing to the intrinsic atomic economy and low environmental impact of the reaction. Taking into account that many l‐amino acids are available through fermentation from inexpensive and renewable natural sources, and l‐amino acid oxidases catalyze their conversion to the 2‐oxo acids, we envisaged that the two‐step enzymatic reactions with l‐amino acid oxidase and d‐aminoacid dehydrogenase would offer economically effective and environmentally benign methods for the synthesis of d‐aminoacids from the corresponding l‐amino acids. For d‐2‐aminobutyric acid, although l‐2‐aminobutyric acid is an unnatural amino acid and not readily available as the natural counterparts, 2‐oxobutyric acid can be easily obtained from l‐threonine by using l‐threonine ammonia lyase (l‐TAL, EC 4.3.1.19). The enzyme usually shows high reactivity and threonine can be readily produced by fermentation.16 Tao et al. reported the enzymatic synthesis of l‐2‐aminobutyric acid from threonine by using l‐TAL combination with l‐amino acid dehydrogenase (l‐AADH) and formate dehydrogenase (FDH).17 Therefore, similar multiple enzymatic reactions should provide an efficient approach for the biosynthesis of d‐amino acids by employing a d‐amino acid dehydrogenase. Recently, it has been reported that meso‐2,6‐diaminopimelic acid dehydrogenase (DAPDH, EC 1.4.1.16) and its mutant enzymes can catalyze the reductive amination of 2‐keto acids to give d‐α‐amino acids.18 For example, meso‐diaminopimelate dehydrogenase from Symbiobacterium thermophilum (StDAPDH) can transform 2‐keto acids to the corresponding d‐α‐amino acid, and its excellent thermo‐stability provides one‐step heat‐treatment purification.18c The crystal structure of StDAPDH revealed the structural basis of its unique catalytic properties,19 and site saturation mutagenesis resulted in mutant H227V with 35‐fold activity enhancement over the wild‐type enzyme for phenyl pyruvic acid as the substrate.20
With this unique d‐amino acid dehydrogenase (d‐AADH) in hand, we combined it with l‐threonine ammonia lyase (l‐TAL) and formate dehydrogenase (FDH) to establish a tri‐enzymatic catalytic system for the synthesis of d‐2‐aminobutyric acid from l‐threonine (Scheme 1).
Scheme 1.
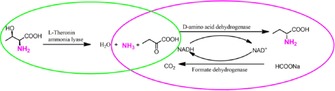
Tri‐enzymatic synthesis of d‐2‐aminobutyric acid from l‐ threonine.
2. Results and Discussion
2.1. Expression and Purification of l‐TAL, d‐AADH and FDH
The enzymes l‐TAL (EcTAL) from Escherichia coli, d‐AADH (M‐StDAPDH) from Symbiobacterium thermophilum, FDH (PFDH, CdFDH) from Pseudomonas sp. 101 and Candida boidinii have all previously been reported and produced in E. coli.16, 18c, 21 Enzymes were purified by heat treatment or precipitated with ammonium sulfate, except CbFDH, which was purified by using ÄKTA purifier. CbFDH lost >90 % enzyme activity when incubated at 50 °C for 1 h, whereas 90 % activity of PFDH was retained when heated at 57 °C for 1 h. PFDH was more heat tolerant and could be purified by heat treatment. As such, PFDH was used in the following studies.
2.2. pH Influence on the Enzyme Activity of the M‐StDAPDH and PFDH
A previous study showed that the activity of EcTAL was high enough (specific activity >200 U m− at pH 7.5)16 for the transformation, so we chose the enzyme as the biocatalyst for the ammonia lysis reaction of threonine. Because of the relatively low specific activity of M‐StDAPDH and PFDH, the pH profiles of their activity were studied. As shown in Figure 1 A, PFDH showed increased activity with pH from 5.0 to 10.0, and the activity was also affected by the nature of the buffers. A similar pH‐dependent trend was observed for glucose dehydrogenases22 and alcohol dehydrogenases23 for the oxidation reaction. For M‐StDAPDH, the enzyme activity was affected by the buffer and pH value (Figure 1 B), and the activity was higher in basic buffer. In phosphate buffer, M‐StDAPDH showed the lowest enzymatic activity; whereas, in the Na2CO3‐NaHCO3 buffer (pH 9.0), the specific activity of M‐StDAPDH was 0.55 U mg− and K m and k cat for 2‐oxobutyric acid were 12.3±0.4 mm and 1.6±0.2 s−1, respectively.
Figure 1.
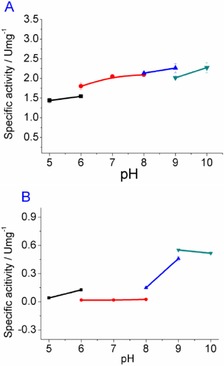
A) pH profile of PFDH; B) pH profile of M‐StDAPDH. The buffers (100 mm) were NaOAc‐HOAc (pH 5.0, 6.0), Na2HPO4‐NaH2PO4 (pH 6.0, 7.0, 8.0), Tris‐HCl (pH 8.0, 9.0), and Na2CO3‐NaHCO3 (pH 9.0, 10.0).
2.3. Influence of the Reaction Components on the Enzyme Activity
The influences of the reaction components on the enzyme activity were studied, and the results are presented in Figure 2 and Figure 3. For EcTAL, the conversion of l‐threonine was determined when different compounds were added into Na2CO3‐NaHCO3 (100 mm, pH 9.0) buffer at 30 °C. When 120 mm 2‐oxobutyric acid, d‐2‐aminobutyric acid, or 240 mm ammonium formate was added, the conversion of l‐threonine was not affected (Figure 2 A). The existence of l‐threonine, 2‐oxobutyric acid, and ammonium chloride in the reaction mixture showed little influence on the enzyme activity of PFDH, except at 480 mm of d‐2‐aminobutyric acid (Figure 2 B).
Figure 2.
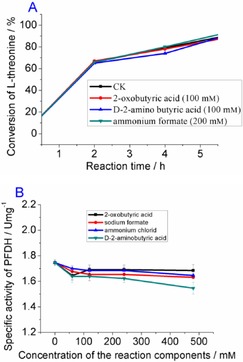
A) Conversion rate of l‐threonine catalyzed by EcTAL when a different reaction component was added to the reaction mixture (CK: no additive was added). B) Effects of different reaction components on the enzyme activity of PFDH.
Figure 3.
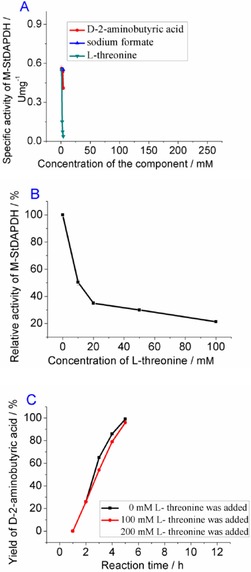
A) Effects of different reaction components on the activity of M‐StDAPDH. B) Relative activity of M‐StDAPDH when different concentrations of l‐threonine were added to the reaction mixture; the specific activity toward the 2‐oxobutyric acid without the addition of l‐threonine was defined as 100 %. C) Time course for the reductive amination of 2‐oxobutyric acid when 0, 100, or 200 mm l‐threonine was added into the reaction mixture.
For M‐StDAPDH, it was found that the enzymatic activity was affected by addition of d‐2‐aminobutyric acid or l‐threonine (Figure 3 A, 3B). In particular, the activity of M‐StDAPDH was reduced to nearly 50 % when only 10 mm l‐threonine was added (Figure 3 B). However, it is interesting that the reductive amination of 2‐oxobutyric acid proceeded smoothly and was completed in 12 h with 200 mm l‐threonine (Figure 3 C), although l‐threonine greatly affected the specific activity of the enzyme, as measured by using spectrometric methods. Monitoring of the conversion rate of 2‐oxobutyric acid showed that, even when 200 mm l‐threonine was added to the reaction mixture, the yield of d‐2‐aminobutyric acid did not show much difference to that of the control reaction without addition of l‐threonine (Figure 3 C).
2.4. Influence of the Biocatalyst Type on the Yield and ee Value
In previous studies, cell lysates could be used as the catalyst for the synthesis of l‐2‐aminobutyric acid.17 However, for the d‐configured counterpart, the use of whole cells or cell lysates might reduce the ee value of the product, owing to the endogenous l‐amino acid dehydrogenases, amino acid racemases, or amino transferase.24 As such, the different forms of the biocatalyst were employed to examine their effect on the optical purity of the product.
The reductive amination of 2‐oxobutyric acid was carried out with same activity units of whole cells, cell‐free extract, or purified enzyme in Na2CO3‐NaHCO3 (100 mm, pH 9.0) buffer. The yield and ee value of d‐2‐aminobutyric acid were measured, and the results are summarized in Table 1. When the cell‐free extract or purified enzyme was used as the catalyst, the yield of the product was >95 %, whereas the yield with whole cells as the catalyst was <5 %. This may be attributed to the fact that the cell membrane prevents transfer of the substrate into the cell for the enzymatic reaction. The ee value of the product for the reaction with cell‐free extract was slightly lower than that with purified enzyme. The endogenous enzyme in the cell‐free extract may be responsible for the slightly reduced ee value of the product.
Table 1.
Yield and ee values of the product with three different types of biocatalyst.
| Biocatalyst | Yield [%] | ee [%] |
|---|---|---|
| whole cells | <5 | not measured |
| cell‐free extract | >95 | 99.0 |
| purified enzymes | >98 | >99.5 |
2.5. Optimization of the Reaction Conditions
The purified enzymes EcTAL, M‐STDAPDH, and PFDH were used as the catalysts for the reactions with different amounts of the enzymes and varied concentrations of substrates. The results are summarized in Figure 4. It can be seen that at condition I with 0.7 U EcTAL, 1 U M‐StDAPDH, 2 U PFDH, 100 mm l‐threonine, and 150 mm ammonium formate in Na2CO3‐NaHCO3 buffer (pH 9.0), the yield of product was close to 90 % in 24 h. Enhancing the amount of EcTAL (Figure 4, I and II) increased the reaction rate, as evidenced by the fact that the yield of d‐2‐aminobutyric acid was improved from 52 to 92 % at 8 h when 2.0 U EcTAL was added instead of 0.7 U enzyme. Further enhancing the amount of EcTAL to 3.0 U did not increase the reaction rate (data not shown). As expected, when more PFDH and M‐StDAPDH were added, the reaction rate was significantly increased (II and III). The yield of product reached 90 % at 6 h when 2 U M‐StDAPDH and 4 U PFDH were added (condition III), compared to about 60 % at condition II after 6 h. Under these conditions, when the concentration of l‐threonine increased to 200 and 300 mm, the reaction still proceeded successfully (Figure 4 IV and VI, 95 % yield at 8 h and 92 % yield at 24 h, respectively). When no extra ammonia was added, the reaction was completed with 200 mm of l‐threonine (Figure 4, V). However, without the addition of extra ammonia, the transformation at the l‐threonine concentration of 300 mm was not completed within 24 h, and the yield of the product was less than 80 % (Figure 4, VII).
Figure 4.
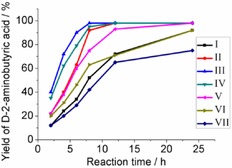
Time course for the formation of d‐2‐aminobutyric acid with different amounts of enzymes and varied concentrations of substrates. The reactions were conducted in Na2CO3–NaHCO3 buffer (100 mm, pH 9.0), and monitored at different time intervals. I: 0.7 U EcTAL, 1 U M‐StDAPDH, 2 U PFDH, 100 mm l‐threonine, 150 mm ammonium formate; II: 2 U EcTAL, 1 U M‐StDAPDH, 2 U PFDH, 100 mm l‐threonine, 150 mm ammonium formate; III: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 100 mm l‐threonine, 150 mm ammonium formate; IV: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 200 mm l‐threonine, 300 mm ammonium formate; V: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 200 mm l‐threonine, 300 mm sodium formate; VI: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 300 mm l‐threonine, 450 mm ammonium formate; VII: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 300 mm l‐threonine, 450 mm sodium formate.
2.6. Preparation of d‐2‐Aminobutyric Acid
In order to test the applicability of this enzymatic system, the reaction was conducted in 50 mL reaction solution, which contained 200 mm of l‐threonine, 300 mm of ammonium formate, or sodium formate. By monitoring the change in the concentrations of substrate and product, it was found that the deamination of l‐threonine was completed within 4 h, and the quick consumption of l‐threonine reduced the substrate inhibition effect during the second reaction. More importantly, although the reaction rate of the second step was decreased with addition of sodium formate, the transformation was nearly complete in 20 h, and the yield of the product reached 90 % after ion‐exchange purification. Therefore, the ammonia, which is produced in the first reaction catalyzed by EcTAL, can be used as the amino source in the reductive amination reaction. The multi‐enzymatic transformation proceeded smoothly without addition of external ammonia, making it more atomically economic.
3. Conclusions
A new effective enzymatic method has been developed for the synthesis of d‐2‐aminobutyric acid from commercially and readily available l‐threonine. In this multi‐enzymatic approach, l‐threonine ammonia lyase catalyzes the deamination of l‐threonine to give 2‐oxobutyric acid, which is reductively aminated by d‐amino acid dehydrogenase with formate dehydrogenase for co‐factor regeneration to produce d‐2‐aminobutyric acid. Particularly, this biotransformation can proceed smoothly at an l‐threonine concentration of 200 mm without extra addition of ammonia, rendering it a highly atom‐efficient reaction. The chosen thermostable enzymes are well suited for industrial applications, owing to their easy purification by heat‐treatment and robustness, although their specific activity needs to be improved for higher efficiency. The (co‐)immobilization of these enzymes will also increase the economic advantage of this tri‐enzymatic catalytic system and these studies are under way. In general, a similar approach should be applicable for the preparation of a wide range of d‐amino acids by combining a suitable l‐amino acid oxidase with a d‐amino acid dehydrogenase, and this will be further explored in our laboratory.
Experimental Section
General
All chemicals were obtained from commercial sources and the cofactors (NADH and NAD+) were obtained from Roche. Materials used for culture media including peptone, yeast extract, and agar were purchased from Becton, Dickinson, and Company (BDX). The product yield and ee value were determined by HPLC analysis on an Eclipse plus C18 column (4.6×250 mm, Agilent, USA) after being derived with 1‐fluoro‐2–4‐dinitrophenyl‐5‐l‐alanine amide (FDAA).
Expression of the EcTAL, M‐StDAPDH, and FDH
All of the selected enzymes have previously been reported. EcTAL gene (Gene ID: 948287) was amplified from the genome of E. coli.21 The EcTAL fragment was digested at the sites of BamHI and HindIII, and ligated into plasmid vector pRSFduet™‐1. FDH gene (Gene ID: 4033692) from Pseudomonas sp. 101 was synthesized by Shanghai Xuguan Biotechnological Development Co., Ltd. (China) and cloned into the NdeI/XhoI sites of pET21a. The pET21d‐CbFDH plasmid with FDH gene (Gene ID: 74654561) from Candida boidinii was available in our laboratory. The mutant (R35E/R36V/Y76V) of StDAPDH (Gene ID: 2979910) from Symbiobacterium thermophilum (M‐StDAPDH) was preserved in our laboratory, and the cofactor specificity of M‐StDAPDH has been changed from NADP(H) to NAD(H).
The plasmids pRSFduet‐EcTAL, pET‐32a(+)‐M‐StDAPDH, and pET21a‐PFDH were transformed into E. coli BL21(DE3) cells. Recombinant E. coli BL21(DE3) was propagated in 1 L of Luria–Bertani medium containing 100 μg mL−1 ampicillin (50 μg mL−1 kanamycin for pRSFduet‐EcTAL) at 37 °C. The culture was induced by addition of isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) with a final concentration of 0.1 mm when the optical density (λ=600 nm) reached 0.6–0.8 and then incubated for an additional 16 h at 25 °C (pET21d‐CbFDH at 20 °C) with 200 rpm. After centrifugation at 6000×g, 4 °C for 20 min, the recombinant cells were washed with potassium phosphate buffer and cryopreserved at −20 °C without loss of activity over 2 weeks.
Purification of the EcTAL, M‐StDAPDH, and FDHs
The enzymes were purified by following the procedures described herein. For EcTAL, the recombinant cells were suspended in 100 mm potassium phosphate buffer (pH 7.0) and lysed by French press at an operating pressure of 12 000 psi. The cell debris was removed by centrifugation at 10 000×g for 30 min at 4 °C. The resulting cell‐free extract was mixed with 0.025 % PEI solution. After centrifugation, the supernatant was precipitated with 40–60 % ammonium sulfate. The resulting precipitate was collected after centrifugation and dissolved in potassium phosphate buffer (100 mm, pH 7.0).
For M‐StDAPDH, PFDH, and CbFDH, The recombinant cells were suspended in 100 mm potassium phosphate buffer (pH 8.0) and lysed by French press at an operating pressure of 12 000 psi. The cell debris was removed by centrifugation at 10 000×g for 30 min at 4 °C. The resulting cell‐free extract was mixed with 0.025 % PEI solution. After centrifugation, the supernatant of M‐StADPDH was heat‐treated at 70 °C for 80 min; the supernatant of PFDH was heat‐treated at 57 °C for 60 min. After centrifugation, the supernatant of M‐StDAPDH and PFDH was concentrated by ultrafiltration (Amicon Ultra 30 kDa, Millipore) and stored in 5 % glycerol at −80 °C for further use. For CbFDH, after centrifugation, the supernatant was precipitated with 55 % ammonium sulfate and the supernatant was then loaded into a 20 mL Phenyl Sepharose 6 Fast Flow column (high sub) (GE Healthcare) on ÄKTA purifier. The target protein was washed with elution buffer (100 mm potassium phosphate buffer, pH 8.0) and dialyzed against potassium phosphate buffer (100 mm, pH 8.0). Then, the protein was concentrated by ultrafiltration (Amicon Ultra 30 kDa, Millipore) and stored in 5 % glycerol at −80 °C.
Activity Assay of the EcTAL, M‐StDAPDH and FDH
The activity of EcTAL toward l‐threonine was determined by measuring the decrease of threonine in the reaction mixture through HPLC analysis. Into 500 μL reaction solution, which contained 100 mm l‐threonine and 10 μm pyridoxal phosphate, an appropriate amount of EcTAL was added. After a certain time, the remaining amount of l‐threonine was measured. The specific activity was defined as the number of micromoles of l‐threonine converted by 1 mg of enzyme in 1 min (μmol min−1 mg−1).
The activity of M‐StDAPDH toward the reductive amination of 2‐oxobutyric acid and the activity of the FDH toward the oxidation of formate were determined by measuring the oxidation of NADH or reduction of NAD+ at 340 nm (ϵ=6.22 mm −1 cm−1), respectively. The reductive amination activity was measured at room temperature in a 96‐well plate; each well contained 2‐oxobutyric acid (20 mm), NH4Cl (60 mm), and NADH (0.40 mm) in Na2CO3–NaHCO3 (100 mm, pH 9.0, 190 μL) solution. The reaction was initiated by addition of M‐StDAPDH (10 μL solution containing appropriate amount of enzyme).The kinetic parameters of M‐StDAPDH towards 2‐oxobutyric acid were determined with the final concentration of 2‐oxobutic acid being between 1 and 60 mm. The oxidative activity of FDH was measured at room temperature in a 96‐well plate, in which each well contained sodium formate (20 mm) and NAD+ (0.50 mm) in Na2CO3‐NaHCO3 (100 mm, pH 9.0, 190 μL) solution. The reaction was initiated by the addition of the FDH (10 μL solution containing appropriate amount of enzyme). The specific activity was defined as the number of micromoles of NAD+ or NADH converted by 1 mg of enzyme in 1 min (μmol min−1 mg−1).
The pH Influence on the Enzyme Activity
The influences of pH on the enzyme activities were measured at various pH by activity assay methods mentioned above. The buffers (100 mm) were NaOAc‐HOAc (pH 5.0, 6.0), Na2HPO4‐NaH2PO4 (pH 6.0, 7.0, 8.0), Tris‐HCl (pH 8.0, 9.0) and Na2CO3‐NaHCO3 (pH 9.0, 10.0).
The Influence of the Reaction Components on the Enzyme Activity
For EcTAL, a final concentration of 100 mm 2‐oxobutyric acid, 100 mm d‐2‐aminobutyric acid, or 200 mm ammonium formate was added to the reaction mixture, which contained EcTDA (9 μg), l‐threonine (100 mm), and pyridoxal phosphate (10 μm). The activity of EcTDA was determined through the assay method mentioned above. For PFDH, a different final concentration (60, 120, 240, and 480 mm) of l‐threonine, ammonium chloride, 2‐oxobutyric acid, or d‐2‐aminobutyric acid was added into the reaction mixture, which contained PFDH (1 mg), sodium formate (20 mm), and NAD+ (0.50 mm). The activity of PFDH was determined by the assay method mentioned above. For M‐StDAPDH, a different final concentration (60, 120, 240, 480 mm) of sodium formate, d‐2‐amino butyric acid, or l‐threonine was added into the reaction mixture, which contained M‐StDAPDH (1 mg), 2‐oxobutyric acid (20 mm), NH4Cl (60 mm), and NADH (0.40 mm). The activity was determined by the assay procedure mentioned above. To further study the effect of l‐threonine on the M‐StDAPDH‐catalyzed transformation, 100 mm l‐threonine was added into the reaction mixture (1 mL) containing 2 U PFDH, 1 U M‐StDAPDH, 100 mm 2‐oxobytyric acid, and 200 mm ammonium formate. The reaction was carried out at 30 °C, and the yield of d‐2‐amino butyric acid was measured by HPLC analysis after FDAA derivatization every 3 h. The retention times of d‐ and l‐2‐amino butyric acid were 13.0 and 8.1 min, respectively.
The Influence of the Biocatalyst Type on the ee Value
Wet recombinant cells expressing the PFDH or M‐StDAPDH gene (30 mg mL−1) were suspended in Na2CO3‐NaHCO3 (100 mm, pH 9.0) and lysed by sonication; the cell debris was removed by centrifugation at 10 000×g for 2 min. The specific activity of the resulting crude PFDH enzyme solution was 32 U m−, corresponding to 1.6 U m− wet whole cells. The specific activity of crude enzymatic solution of M‐StDAPDH was 8.2 U m−, corresponding to 0.41 U m− wet whole cells. The reaction mixture (1 mL) containing 2 U of PFDH wet whole cells (or enzyme solution or purified enzyme), 1 U of M‐StDAPDH wet whole cells (or enzyme solution or purified enzyme), 100 mm 2‐oxobutyric acid, and 200 mm ammonium formate was shaken at 30 °C for 16 h. The yield and ee value of d‐2‐aminobutyric acid was determined by HPLC after FDAA derivation.
Optimization of Tri‐enzymatic Reaction System
In 1 mL Na2CO3‐NaHCO3 buffer (100 mm, pH 9.0), 0.7 U EcTAL, 1 U M‐StDAPDH, 2 U PFDH, 100 mm l‐threonine, and 150 mm ammonium formate were added (I), and the reaction was monitored by measuring the yield of d‐2‐aminobutyric acid at 2, 4, 6, 8, 12, and 24 h. The same procedure was followed for the reactions with different amount of enzymes and substrates, which were as follows. II: 2 U EcTAL, 1 U M‐StDAPDH, 2 U PFDH, 100 mm l‐threonine, and 150 mm ammonium formate; III: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 100 mm l‐threonine, and 150 mm ammonium formate; IV: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 200 mm l‐threonine, and 300 mm ammonium formate; V: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 200 mm l‐threonine, and 300 mm sodium formate; VI: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 300 mm l‐threonine, and 450 mm ammonium formate; VII: 2 U EcTAL, 2 U M‐StDAPDH, 4 U PFDH, 300 mm l‐threonine, and 450 mm sodium formate.
Preparation of d‐2‐Aminobutyric Acid
l‐Threonine (200 mm), EcTAL (100 U), PFDH (200 U), M‐StDAPDH (100 U), and sodium formate (or ammonium formate, 300 mm) were added into 50 mL Na2CO3‐NaHCO3 buffer (100 mm, pH 9.0); the yield and ee value of d‐2‐amino butyric acid were detected by HPLC analysis at the time intervals shown in Figure 5. After 24 h, the pH of the reaction mixture was adjusted to below 2 and the precipitated enzymes were removed by centrifugation. The product was purified on a cation‐exchange resin column and eluted by 3 % NH3⋅H2O. 0.92 g (99.6 % ee, 90 % yield for the reaction with sodium formate) and 0.95 g (99.6 % ee, 92 % yield for the reaction with ammonium formate) d‐2‐amino butyric acid were obtained, respectively. 1H NMR (D2O, 400 MHz) δ (ppm) 3.62 (t, J=6.0 Hz, 1 H), 1.80 (m, 2 H), 0.89 (m, J=8.4 Hz, 3 H).
Figure 5.
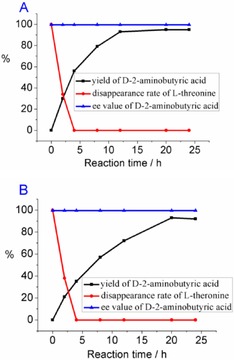
Monitoring of the yield (HPLC) of d‐2‐aminobutyric acid and disappearance of l‐threonine in the reaction at 50 mL scale, with addition of 300 mm ammonium formate (A) or sodium formate (B).
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21472232) and Tianjin Municipal Science and Technology Commission (No 15PTGCCX00060).
X. Chen, Y. Cui, X. Cheng, J. Feng, Q. Wu, D. Zhu, ChemistryOpen 2017, 6, 534.
References
- 1. Chenault H. K., Kim M.-J., Akiyama A., Miyazawa T., Simon E. S., Whitesides G. M., J. Org. Chem. 1987, 52, 2608–2611. [Google Scholar]
- 2. Friedman M., Chem. Biodiversity 2010, 7, 1491–1530. [DOI] [PubMed] [Google Scholar]
- 3. Fujii N., Biol. Pharm. Bull. 2005, 28, 1585–1589. [DOI] [PubMed] [Google Scholar]
- 4. Pollegioni P., Servi S., Methods Mol. Biol. 2012, 794, Humana Press. [Google Scholar]
- 5.
- 5a. Ondetti M. A., Thomas P. L., J. Am. Chem. Soc. 1965, 87, 4373–4380; [DOI] [PubMed] [Google Scholar]
- 5b. Ley S. V., Priour A., Eur. J. Org. Chem. 2002, 3995–4004; [Google Scholar]
- 5c. Shaginian A., Rosen M. C., Binkowski B. F., Belshaw P. J., Chemistry 2004, 10, 4334–4340. [DOI] [PubMed] [Google Scholar]
- 6. Deaton D. N., Gao E. N., Graham K. P., Gross J. W., Miller A. B., Strelow J. M., Bioorg. Med. Chem. Lett. 2008, 18, 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Aubele D. L., Hom R. K., Adler M., R. A. Galemmo, Jr. , Bowers S., Truong A. P., Pan H., Beroza P., Neitz R. J., Yao N., Lin M., Tonn G., Zhang H., Bova M. P., Ren Z., Tam D., Ruslim L., Baker J., Diep L., Fitzgerald K., Hoffman J., Motter R., Fauss D., Tanaka P., Dappen M., Jagodzinski J., Chan W., Konradi A. W., Latimer L., Zhu Y. L., Sham H. L., Anderson J. P., Bergeron M., Artis D. R., ChemMedChem 2013, 8, 1295–1313; [DOI] [PubMed] [Google Scholar]
- 7b. Scharow A., Knappe D., Reindl W., Hoffmann R., Berg T., ChemBioChem 2016, 17, 759–767. [DOI] [PubMed] [Google Scholar]
- 8. Behrends M., Wagner S., Kopka K., Schober O., Schafers M., Kumbhar S., Waller M., Haufe G., Bioorg. Med. Chem. 2015, 23, 3809–3818. [DOI] [PubMed] [Google Scholar]
- 9.P. M. Fisher, M. Jarman, E. Mcdonald, B. Nutley, F. Raynaud, S. Wilson, P. Workman, US20090325983, 2009.
- 10. Youshko M. I., van Langen L. M., Sheldon R. A., Švedas V. K., Tetrahedron: Asymmetry 2004, 15, 1933–1936. [Google Scholar]
- 11. Miyazawa T., Imagawa K., Minowa H., Miyamoto T., Yamada T., Tetrahedron 2005, 61, 10254–10261. [Google Scholar]
- 12.
- 12a. Soloshonok V. A., Ellis T. K., Ueki H., Ono T., J. Am. Chem. Soc. 2009, 131, 7208–7209; [DOI] [PubMed] [Google Scholar]
- 12b. Yajima T., Aizawa Y., Nishida M., Sakaguchi Y., Shiraiwa T., Biosci. Biotechnol. Biochem. 2007, 71, 1338–1341. [DOI] [PubMed] [Google Scholar]
- 13. Malik M. S., Park E.-S., Shin J.-S., Green Chem. 2012, 14, 2137–2140. [Google Scholar]
- 14.
- 14a. Myers A. G., Gleason J. L., Yoon T., Kung D. W., J. Am. Chem. Soc. 1997, 119, 656–673; [Google Scholar]
- 14b. Ueda M., Miyabe H., Teramachi M., Miyata O., Naito T., Chem. Commun. 2003, 426–427; [PubMed] [Google Scholar]
- 14c. Basra S., Fennie M. W., Kozlowski M. C., Org. Lett. 2006, 8, 2659–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alexandre F.-R., Pantaleone D. P., Taylor P. P., Fotheringham I. G., Agerc D. J., Turner N. J., Tetrahedron Lett. 2002, 43, 707–710. [Google Scholar]
- 16. Eisenstein E., J. Biol. Chem. 1991, 266, 5801–5807. [PubMed] [Google Scholar]
- 17. Tao R., Jiang Y., Zhu F., Yang S., Biotechnol. Lett. 2014, 36, 835–841. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Vedha-Peters K., Gunawardana M., Rozzell J. D., Novick S. J., J. Am. Chem. Soc. 2006, 128, 10923–10929; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Akita H., Doi K., Kawarabayasi Y., Ohshima T., Biotechnol. Lett. 2012, 34, 1693–1699; [DOI] [PubMed] [Google Scholar]
- 18c. Gao X., Chen X., Liu W., Feng J., Wu Q., Hua L., Zhu D., Appl. Environ. Microbiol. 2012, 78, 8595–8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu W., Li Z., Huang C. H., Guo R. T., Zhao L., Zhang D., Chen X., Wu Q., Zhu D., ChemBioChem 2014, 15, 217–222. [DOI] [PubMed] [Google Scholar]
- 20. Gao X., Huang F., Feng J., Chen X., Zhang H., Wang Z., Wu Q., Zhu D., Appl. Environ. Microbiol. 2013, 79, 5078–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tishkov V. I., Galkin A. G., Marchenko G. N., Egorova O. A., Sheluho D. V., Bulakova L. B., Dementieva L. A., Egorov A. M., Biochem. Biophys. Res. Commun. 1993, 192, 976–981. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Shatton J., Halver E., Weinhouse S., J. Biol. Chem. 1971, 246, 4878–4885; [PubMed] [Google Scholar]
- 22b. Fujita Y., Ramaley R., Freese E., J. Bacteriol. 1977, 132, 282–293; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22c. Ding H. T., Du Y. Q., Liu D. F., Li Z. L., Chen X. J., Zhao Y. H., Bioresour. Technol. 2011, 102, 1528–1536; [DOI] [PubMed] [Google Scholar]
- 22d. Kobayashi Y., Ueyama H., Horikoshi K., Agric. Biol. Chem. 1980, 44, 2837–2841. [Google Scholar]
- 23.
- 23a. Juan E., Gonzàlez-Duatre R., Biochem. J. 1987, 195, 61–69; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Julià P., Farrés J., Parés X., Eur. J. Biochem. 1987, 162, 179–189; [DOI] [PubMed] [Google Scholar]
- 23c. Pennacchio A., Pucci B., Secundo F., La Cara F., Rossi M., Raia C. A., Appl. Environ. Microbiol. 2008, 74, 3949–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanson R. L., Johnston R. M., Goldberg S. L., Parker W. L., Goswami A., Org. Process Res. Dev. 2013, 17, 693–700. [Google Scholar]