

Abstract
Context
Berardinelli-Seip Congenital Lipodystrophy (BSCL) is a rare autosomal recessive syndrome characterized by a difficulty in storing lipids in adipocytes, low body fat mass, hypoleptinemia, and hyperinsulinemia. Sclerostin is a product of SOST gene that blocks the Wnt/β-catenin pathway, decreasing bone formation and enhancing adipogenesis. There are no data about sclerostin in people with BSCL.
Objective
We aimed to evaluate serum sclerostin, bone mineral density (BMD), and L1-L4 Trabecular Bone Score (TBS) in BSCL patients, generating new knowledge about potential mechanisms involved in the bone alterations of these patients.
Design, Setting, and Patients
In this cross-sectional study, we included 11 diabetic patients with BSCL (age 24.7±8.1 years; 6 females). Sclerostin, leptin, L1-L4 TBS, BMD were measured. Potential pathophysiological mechanisms have been suggested.
Results
Mean serum sclerostin was elevated (44.7±13.4 pmol/L) and was higher in men than women (55.3±9.0 vs. 35.1±8.4 pmol/L, p=0.004). Median of serum leptin was low [0.9 ng/mL (0.5-1.9) ]. Seven out of 11 patients had normal BMD, while four patients had high bone mass (defined as Z-score>+2.5SD). Patients on insulin had lower sclerostin (37.3±9.2 vs. 52.6±13.4 pmol/L, p=0.05). The mean TBS was 1.402±0.106, and it was higher than 1.300 in nine patients.
Conclusions
Patients with lipoatrophic diabetes (BSCL) have high serum concentrations of sclerostin, normal or high BMD, and reasonable trabecular bone mass measured by TBS. This is the first report of high sclerostin and good bone microarchitecture (TBS) in BSCL patients.
Keywords: sclerostin, lipodystrophy, bone, diabetes
Introduction
Congenital generalized lipodystrophy is a rare autosomal recessive disease first described in Brazil by Waldemar Berardinelli [1], and five years later by Martin Seip in Norway [2], named later as Berardinelli-Seip Congenital Lipodystrophy (BSCL). Patients with this syndrome lack the capacity to store lipids in adipocytes, causing severe insulin resistance diabetes, hepatomegaly, hypertriglyceridemia, and pancreatitis.
There are currently four types of BSCL described [3-6], and Types 1 and 2 are the most prevalent, accounting for 95% of cases [7]. In the Brazilian BSCL cohort, from the state of Rio Grande do Norte, there are very high prevalence rates of Type 2 as described previously [8] due to consanguineous marriages.
We have previously shown that patients with BSCL have normal or even elevated BMD [9]. Hyperinsulinemia, hypoleptinemia, muscular hypertrophy are some potential mechanisms involved [10]. Sclerostin is a product of SOST gene, generated almost exclusively by osteocytes. It binds to the LRP5 or LRP6 receptor, preventing the action of the Wnt/β-catenin pathway, and decreasing bone formation. Recent reports suggest that sclerostin may also enhance adipogenesis through inhibition of the Wnt/β-catenin signaling system [11]. It is often reported to be high in type 2 diabetes (T2D) but not in Type 1 [12]. There are no data on serum sclerostin of Berardinelli-Seip Congenital Lipodystrophy (BSCL) patients. Once these patients have diabetes with severe insulin resistance and very low body fat, sclerostin should be altered in some way.
In the last years, the acknowledgment about sclerostin is increasing, and it is no longer considered with only a bone function. The aim of our paper was to generate new data of sclerostin in a group of patients with diabetes (BSCL) and, differently of type 2 diabetes, with normal bone density and low adipogenesis. These data help to understand better the several functions of sclerostin.
Materials and Methods
Study population
In this descriptive cross-sectional non-interventional study, we enrolled 11 patients with BSCL treated at the outpatient endocrine clinic of the Hospital Universitário Onofre Lopes, Natal, RN, Brazil (Table 1). Inclusion criteria were a diagnosis of BSCL without other diseases or use of drugs that interfere with BMD or bone metabolism (except diabetes and its treatments). Clinical (acromegaloid facies, prognathism, atrophic cheeks [loss of Bichat's fat ball], prominence of the umbilicus, muscle hypertrophy, phlebomegaly, and acanthosis nigricans), and laboratory criteria (hypertriglyceridemia, hypoleptinemia, hyperinsulinemia) were considered for BSCL diagnosis. The presence of mutations in genes AGPAT2 or BSCL2 confirmed the syndrome.
Table 1. Clinical, laboratorial and densitometric results of each BSCL patient.
| Patient # | Gene mutated | Age (years) | Gender | BMI (kg/m2) | Treatment | HbA1c (%) | Leptin (ng/mL) | Sclerostin (pmol/L) | L1-L4 BMD (Z-Score) | Total body BMD (Z-Score) | TBS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | BSCL2 | 27 | Female | 19.3 | Insulin | 9.3 | 1.1 | 31.89 | 0.09 | 0.20 | 1.286 |
| 2 | BSCL2 | 28.9 | Male | 22.6 | Metformin | 7.4 | 0.5 | 62.76 | 1.00 | 1.40 | 1.514 |
| 3 | BSCL2 | 41 | Female | 22.4 | Insulin | 7.9 | 0.5 | 41.67 | 1.08 | 1.00 | 1.423 |
| 4 | BSCL2 | 26.6 | Male | 23.8 | Insulin, metformin | 8.4 | 0.5 | 45.67 | 3.31 | 2.90 | 1.591 |
| 5 | BSCL2 | 33.2 | Male | 27.2 | Diet | 7.9 | NA | 57.26 | 5.59 | 5.60 | 1.478 |
| 6 | AGPAT2 | 15.4 | Female | 16.2 | Insulin, metformin | 12.4 | NA | 30.73 | 1.15 | -0.20 | 1.377 |
| 7 | AGPAT2 | 12.5 | Male | 18.6 | Diet | 6.5 | NA | 45.98 | 1.04 | 1.00 | 1.448 |
| 8 | AGPAT2 | 19.1 | Female | 15.2 | Insulin | 12.4 | NA | 48.57 | 1.12 | 0.00 | 1.310 |
| 9 | BSCL2 | 22.1 | Male | 23.4 | Metformin | 9.3 | 1.1 | 64.66 | 1.58 | -0.30 | 1.440 |
| 10 | BSCL2 | 25.7 | Female | 19.9 | Insulin, linagliptin | 8.9 | 0.8 | 25.46 | 0.86 | 1.60 | 1.331 |
| 11 | BSCL2 | 20 | Female | 18.2 | Diet | 12.3 | 1.9 | 32.31 | 0.65 | 1.20 | 1.234 |
NA = not available. BMI = body mass index. Normal values: leptin 3.7-11.1 (female) and 2.0-5.6 ng/mL (male); sclerostin 12-29 pmol/L.
Serum measurements
Blood samples were collected after an overnight fasting. Serum sclerostin was measured by ELISA (Biomedica, Vienna, Austria, reference range 12 – 29 pmol/L). We used serum albumin to adjust the serum calcium. HOMAIR (homeostasis model assessment) was calculated (fasting glycemia [mmol/L] × fasting insulin [μU/mL]/22.5) [13] and a value higher than 2.7 was considered as indicative of insulin resistance [14]. Osteocalcin, C-telopeptide (CTx), and leptin, due to availability, were only measured in seven patients. Deficiency and insufficiency of vitamin D were defined according to the criteria of the Endocrine Society (25-OH-vitamin D <20 and between 20 and 30 ng/mL, respectively) [15].
Bone density and TBS measurement
BMD was measured in the lumbar spine (LS), femoral neck (FN), total hip (TH) radius ultra distal (UD), radius 33% (R33), and total body less head (TBLH) using the same densitometer (GE Lunar) and was analyzed by one of the authors (JGL). Patients under 20 years of age had the BMD measured only in the LS and TBLH as recommended [16]. We defined low and high bone density as a BMD Z-Score lower than -2.0SD and higher than +2.5SD, respectively [17]. L1-L4 Trabecular Bone Score (TBS, TBS insight v3.0, Medimaps SASU, Merignac, France) was measured by one of the authors (RW) blinded to the clinical and laboratory characteristics of participants. It was used to estimate bone microarchitecture [18].
Statistical analysis
Parametric data are expressed as mean (standard deviation), and non-parametric data are expressed as median (minimum - maximum). Proportions are presented as n (%). The Shapiro-Wilk test was used to analyze normality data distribution. T-Test was used to compare mean of parametric data, and the Mann-Whitney test was used for non-parametric data. A p-value<0.05 was considered statistically significant.
Ethical Considerations
The research protocol was reviewed and approved by the Federal University of Rio Grande do Norte Ethical Committee (CAAE 14070213.3.0000.5537). Written informed consent was obtained from all participants or legal guardians.
Results
Clinical characteristics of Berardinelli-Seip participants
Eight patients had BSCL Type 2 (mutation in BSCL2 gene), and three patients had Type 1 (AGPAT2 gene mutation). These patients are part of a cohort previously published [8]. The mean age was 24.7±8.1 years old, and there were six females. Five patients had menarche at an average age of 14.4±2.3 years, and one had not yet menstruated (15 years old). There was no history of alcohol intake or smoking. The mean BMI of patients was 20.6±3.6 kg/m2, and the total body fat measured by DXA scan was 5.5±0.9%. Muscle hypertrophy detected on physical examination was present in nine patients (81.8%). All patients had diabetes. The mean age of diabetes onset was 15.8±5.5 years old, and the diabetes duration was 11.1±7.2 years. They were on one or more treatments, including insulin (6/11; 54.5%), metformin (4/11; 36.4%), and DPP-4 inhibitor (one patient).
Laboratory findings
The glycemic control was poor, and the kidney function was normal (Table 2). Hypertriglyceridemia was present in ten patients (90.9%), and ten patients also had insulin resistance defined as HOMAIR> 2.7. HDL cholesterol was low (Table 2), and the median of leptin was 0.9 ng/mL (0.5-1.9).
Table 2. Laboratory results of patients with BSCL.
| Test | Result | # Patients altered | Reference |
|---|---|---|---|
| Fasting glycemia (mg/dL) | 220.1±110.1 | 10/11 (90.9%) | <100 |
| HbA1c (%) | 9.3±2.1 | 10/11 (90.9%) | <7.0 |
| Total Cholesterol (mg/dL) | 185±18.1 | 3/11 (27.2%) | <200 |
| HDL cholesterol (mg/dL) | 35.6±5.0 | 11 (100%) | >45 |
| Triglycerides (mg/dL) | 228.0 (80.0-1300.0) | 9/11 (81.8%) | <150 |
| Creatinine (mg/dL) | 0.5±0.1 | 0/11 | <1.3 |
| Calcium (mg/dL) | 10.1±0.5 | 0/11 | 8.8-11.0 |
| Phosphate (mg/dL) | 4.1±1.0 | 0/11 | 2.3-5.5 |
| Alkaline phosphatase (UI/L) | 81.6±46.0 | 0/11 | 27-100 |
| AST (mg/dL) | 24.0±6.1 | 0/11 | <40 |
| ALT (mg/dL) | 28.2±8.5 | 1/11 (9%) | <40 |
| GGT (UI/L) | 32.0 (17.0-356.0) | 3/11 (27.3%) | <36(F)/<64(M) |
| TSH (mIU/L) | 1.2±0.4 | 0/11 | 0.4-4.0 |
| PTH (pg/mL) | 26.0 (10.3-98.6) | 1/11 (9%) | 12-72 |
| 25-OH-vitamin D (ng/mL) | 27.8±9.1 | 7/11 (63.6%) | >30 |
| Insulin (mIU/L) | 15.1 (7.0-115.0) | 5/11 (45.4%) | <16 |
| HOMA-R | 11.3 (2.4-73.8) | 10/11 (90.9%) | <2.7 |
| Osteocalcin (ng/mL) | 16.0 (6.0-23.0) | 5/7 (71.4%) | - |
| C-telopeptide (ng/mL) | 0.348±0.172 | 1/7 (14.2%) | - |
F=female; M=male. AST = aspartate aminotransferase. ALT = alanine aminotransferase. GGT = Gamma-glutamyl transpeptidase.
Number of patients (%) with the exam altered.
Mean serum sclerostin was 44.7±13.4 pmol/L, and ten patients (90.9%) had serum sclerostin higher than reference values (12 – 29 pmol/L). It was higher in men than women (55.3±9.0 vs. 35.1±8.4 pmol/L, p=0.004), and was lower in patients on insulin (37.3±9.2 vs. 52.6±13.4 pmol/L, p=0.05). Figure 1 shows the sclerostin values according to age, sex, and TBS. Three patients (27.3%) had vitamin D deficiency, and four (36.4%) had insufficiency. Five out of 7 patients (71.4%) had low osteocalcin, and two (28.6%) had normal. No patients had low CTx, and this was normal in 6 out of 7 (85.7%) and high in one (14.3%).
Figure 1. Serum sclerostin according to (A) age, (B) gender, and (C) trabecular bone score in patients with BSCL.
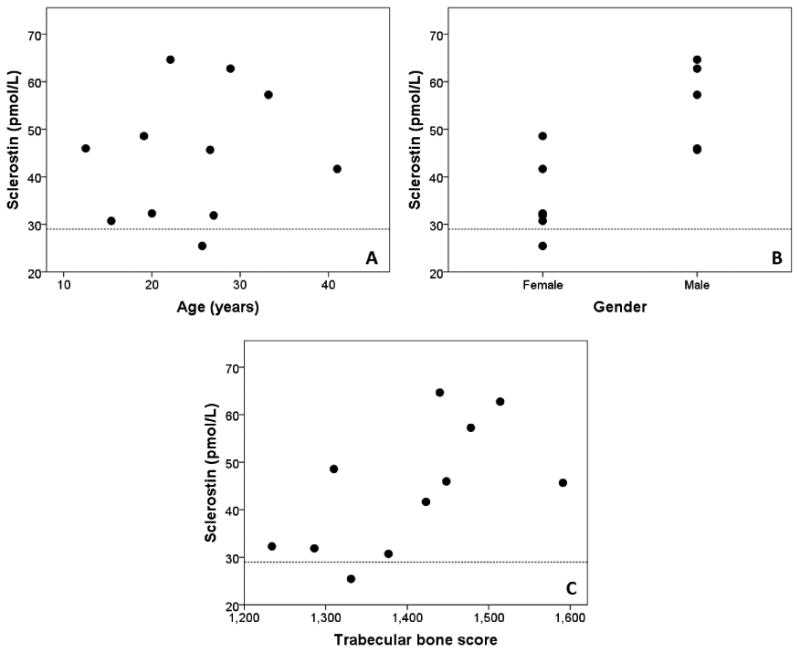
Dashed line indicates the upper normal limit of sclerostin.
Bone mineral density
One patient had low bone mass on Radius 33% (Z-score-2.2SD), and four patients (36.4%) had high bone mass (defined as Z-score>+2.5SD). Sclerostin concentration was not different in these patients (44.2±10.3 vs. 44.3±15.6 pmol/L, p>0.05).
The mean TBS was 1.402±0.106, and nine of 11 patients had values higher than 1.300. Males had higher TBS than female (1.494±0.061 vs. 1.326±0.066, p=0.002). Table 1 shows values of HbA1c, leptin, TBS, Z-Score and serum sclerostin of each patient.
Discussion
The Wnt/β-catenin signaling pathway is essential for bone formation. Sclerostin is a protein encoded by the SOST gene and synthesized by osteocyte. It blocks the Wnt/β-catenin (an anti-anabolic bone effect), reducing bone formation [19]. Sclerostin may also induce adipogenesis in vitro through the same mechanism, i.e. antagonism of the Wnt/β-catenin signaling [11]. Treatment with anti-sclerostin monoclonal antibody increases spine and hip BMD, confirming its potential anti-anabolic bone effect [20].
It has been reported that BSCL patients have high BMD [9, 21]. We first report here a case series of BSCL patients showing that they have normal trabecular and areal bone density (according to TBS values), despite high serum sclerostin.
Patients with insulin resistance, like prediabetes and type 2 diabetes (T2D) patients, usually have elevated values of serum sclerostin [22, 23]. Likewise, SOST gene expression is increased in diabetic rats [24]. BSCL patients have severe insulin resistance due to ectopic fat deposition, and all of our patients reported here had lipoatrophic diabetes. Sclerostin levels were shown to correlate positively with BMD in healthy adults [25], and it was higher than the upper normal limit in almost all BSCL patients, potentially as a compensatory mechanism to prevent further increases in bone formation. Indeed, most patients had low serum osteocalcin (a marker of bone formation), supporting the bone anti-anabolic sclerostin effect. Otherwise, this increase could also indicate a resistance to sclerostin. If so, the lack of action of sclerostin could help in the development of the high BMD.
There are several caveats related to serum sclerostin. First, it is possible that non-skeletal sources can be involved in serum levels of sclerostin. Calcification of vascular smooth muscle cells in vitro up-regulates expression of sclerostin in these cells [26]. These could be the source of sclerostin excess in patients with established atherosclerosis. In T2D, for every increase of 1 pmol/L of sclerostin, there is a 4% increase in the chance of having an atherosclerotic disease (abnormal intima-media thickness, carotid plaque or aortic calcifications) [27]. Patients with BSCL have hypertriglyceridemia, very low HDL (high-density lipoprotein), and probably small-dense LDL (low-density lipoprotein), a very atherogenic triad [28]. Thus, accelerated atherogenesis could be a potential mechanism to explain high sclerostin values in these patients. Indeed, serum sclerostin levels in BSCL are as high as reported in diabetic patients with atherogenesis. However, some of our BSCL patients as young as five years old (not included here) already have high serum sclerostin, even without presenting clinically detectable atherosclerosis (unpublished data). It is also conceivable that serum sclerostin does not reflect local sclerostin actions on the skeleton. A recent abstract suggests that there is no or little relationship between serum sclerostin and skeletal sclerostin in mice (Baldock- ASBMR abstract, 2016). Similarly, a recent study in diabetic rats suggested the sclerostin does not have a major role in skeletal alterations of T2D [29].
TBS reflects the microarchitecture of the vertebrae bone [18]. We first report here the TBS is not low in lipoatrophic diabetes (BSCL). In T2D, sclerostin is high, and BMD is normal or even high, but the TBS is low, indicating a poor bone microarchitecture [30]. Type 2 diabetics are more prone to fractures, even with a higher BMD [12, 31] and elevated sclerostin levels are associated with vertebral fractures in these patients [32]. On the other hand, BSCL patients are not prone to fracture, and in this report, TBS scores were normal (>1.300 in nine of the 11 patients) despite high serum sclerostin. Thus, in BSCL, high serum sclerostin does not seem to be deleterious to bone mass as some authors have suggested for T2D [33]. Metabolic alterations, like insulin resistance, can contribute to explain the rising of sclerostin. Patients with Type 1 diabetes do not have insulin resistance, and serum sclerostin does not differ from controls [12, 34].
Patients on insulin (especially patients 1, 6, and 10 – Table 1) in our study had lower serum sclerostin than patients without insulin therapy. If states of impaired insulin action (insulin resistance – diabetes, prediabetes) are related to sclerostin increase, the use of high daily doses of insulin, as we see in BSCL patients, could minimize the sclerostin increment. Moreover, PTH acts decreasing sclerostin expression [35], and insulin potentiates the effect of the PTH on the bone [36]. Hence, it is conceivable that patients on insulin could be more sensitive to the suppressive effects of PTH.
Usually, serum sclerostin increases with aging, possibly due to a decrease in sclerostin clearance [23]. However, even the young subjects reported here (patients 7 and 8 – Table 1) had high sclerostin levels, indicating that early metabolic changes are more important factors in this population.
Mechanical loading usually inhibits sclerostin expression, and weight loss increases it [37]. Patients with BSCL usually have BMI at the lower limit of normal or even below (patients 6, 7, and 11 – Table 1). The lower body weight of these patients could contribute to the higher values of serum sclerostin. Patients with anorexia nervosa also have low weight and low leptin, but sclerostin is normal [38]. Low BMI does not appear to be a determining factor in the elevation of sclerostin in patients with BSCL.
Table 3 shows differences in some parameters of several clinical conditions. The combination of hyperinsulinemia and low leptinemia, as we see in BSCL patients, looks good to the bone. In clinical conditions with only hyperinsulinemia (T2D), the BMD is high, but the TBS is low. In diseases with low leptinemia (anorexia nervosa and congenital generalized lipodystrophy without insulin resistance) and normal insulin, the BMD is low [39, 40]. If insulin is low and leptin is normal (T1D), there is a tendency to BMD and TBS to be normal or low [41]. Hyperinsulinemia is important to increase BMD. We could hypothesize that low leptin is important to have a high TBS. The replacement of metreleptin for BSCL patients do not change BMD, but there is no data about TBS changes [21].
Table 3. Differences among BSCL and other clinical conditions that can help to understand BMD and TBS results.
| BMD | TBS | Insulin | Leptin | Sclerostin | BMI | Muscle | |
|---|---|---|---|---|---|---|---|
| BSCL [8, 9] | Nl or ↑ | Nl or ↑ | ↑↑ | ↓↓ | ↑ | Nl or ↓ | Hypertrophy |
| T2D [23, 30, 31] | Nl or ↑ | ↓ | ↑ | Nl or ↑ | ↑ | Nl or ↑ | Nl or sarcopenia |
| T1D [12, 34, 41] | Nl or ↓ | Nl or ↓ | ↓ | Nl | Nl | Nl | Nl |
| Anorexia nervosa [38, 39] | ↓ | Nl or ↓ | Nl | ↓ | Nl | ↓↓ | ↓ |
| LCG without insulin resistance [40] | ↓ | Unknown | ↓ | ↓ | Unknown | ↓ | ↓ |
BSCL: Berardinelli-Seip Congenital Lipodystrophy. T2D: type 2 diabetes. T1D: type 1 diabetes. LCG: Lipodystrophy congenital generalized. BMD: bone mineral density. TBS: trabecular bone score. Insulin: serum insulin. Leptin: serum leptin. BMI: body mass index. Nl: Normal. ↑: elevated. ↓: decreased.
In vitro data suggest that muscle cells produce some factor that protects osteocytes by activating the Wnt/β-catenin pathway [42]. Differently from patients with T2D, muscle hypertrophy is present in BSCL patients, and this may contribute to the bone findings of these patients. In contrast, patients with anorexia nervosa or congenital generalized lipodystrophy (CGL) without hyperinsulinemia have decreased muscle mass and low bone density [39, 40].
Our study has some limitations since it is a cross-sectional study, not allowing establishing causality. Also, the number of patients is small, but BSCL is a rare disease with a prevalence of only 1 case in 1 million [7]. TBS is not the gold standard method to assess bone quality, but it is the only reliable and noninvasive indirect method to perform trabecular bone measurements in daily clinical routine. We did not report any case of BSCL Types 3 and 4, but the types here reported account for 95% of all BSCL cases [7].
In conclusion, patients with lipoatrophic diabetes (BSCL) have high serum concentrations of sclerostin, normal or high BMD, and reasonable trabecular bone mass measured by TBS. This is the first report of high serum sclerostin and good bone microarchitecture (TBS) in BSCL patients, and this may inform our understanding of the pathophysiology of the diabetic bone disease. Further prospective studies in humans are warranted to fully elucidate sclerostin actions in patients with insulin resistance.
Highlights.
Patients with BSCL have normal or high BMD, and this may be related to high values of insulin.
Differently of T2D who also have high BMD, the bone microarchitecture estimated by TBS is good, and the patients are not prone to fracture.
Serum sclerostin is high, but this elevation is probably a compensatory mechanism, or it is related to sclerostin functions beyond the bone tissue.
Males tend to have higher, and patients on treatment with insulin have lower serum sclerostin values.
Hypernsulinemia, low leptinemia, muscle hypertrophy are potential mechanisms of bone alterations.
Acknowledgments
We thank the staff from the Hospital Universitário Onofre Lopes, Universidade Federal do Rio Grande do Norte, for their help with subject recruitment. This work was supported in part by grants P50 AI-30639 (SMBJ) from the US National Institutes of Health and Conselho Nacional de Pesquisa (CNPq). The funders had no role in designing the study nor in the writing of the manuscript.
Footnotes
Disclosure statement: RW is a senior scientist at Medimaps SASU, Merignac, France.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berardinelli W. An undiagnosed endocrinometabolic syndrome: report of 2 cases. J Clin Endocrinol Metab. 1954;14(2):193–204. doi: 10.1210/jcem-14-2-193. [DOI] [PubMed] [Google Scholar]
- 2.Seip M. Lipodystrophy and gigantism with associated endocrine manifestations. A new diencephalic syndrome? Acta Paediatr. 1959;48:555–74. [PubMed] [Google Scholar]
- 3.Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, Gurakan F, Kocak N, O'Rahilly S, Taylor SI, Patel SB, Bowcock AM. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84(9):3390–4. doi: 10.1210/jcem.84.9.6103. [DOI] [PubMed] [Google Scholar]
- 4.Magre J, Delepine M, Khallouf E, Gedde-Dahl T, Jr, Van Maldergem L, Sobel E, Papp J, Meier M, Megarbane A, Bachy A, Verloes A, d'Abronzo FH, Seemanova E, Assan R, Baudic N, Bourut C, Czernichow P, Huet F, Grigorescu F, de Kerdanet M, Lacombe D, Labrune P, Lanza M, Loret H, Matsuda F, Navarro J, Nivelon-Chevalier A, Polak M, Robert JJ, Tric P, Tubiana-Rufi N, Vigouroux C, Weissenbach J, Savasta S, Maassen JA, Trygstad O, Bogalho P, Freitas P, Medina JL, Bonnicci F, Joffe BI, Loyson G, Panz VR, Raal FJ, O'Rahilly S, Stephenson T, Kahn CR, Lathrop M, Capeau J. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28(4):365–70. doi: 10.1038/ng585. [DOI] [PubMed] [Google Scholar]
- 5.Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O'Rahilly S, Dugail I, Capeau J, Lathrop M, Magre J. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1129–34. doi: 10.1210/jc.2007-1328. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, Park YE, Nonaka I, Hino-Fukuyo N, Haginoya K, Sugano H, Nishino I. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119(9):2623–33. doi: 10.1172/JCI38660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patni N, Garg A. Congenital generalized lipodystrophies--new insights into metabolic dysfunction. Nat Rev Endocrinol. 2015;11(9):522–34. doi: 10.1038/nrendo.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lima JG, Nobrega LH, de Lima NN, do Nascimento Santos MG, Baracho MF, Jeronimo SM. Clinical and laboratory data of a large series of patients with congenital generalized lipodystrophy. Diabetol Metab Syndr. 2016;8:23. doi: 10.1186/s13098-016-0140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lima JG, Nobrega LH, Lima NN, Dos Santos MC, Baracho MF, Bandeira F, Capistrano L, Freire Neto FP, Jeronimo SM. Bone Density in Patients With Berardinelli-Seip Congenital Lipodystrophy Is Higher in Trabecular Sites and in Type 2 Patients. J Clin Densitom. 2016 doi: 10.1016/j.jocd.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Lima JG, Lima NN, Nobrega LH, Jeronimo SM. Conversations between insulin and bone: Potential mechanism of high bone density in patients with Berardinelli-Seip Congenital Lipodystrophy. Med Hypotheses. 2016;97:94–97. doi: 10.1016/j.mehy.2016.10.025. [DOI] [PubMed] [Google Scholar]
- 11.Ukita M, Yamaguchi T, Ohata N, Tamura M. Sclerostin Enhances Adipocyte Differentiation in 3T3-L1 Cells. J Cell Biochem. 2016;117(6):1419–28. doi: 10.1002/jcb.25432. [DOI] [PubMed] [Google Scholar]
- 12.Gennari L, Merlotti D, Valenti R, Ceccarelli E, Ruvio M, Pietrini MG, Capodarca C, Franci MB, Campagna MS, Calabro A, Cataldo D, Stolakis K, Dotta F, Nuti R. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J Clin Endocrinol Metab. 2012;97(5):1737–44. doi: 10.1210/jc.2011-2958. [DOI] [PubMed] [Google Scholar]
- 13.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 14.Geloneze B, Vasques AC, Stabe CF, Pareja JC, Rosado LE, Queiroz EC, Tambascia MA. HOMA1-IR and HOMA2-IR indexes in identifying insulin resistance and metabolic syndrome: Brazilian Metabolic Syndrome Study (BRAMS) Arq Bras Endocrinol Metabol. 2009;53(2):281–7. doi: 10.1590/s0004-27302009000200020. [DOI] [PubMed] [Google Scholar]
- 15.Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, Murad MH, Weaver CM. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(7):1911–30. doi: 10.1210/jc.2011-0385. [DOI] [PubMed] [Google Scholar]
- 16.Bishop N, Braillon P, Burnham J, Cimaz R, Davies J, Fewtrell M, Hogler W, Kennedy K, Makitie O, Mughal Z, Shaw N, Vogiatzi M, Ward K, Bianchi ML. Dual-energy X-ray aborptiometry assessment in children and adolescents with diseases that may affect the skeleton: the 2007 ISCD Pediatric Official Positions. J Clin Densitom. 2008;11(1):29–42. doi: 10.1016/j.jocd.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 17.Whyte MP. Misinterpretation of osteodensitometry with high bone density: BMD Z > or = + 2.5 is not “normal”. J Clin Densitom. 2005;8(1):1–6. doi: 10.1385/jcd:8:1:001. [DOI] [PubMed] [Google Scholar]
- 18.Hans D, Barthe N, Boutroy S, Pothuaud L, Winzenrieth R, Krieg MA. Correlations between trabecular bone score, measured using anteroposterior dual-energy X-ray absorptiometry acquisition, and 3-dimensional parameters of bone microarchitecture: an experimental study on human cadaver vertebrae. J Clin Densitom. 2011;14(3):302–12. doi: 10.1016/j.jocd.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Yavropoulou MP, Xygonakis C, Lolou M, Karadimou F, Yovos JG. The sclerostin story: from human genetics to the development of novel anabolic treatment for osteoporosis. Hormones (Athens) 2014;13(4):323–37. doi: 10.14310/horm.2002.1552. [DOI] [PubMed] [Google Scholar]
- 20.Recker RR, Benson CT, Matsumoto T, Bolognese MA, Robins DA, Alam J, Chiang AY, Hu L, Krege JH, Sowa H, Mitlak BH, Myers SL. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res. 2015;30(2):216–24. doi: 10.1002/jbmr.2351. [DOI] [PubMed] [Google Scholar]
- 21.Christensen JD, Lungu AO, Cochran E, Collins MT, Gafni RI, Reynolds JC, Rother KI, Gorden P, Brown RJ. Bone mineral content in patients with congenital generalized lipodystrophy is unaffected by metreleptin replacement therapy. J Clin Endocrinol Metab. 2014;99(8):E1493–500. doi: 10.1210/jc.2014-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daniele G, Winnier D, Mari A, Bruder J, Fourcaudot M, Pengou Z, Tripathy D, Jenkinson C, Folli F. Sclerostin and Insulin Resistance in Prediabetes: Evidence of a Cross Talk Between Bone and Glucose Metabolism. Diabetes Care. 2015;38(8):1509–17. doi: 10.2337/dc14-2989. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Martin A, Rozas-Moreno P, Reyes-Garcia R, Morales-Santana S, Garcia-Fontana B, Garcia-Salcedo JA, Munoz-Torres M. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(1):234–41. doi: 10.1210/jc.2011-2186. [DOI] [PubMed] [Google Scholar]
- 24.Nuche-Berenguer B, Moreno P, Portal-Nunez S, Dapia S, Esbrit P, Villanueva-Penacarrillo ML. Exendin-4 exerts osteogenic actions in insulin-resistant and type 2 diabetic states. Regul Pept. 2010;159(1-3):61–6. doi: 10.1016/j.regpep.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Amrein K, Amrein S, Drexler C, Dimai HP, Dobnig H, Pfeifer K, Tomaschitz A, Pieber TR, Fahrleitner-Pammer A. Sclerostin and its association with physical activity, age, gender, body composition, and bone mineral content in healthy adults. J Clin Endocrinol Metab. 2012;97(1):148–54. doi: 10.1210/jc.2011-2152. [DOI] [PubMed] [Google Scholar]
- 26.Zhu D, Mackenzie NC, Millan JL, Farquharson C, MacRae VE. The appearance and modulation of osteocyte marker expression during calcification of vascular smooth muscle cells. PLoS One. 2011;6(5):e19595. doi: 10.1371/journal.pone.0019595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morales-Santana S, Garcia-Fontana B, Garcia-Martin A, Rozas-Moreno P, Garcia-Salcedo JA, Reyes-Garcia R, Munoz-Torres M. Atherosclerotic disease in type 2 diabetes is associated with an increase in sclerostin levels. Diabetes Care. 2013;36(6):1667–74. doi: 10.2337/dc12-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Girard-Mauduit S. The lipid triad, or how to reduce residual cardiovascular risk? Ann Endocrinol (Paris) 2010;71(2):89–94. doi: 10.1016/j.ando.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Pereira M, Gohin S, Lund N, Hvid A, Smitham PJ, Oddy MJ, Reichert I, Farlay D, Roux JP, Cleasby ME, Chenu C. Sclerostin does not play a major role in the pathogenesis of skeletal complications in type 2 diabetes mellitus. Osteoporos Int. 2016 doi: 10.1007/s00198-016-3718-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harvey NC, Gluer CC, Binkley N, McCloskey EV, Brandi ML, Cooper C, Kendler D, Lamy O, Laslop A, Camargos BM, Reginster JY, Rizzoli R, Kanis JA. Trabecular bone score (TBS) as a new complementary approach for osteoporosis evaluation in clinical practice. Bone. 2015;78:216–24. doi: 10.1016/j.bone.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leslie WD, Rubin MR, Schwartz AV, Kanis JA. Type 2 diabetes and bone. J Bone Miner Res. 2012;27(11):2231–7. doi: 10.1002/jbmr.1759. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto M, Yamauchi M, Sugimoto T. Elevated sclerostin levels are associated with vertebral fractures in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2013;98(10):4030–7. doi: 10.1210/jc.2013-2143. [DOI] [PubMed] [Google Scholar]
- 33.Gaudio A, Privitera F, Battaglia K, Torrisi V, Sidoti MH, Pulvirenti I, Canzonieri E, Tringali G, Fiore CE. Sclerostin levels associated with inhibition of the Wnt/beta-catenin signaling and reduced bone turnover in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(10):3744–50. doi: 10.1210/jc.2012-1901. [DOI] [PubMed] [Google Scholar]
- 34.Catalano A, Pintaudi B, Morabito N, Di Vieste G, Giunta L, Bruno ML, Cucinotta D, Lasco A, Di Benedetto A. Gender differences in sclerostin and clinical characteristics in type 1 diabetes mellitus. Eur J Endocrinol. 2014;171(3):293–300. doi: 10.1530/EJE-14-0106. [DOI] [PubMed] [Google Scholar]
- 35.Kramer I, Keller H, Leupin O, Kneissel M. Does osteocytic SOST suppression mediate PTH bone anabolism? Trends Endocrinol Metab. 2010;21(4):237–44. doi: 10.1016/j.tem.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki K, Miyakoshi N, Tsuchida T, Kasukawa Y, Sato K, Itoi E. Effects of combined treatment of insulin and human parathyroid hormone(1-34) on cancellous bone mass and structure in streptozotocin-induced diabetic rats. Bone. 2003;33(1):108–14. doi: 10.1016/s8756-3282(03)00169-8. [DOI] [PubMed] [Google Scholar]
- 37.Armamento-Villareal R, Sadler C, Napoli N, Shah K, Chode S, Sinacore DR, Qualls C, Villareal DT. Weight loss in obese older adults increases serum sclerostin and impairs hip geometry but both are prevented by exercise training. J Bone Miner Res. 2012;27(5):1215–21. doi: 10.1002/jbmr.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Faje AT, Fazeli PK, Katzman DK, Miller KK, Breggia A, Rosen CJ, Mendes N, Klibanski A, Misra M. Sclerostin levels and bone turnover markers in adolescents with anorexia nervosa and healthy adolescent girls. Bone. 2012;51(3):474–9. doi: 10.1016/j.bone.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donaldson AA, Feldman HA, O'Donnell JM, Gopalakrishnan G, Gordon CM. Spinal Bone Texture Assessed by Trabecular Bone Score in Adolescent Girls With Anorexia Nervosa. J Clin Endocrinol Metab. 2015;100(9):3436–42. doi: 10.1210/jc.2015-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajab A, Khaburi M, Spranger S, Kunze J, Spranger J. Congenital generalized lipodystrophy, mental retardation, deafness, short stature, and slender bones: a newly recognized syndrome? Am J Med Genet A. 2003;121A(3):271–6. doi: 10.1002/ajmg.a.20245. [DOI] [PubMed] [Google Scholar]
- 41.Neumann T, Lodes S, Kastner B, Lehmann T, Hans D, Lamy O, Muller UA, Wolf G, Samann A. Trabecular bone score in type 1 diabetes--a cross-sectional study. Osteoporos Int. 2016;27(1):127–33. doi: 10.1007/s00198-015-3222-y. [DOI] [PubMed] [Google Scholar]
- 42.Jahn K, Lara-Castillo N, Brotto L, Mo CL, Johnson ML, Brotto M, Bonewald LF. Skeletal muscle secreted factors prevent glucocorticoid-induced osteocyte apoptosis through activation of beta-catenin. Eur Cell Mater. 2012;24:197–209. doi: 10.22203/ecm.v024a14. discussion 209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]