

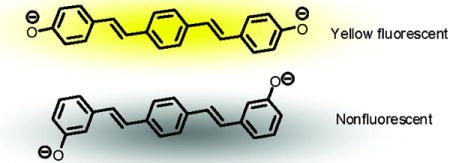
Graphical abstract
The dianions of two isomeric bis(hydroxystyryl)benzenes show dramatically different photophysical properties.
Stilbenes and distyrylbenzenes represent the first two oligomers leading to poly(para-phenylenevinylene) (PPV) and thus are important model compounds for conjugated polymers.1 As a consequence there is a significant fundamental interest in the excited-state behavior of these materials. Curiously, trans-stilbene (TS) is poorly fluorescent, the result of its well-known propensity for isomerization in the excited state.2 In contrast, trans-trans-distyrylbenzene (TTSB) and certain derivatives resist isomerization and are strongly fluorescent.3,4, Recently, Lewis et al.5 investigated the photoinduced processes in 4-hydroxystilbene (2), 3-hydroxystilbene (4) and several of their derivatives. The authors observed strikingly different behavior upon excitation, concluding that 4 is a strong photoacid in water, while 2 does not undergo efficient ESPT (excited state proton transfer) because of much faster photoisomerization. As a result, both compounds exhibit very weak fluorescence in aqueous solutions.
Given the stronger emissive properties of TTSB vs. TS, we speculated that hydroxyarenes based upon the former might show interesting excited-state proton transfer (ESPT) properties. In this communication we demonstrate that the excited-state properties of the homologues 1 and 3 are different both from each other and from those of 2 and of 4, coinciding with the differences between TS and TTSB in some respects but not in others. Specifically, we show that neither 1 nor 3 is a photoacid. Surprisingly, there is no published literature on the spectroscopy and photoinduced phenomena of either 1 or 3. In methanol both 1 and 3 display intensive, single-peak blue fluorescence with a Φfl of 0.37 and 0.62, respectively. Upon preparative photolysis with 355 nm light for 1 h, 3% of the compound 1 interconverted into its cis,trans-isomer, while 3 remained unchanged. Similar to hydroxystilbenes, the bis-para isomer 1 has somewhat red-shifted spectroscopic features in absorption and emission compared to the bis-meta isomer 3 (Table 1). Both 1 and 3 are soluble in methanol but begin to aggregate in solutions with more than 50 vol% water. To obtain pKa values, all measurements were performed in a 2:1 methanol/water (v/v) mixture,6 in which both compounds were soluble without apparent aggregation. Figure 1 shows the absorption and emission spectra of 1 and 3. Upon addition of KOH the absorption maximum of 1 shifts from 362 nm in the neutral compound to 393 nm in the bis-deprotonated form of 1 (12−) while λmax of 3 shifted from 355 to 363 nm.
Table 1.
Thermodynamic and Photophysical Properties of 1 and 3 in MeOH/H2O (2:1 v/v) at 298 K.
| Compound | 1 | 3 | ||||
|---|---|---|---|---|---|---|
| pKa1, pKa2 | 10.1±0.1, 12.0±0.1 | 10.6±0.1, 11.2±0.3 | ||||
| Species | H2A | HA− | A2− | H2A | HA– | A2− |
| Absorption (nm) | 362 | 388 | 393 | 355 | 359 | 363 |
| Emission (nm) | 428 | 575 | 533 | 412 | quenched | |
| Φ fl | 0.34 | n/d | 0.26 | 0.46 | < 0.001 | |
| τ(ns) | 0.91 | n/d | 1.0 | 1.41 | ||
Figure 1.
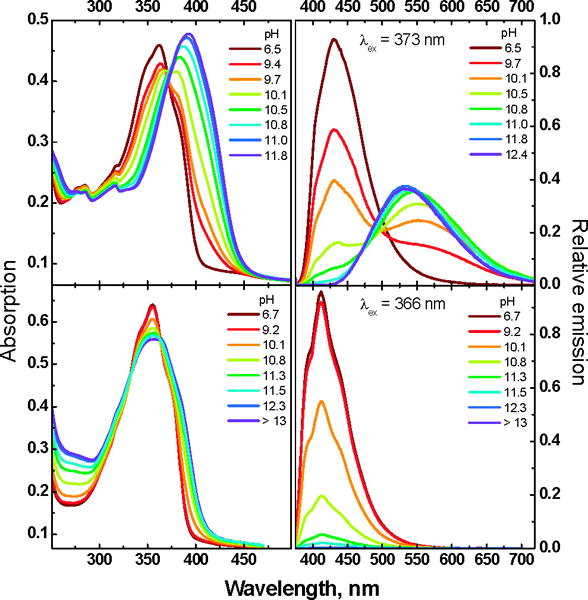
Absorption (left column) and emission (right column) spectra of 1 (top row) and 3 (bottom row) in the 2:1 mixture of MeOH/H2O (v/v) at different pH values.
To obtain more information about the deprotonation-dependent properties of the two distyrylbenzenes 1 and 3 we analyzed the obtained absorption data (1 and 3) using the principal component analysis program SPECFIT.7 Figure 2 shows the results. The pKa values for 1 are pKa1 = 10.1 (±0.1) and pKa2 = 12.0 (±0.1) while those for 3 are pKa1 = 10.6 (±0.1) and pKa2 = 11.2 (±0.3). The absorption data show nicely that the first deprotonation of 1 is easier than that of 3, however, the second pKa of 1 is almost 0.8 units higher than that of 3. The difference must be due to the conjugative, mesomeric interactions effective in 1 and its anions, as opposed to the purely electrostatic interactions in 3, that render its two pKa’s more alike.
Figure 2.
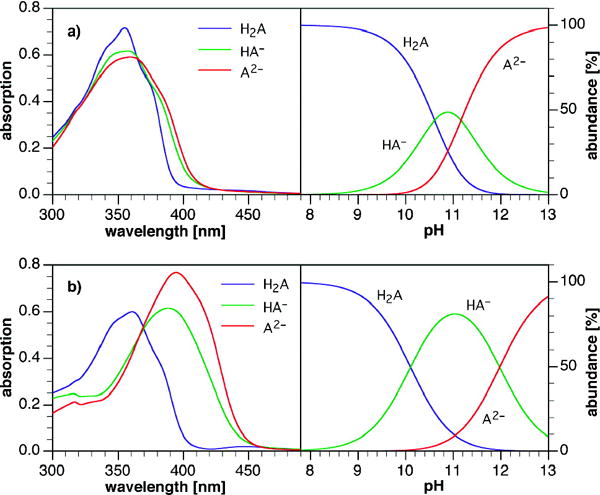
Deconvoluted absorption spectra (left) and species distribution diagram (right) for compounds 3 (row a) and 1 (row b).
The excited state pKa1* of 1 estimated using a modified Förster method8, is 1.9 and unlike in more condensed hydroxyarenes, such moderate thermodynamic photoacidity does not lead to detectable ESPT in neutral methanol/water solutions that can compete effectively with the 0.91 ns decay.9 Therefore for 1 we do not see any appreciable ESPT, and the pKa determined from the fluorescence pH-titration simply reflects the ground-state acid-base equilibrium.
From Figure 2 it is clear that upon the first deprotonation of 1 an absorption spectrum results that is close in appearance to that of the dianion 12−. However, in emission, the monoanion 1− emits red-shifted and at a different wavelength (575 nm) from both the neutral compound (428 nm) and the dianion 12− (533 nm). The monoanion of 1 is interesting, as its absorption is blue shifted from that of 12−, but its emission red-shifted. We assume that the anion experiences an intramolecular charge transfer stabilization in the excited state as the monodeprotonated species is formally a donor acceptor system, leading to the observed red-shifted emission.
The same experiment, i.e. deprotonation of 3 (λmax 355 nm) to 32− leads to a broadening of the absorption and a slight red-shift to 363 nm with a red-shifted absorption edge. The emission of neutral 3 is centered at 412 nm. Upon deprotonation its fluorescence is not shifted but quenched. A reliable determination of pKa* for 3 is problematic due to the complete absence of anion fluorescence.
These observations, i.e. the quenching of the fluorescence of 3 upon deprotonation and the large red shift of the fluorescence of 1 upon exposure to aqueous base are in stark contrast to the effects visible upon deprotonation of 2 and 4.5 On the one hand, 1 shows a much larger red-shift upon deprotonation and its dianion 12− is highly fluorescent. Pathways that lead to radiationless deactivation of the excited state of 12− are blocked. On the other hand, dianion 32− is weakly fluorescent but its absorption spectrum does not show a significant shift upon exposure to base, similar to the observation for other m-hydroxybenzylidene derivatives (m-hydroxystilbene5a and m-hydroxybenzylidene-imidazolinone10). It is tempting to conclude that the reason for the fluorescence quenching involves twisting about the formal double bond. However, Sandros3 and Motoyoshiya4 have observed that distyrylbenzenes undergo adiabatic one-way cis/trans isomerization, producing emission spectra corresponding to the E/E forms only. More recent, time-resolved studies indicate the formation of an intermediate but largely planar excited state.11 Thus we conclude that twisting leading to quenching does not occur. In the case of stilbene, twisting leads to such decay pathways.
In order to investigate this phenomenon further, we performed quantum chemical calculations (B3LYP/6-311+G(2d,2p)// B3LYP//6-311+G(2d,2p)) upon 1, 3 and their respective dianions 12− and 32−. Figures 3,4 and Table 2 show the most salient results. While the neutral compounds 1 and 3 show frontier molecular orbitals (FMO) that are very similar to those calculated for distyrylbenzene (see SI), the FMOs for 12− show larger amplitudes in the two peripheral rings, as a consequence of the delocalized phenolate moieties. According to these calculations the HOMO-LUMO gap decreases upon deprotonation from 3.27 to 2.48 eV.
Figure 3.
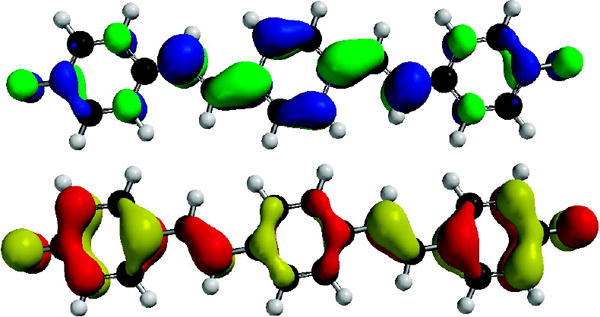
LUMO (Top) and HOMO (Bottom) of 12−.
Figure 4.
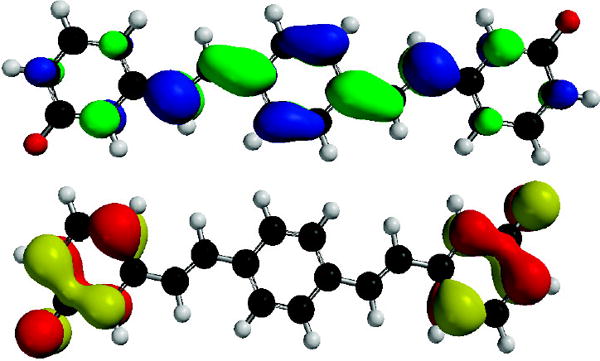
LUMO (Top) and HOMO (Bottom) of 32−.
Table 2.
Gas Phase Computational Data for compounds 1 and 3a
| Compound | 1 | 3 | ||
|---|---|---|---|---|
| Species | H2A | A2− | H2A | A2− |
| S1 (eV) | 3.18 | 2.48 | 3.25 | 1.91 |
| 7Bg –> 7Au c | (2.33)b | (2.39)b | (2.21)b | (0.061)b |
| S6 (eV) | 3.13 | |||
| 6Bg –> 7Au c | (1.74) b | |||
| Exp (eV)d | 3.42 (S1) | 3.15 (S1) | 3.49 (S1) | 3.04 (S1)e |
| 3.42 (S6) | ||||
| HOMO (eV) (7Bg) | −5.22 | 0.73 | −5.50 | 0.44 |
| LUMO (eV) (7Au) | −1.96 | 3.21 | −2.18 | 2.69 |
| HOMO-LUMO Gap (eV) | 3.27 | 2.48 | 3.32 | 2.26 |
TDDFT/B3LYP/6-311+G(2d,2p)//B3LYP/6-311+G(2d,2p) level of theory.
Oscillator strength in parantheses.
Major component of the CI description.
Experimental vertical absorption energy.
from excitation spectrum.
In the case of 3 the situation is dramatically different. The HOMO and the HOMO-1 are almost degenerate and localized on the two phenolate rings. In valence bond terms, the two phenolates are disjoint12 and are electronically only weakly coupled, while the LUMO is extended over the whole π-system but has larger coefficients in the central ring.13 Given the poor orbital overlap, the HOMO-LUMO transition leading to the lowest singlet excited state S1 is expected to exhibit a negligible oscillator strength despite the fact that a Bg–>Au transition is symmetry allowed in the C2h point group.
A closer inspection of the excited state manifold obtained from time-dependent density functional theory (TD-DFT) calculations indeed revealed a strong S1 oscillator strength for neutral 1 and 3 as well as 12− but not the meta-substituted dianion 32− (Table 2). The latter is preferably excited into S6, while all lower states exhibit neglible oscillator strengths. Although the quantum chemical calculations offer only estimates for gas phase vertical excitation energies at 0 K, the dominant lowest energy transitions scale linearly with the solution phase experimental data (correlation coefficient 0.994, mean unsigned error 0.01 eV). In agreement with the experiment, the calculations predict a strong bathochromic shift upon deprotonation of 1, but only a small shift for 3 due to excitation into S6 rather S1 (Table 2). The TD-DFT results furthermore indicate a possible non-radiative deactivation pathway through a lower lying triplet 3(n-π*) state (Figure 5). According to the El-Sayed rule,14 intersystem crossing from 1(π−π*) to 3(n-π*) is very rapid and typically results in fluorescence quenching due to an increased nonradiative deactivation rate.15 As illustrated with the electron detachment-attachment densities16 in Figure 5, the triplet states T3 and T4 together with their parent states S3 and S4 exhibit n-π* character involving excitation of a non-bonding oxygen lone-pair electron, thus offering an efficient non-radiative deactivation channel from S6 through T3 and T4. Because the calculations indicate that the two 3(n-π*) states lie above the lowest energy 1(π−π*) state, this nonradiative pathway should not be accessible upon excitation into S1. Excitation at the red-edge in the absorption spectrum of 32− revealed indeed a weak emission band centered around 541 nm, which was not visible with excitation at the absorption maximum (Supporting Information). The corresponding excitation trace acquired at 541 nm peaked at 407 nm and lacked the major higher energy band visible in the absorption spectrum, thus confirming that excitation into S6 results in non-radiative deactivation without detectible emission from S1.
Figure 5.
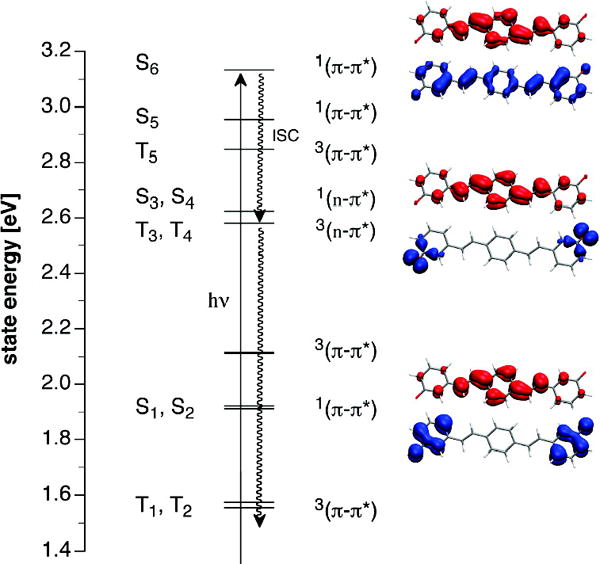
Excited state manifold for dianion 32− based on TD-DFT calculations (B3LYP/6-311+G(2d,2p)//B3LYP/6-311+G(2d,2p)). Upon excitation into S6, nonradiative deactivation may occur through rapid intersystem crossing (ISC) to the 3(n-π*) states T3 and T4. The surface plots to the right illustrate theπ-π* and n-π* nature of S1, S3 and S6 with the corresponding electron detachment (blue) and attachment (red) densities.
In conclusion we have demonstrated that the two bis(hydroxystyryl)benzenes 1 and 3 show photophysical properties that are distinct from each other and also distinct from the smaller 3- and 4-hydroxystilbenes 2 and 4. It is remarkable that the dianion of 1 is highly fluorescent, while the dianion of its isomer 3 is completely nonfluorescent. The large fluorescence quantum yield of 1, and its dianion presumably reflects a planarized and quite rigid excited-state with quinoidal resonance contributions,3 while the quenching of the dianion of 3 may be explained by the presence of an intermediate 3(n-π*) state combined with a poor Franck-Condon overlap between the HOMO and LUMO of this double phenolate. We recently observed similar phenomena in the case of para- vs. meta-dihydroxycruciforms.17,18 Overall we find it remarkable that a consanguine group of styryl-based phenols 1–4 display such disparate –and fundamentally interesting– photoinduced effects, not easily predicted by simply examining the structural motifs involved. Such effects, when understood, help illuminate the rather unusual properties of the related cruciforms18 and may aid in the design of other conjugated fluorophores.19
Supplementary Material
Acknowledgments
We thank the Center for Computational Molecular Science and Technology (Gatech), the National Science Foundation (CHE-0456892, LMT, KMS; CHE-0750275 UB, PM; CRIF CHE-0443564, CJF), and the National Institutes for Health (DK 68096, CJF) for financial support.
Footnotes
Supporting Information Available: Synthetic details for 1 and 3, a description of the experimental procedures and titration curves. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Benfaremo N, Sandman DJ, Tripathy S, Kumar J, Yang K, Rubner MF, Lyons C. Macromolecules. 1998;31:3595–3599. [Google Scholar]
- 2.For leading references, see; (a) Saltiel J, Sun YP. J Phys Chem. 1989;93:6246–50. [Google Scholar]; (b) Meier H. Angew Chem Int Ed. 1992;31:1399–1440. [Google Scholar]; (c) Waldeck DH. J Mol Liq. 1993;57:127–148. [Google Scholar]
- 3.Sandros K, Sundahl M, Wennerström O, Norinder U. J Am Chem Soc. 1990;112:3082–3086. [Google Scholar]
- 4.Fengqiang Z, Motoyoshiya J, Nakamura J, Nishii Y, Aoyama H. Photochem Photobiol. 2006;82:1645–1650. doi: 10.1562/2006-01-17-RA-780. [DOI] [PubMed] [Google Scholar]
- 5.(a) Lewis FD, Crompton EM. J Am Chem Soc. 2003;125:4044–4045. doi: 10.1021/ja029873e. [DOI] [PubMed] [Google Scholar]; (b) Crompton EM, Lewis FD. Photochem Photobiol Sci. 2004;3:660–668. doi: 10.1039/b403661a. [DOI] [PubMed] [Google Scholar]; (c) Lewis FD, Sinks LE, Weigel W, Sajimon MC, Crompton EM. J Phys Chem A. 2005;109:2443–2451. doi: 10.1021/jp044942s. [DOI] [PubMed] [Google Scholar]
- 6.Canals I, Oumada FZ, Roses M, Bosch E. J Chromatogr A. 2001;911:191–202. doi: 10.1016/s0021-9673(00)01271-1. [DOI] [PubMed] [Google Scholar]
- 7.Binstead RA, Zuberbühler AD. SPECFIT v. 3.0.40. Spectrum Software Associates; Marlborough MA: 2007. [Google Scholar]
- 8.Grabowski ZR, Grabowska A. Z Phys Chem N F. 1976;101:197–208. [Google Scholar]
- 9.Tolbert LM, Solntsev KM. Acc Chem Res. 2002;35:19–27. doi: 10.1021/ar990109f. [DOI] [PubMed] [Google Scholar]
- 10.Solntsev KM, Poizat P, Dong J, Rehault J, Lou Y, Burda C, Tolbert LM. J Phys Chem B. 2008;112:2700–2711. doi: 10.1021/jp077707t. [DOI] [PubMed] [Google Scholar]
- 11.Hsu FC, Hayashi M, Wang HW, Lin SH, Wang JK. J Phys Chem. 2007;111:759–763. doi: 10.1021/jp0652897. [DOI] [PubMed] [Google Scholar]
- 12.Borden WT, Davidson ER. J Am Chem Soc. 1977;99:4587–4594. [Google Scholar]; (b) Properly speaking, “disjoint” refers to diradicals, but the description applies to radical anions and dianions as well.
- 13.(a) Wilson JN, Bunz UHF. J Am Chem Soc. 2005;127:4124–4125. doi: 10.1021/ja050017n. [DOI] [PubMed] [Google Scholar]; (b) Zucchero AJ, Wilson JN, Bunz UHF. J Am Chem Soc. 2006;128:11872–11881. doi: 10.1021/ja061112e. [DOI] [PubMed] [Google Scholar]
- 14.(a) Lower SK, El-Sayed MA. Chem Rev. 1966;66:199–241. [Google Scholar]; (b) El-Sayed MA. Acc Chem Res. 1968;1:8–16. [Google Scholar]
- 15.(a) Brederek K, Forster T, Oesterlin HG. In: Luminescence of Organic and Inorganic Materials. Kallman HP, Sprunch GM, editors. Wiley; New York: 1962. p. 161. [Google Scholar]; (b) Young V, Jr, Quiring HL, Sykes AG. J Am Chem Soc. 1997;119:12477–12480. [Google Scholar]; (c) Leray I, Lefevre JP, Delouis JF, Delaire J, Valeur B. Chem Eur J. 2001;7:4590–4598. doi: 10.1002/1521-3765(20011105)7:21<4590::aid-chem4590>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]; (d) Zhou Z, Fahrni CJ. J Am Chem Soc. 2004;126:8862–8863. doi: 10.1021/ja049684r. [DOI] [PubMed] [Google Scholar]
- 16.Head-Gordon M, Grana AM, Maurice D, White CA. J Phys Chem. 1995;99:14261–14270. [Google Scholar]
- 17.IUPAC names: 4,4′-(1E,1′E)-2,2′-(2,5-bis((4-tert-butylphenyl)ethynyl)-1,4-phenylene)bis(ethene-2,1-diyl)diphenol and 3,3′-(1E,1′E)-2,2′-(2,5-bis((4-tert-butylphenyl)ethynyl)-1,4-phenylene)bis(ethene-2,1-diyl)diphenol.
- 18.McGrier PL, Solntsev KM, Schönhaber J, Brombosz SM, Tolbert LM, Bunz UHF. Chem Commun. 2007:2127–2129. doi: 10.1039/b702883k. [DOI] [PubMed] [Google Scholar]
- 19.Hudson B, Kohler B. Synth Met. 1984;9:241–53. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.