

SUMMARY
Diacylglycerol kinases (DGKs) are integral components of signal transduction cascades that regulate cell biology through ATP-dependent phosphorylation of the lipid messenger diacylglycerol. Methods for direct evaluation of DGK activity in native biological systems are lacking and needed to study isoform-specific functions of these multidomain lipid kinases. Here, we utilize ATP acyl phosphate activity-based probes and quantitative mass spectrometry to define, for the first time, ATP- and small molecule-binding motifs of representative members from all five DGK subtypes. We use chemical proteomics to discover an unusual binding mode for the DGK-alpha (DGKα) inhibitor ritanserin, including interactions at the atypical C1 domain distinct from the ATP binding region. Unexpectedly, deconstruction of ritanserin yielded a fragment compound that blocks DGKα activity through a conserved binding mode and enhanced selectivity against the kinome. Collectively, our studies illustrate the power of chemical proteomics to profile protein-small molecule interactions of lipid kinases for fragment-based lead discovery.
Keywords: lipid kinase inhibitors, diacylglycerol kinase, activity based protein profiling, chemical proteomics, fragment based drug discovery, ligand deconstruction, phosphatidic acid signaling, diacylglycerol signaling, lipid phosphorylation, cancer immunotherapy
eTOC BLURB
Franks et al. report the first chemical proteomic map of ligand binding sites of diacylglycerol kinases (DGKs) that reveals key molecular features that distinguish lipid and protein kinases to guide fragment-based discovery of DGK isoform-selective inhibitors.
INTRODUCTION
Diacylglycerols (DAGs) and phosphatidic acid (PA) play fundamental roles in biology as basic components of membranes, intermediates in lipid metabolism, and secondary messengers in cellular signaling (Carrasco and Merida, 2007; Fang et al., 2001). Cells regulate intracellular DAG and PA levels through metabolic networks that utilize distinct enzymes to produce or consume these secondary messengers/metabolites (Brown et al., 2017; Carrasco and Merida, 2007; Hsu et al., 2012; Shulga et al., 2011). One such enzymatic pathway that is central to signal transduction is adenosine triphosphate (ATP)-dependent phosphorylation of DAGs to biosynthesize phosphatidic acid (PA, Figure 1A) by a set of lipid kinases collectively known as diacylglycerol kinases (Shulga et al., 2011) (DGKs). DAG and PA are important lipid messengers that alter localization (Takai et al., 1979), activation (Newton and Koshland, 1989), and protein-protein interactions (Fang et al., 2001) of distinct sets of receptor proteins. Consequently, disruption of the same DGK protein in different cell types can result in opposing effects that can be leveraged, for example, in cancer to simultaneously block tumor growth and activate antitumor immunity (Merida et al., 2017; Sakane et al., 2016). Since DAG and PA serve as key intermediates in lipid metabolism, DGKs are uniquely positioned as key regulators of the structural, bio-energetic, and signaling demands of cells.
Figure 1. Mammalian diacylglycerol kinase (DGK) superfamily.

(A) DGK enzymes catalyze transfer of phosphate from ATP to diacylglycerol (DAG) to biosynthesize phosphatidic acid (PA). The molecular structure of fatty acyl chains regulates functional properties of DAG and PA lipids. (B) The ten DGK isoforms identified to date are divided into five principal subtypes based on organization of structural motifs. Alternative splicing of certain DGK subtypes can generate additional structural diversity. RVH: recoverin homology domain; EF: EF Hands motif; C1: atypical/typical C1 domain; PH: pleckstrin homology domain; SAM: sterile alpha motif; EPAP: Glu-Pro-Ala-Pro repeats; PDZ: protein-protein interactions; HD: hydrophobic domain; MARCKS: myristolated alanine-rich C kinase substrate domain; ANK: Ankyrin repeats; PR: proline-rich region.
Ten mammalian DGKs have been identified and classified into five subtypes based on structural features elucidated from primary sequence analysis (Figure 1B). At the N-terminus, DGKs contain at least two cysteine-rich zinc finger-like motifs similar to C1 domains found in protein kinase C (Carrasco and Merida, 2007) (PKC). DGKs contain a C-terminal catalytic domain composed of a conserved catalytic region (SMART domain (Schultz et al., 1998) DAGKc, SM000046), which is present in other eukaryotic lipid kinases and DGKs from Gram-positive bacteria (Adams et al., 2016), followed by an accessory subdomain (DAGKa, SM000045) of unknown function (Merida et al., 2017). While DGKs share the same basic domain organization, individual subtypes differ widely in regulatory domains proposed to mediate metal binding (EF hand motifs), oligomerization (SAM domain), membrane association (PH domain), subcellular localization (MARCKS domain), or protein-protein interactions (ankyrin repeats, PDZ domain) (Shulga et al., 2011). Given the enormous chemical diversity of DAG and PA lipids (Yetukuri et al., 2008), understanding the cross-talk between regulatory and catalytic domains of DGKs will be critical for assigning metabolic and signaling functions to individual isoforms.
Attempts to define the function of individual DGK domains have resulted in inconclusive results. ATP-binding motifs corresponding to the glycine-rich loops found in protein kinases (GxGxxG consensus sequence (Hanks et al., 1988; Hemmer et al., 1997)) were identified in the first C1 and catalytic domains of DGKs (Sakane et al., 1990; Schaap et al., 1994). Mutation of lysines in these motifs, which abolishes ATP binding and protein kinase activity, did not affect catalytic function of DGKs and led others to hypothesize the existence of a DGK-specific ATP binding motif that remains to be defined (Sakane et al., 1996; Schaap et al., 1994). The role of C1 domains in DGK function is also enigmatic. With the exception of gamma and beta isoforms (Shindo et al., 2003), the C1 domains of DGKs lack conserved residues identified as being required for DAG binding in other proteins including PKC (Hurley and Misra, 2000). In vitro biochemical studies measuring activity of C1 truncation mutants have produced conflicting reports with regards to whether C1 motifs are required (Abe et al., 2003; Houssa et al., 1997; Santos et al., 2002) or dispensable (Merino et al., 2007; Sakane et al., 1996) for maximal DGK catalytic activity.
Thus, DGK active sites remain ill-defined and, combined with the lack of crystal structures for mammalian DGKs, have limited our understanding of substrate and inhibitor binding. As a result, current DGK inhibitors consist of compounds with poor specificity within the DGK superfamily (de Chaffoy de Courcelles et al., 1989; de Chaffoy de Courcelles et al., 1985) or lack selectivity measurements against other lipid and protein kinases (Boroda et al., 2017; Liu et al., 2016; Purow, 2015). Thus, methods that provide information on small molecule binding mode and selectivity are needed to guide development of isoform-selective DGK inhibitors. Selective DGK inhibitors are needed to study isoforms where knockout mice viability is an issue (Crotty et al., 2006) and to help realize the translational potential of targeting specific forms, e.g. DGK-alpha (DGKα), for anticancer (Dominguez et al., 2013) and immunotherapy applications (Prinz et al., 2012).
Here, we use ATP acyl phosphate activity-based probes (Patricelli et al., 2011; Patricelli et al., 2007) and quantitative mass spectrometry to discover ATP- and inhibitor-binding sites of representative members of all five principal DGK subtypes. Our findings define, for the first time, the ATP binding motif of DGKs that is distinct from protein kinases and identifies the DAGKa subdomain as a novel region mediating ATP binding. We discovered a fragment of the DGKα inhibitor ritanserin that shows conservation of binding mode and enhanced selectivity against protein kinases, supporting the concept that the atypical C1 and accessory region of the catalytic domain (DAGKa) are key ligand-binding sites for developing DGKα-selective inhibitors. Our studies demonstrate the utility of chemical proteomics to map ligand binding sites for fragment-based discovery of lipid kinase inhibitors.
RESULTS
ATP acyl phosphates function as activity-based probes of DGKα
To test whether ATP acyl phosphates (Figure 2A) can be used to profile DGK activity, our strategy was to transiently express DGKs and test recombinant enzymes directly in cell proteomes without the need for protein purification. We reasoned this approach would mitigate challenges with detection due to differences in endogenous DGK levels while permitting analyses on a proteomic scale. Our initial studies focused on the alpha isoform (DGKα, Figure 1B) given the availability of inhibitors and matching negative control compounds (Boroda et al., 2017) for our proof-of-principle experiments (Figure 2B). We confirmed overexpression of recombinant FLAG-tagged DGKα by western blot (Figure S1) and used published DAG phosphorylation substrate assays (Sato et al., 2013) to measure recombinant DGKα activity (Figure S2A). We observed significantly higher DAG phosphorylation activity (~6-fold on average) in DGKα-compared with mock-transfected or heat-denatured proteomes (Figure S2B). Furthermore, recombinant DGKα activity was blocked in a concentration-dependent manner using the DGKα inhibitor ritanserin (Boroda et al., 2017) compared with dimethyl sulfoxide (DMSO) vehicle-treated controls (IC50 = 25 μM, Figure 3A). Since ritanserin exhibits 5-HT2 receptor (5-HT2R) inhibitory activity (Barone et al., 1986), we included another 5-HT2R antagonist ketanserin (Boroda et al., 2017) (Figure 2B) to control for non-specific effects in our substrate assay. Ketanserin showed negligible activity against DGKα in our substrate assay, confirming the use of ritanserin and ketanserin as paired probes (i.e. DGK active and inactive inhibitors, respectively, at 100 μM; Figure S2B) suitable for testing in our chemical proteomics assay.
Figure 2. Activity-based probes and inhibitors for functional analysis of DGKs.

(A) Chemical structure and mechanism of ATP acyl phosphate probe labeling. The ATP binding group mediates interactions with lipid and protein kinases to place the reactive acyl phosphate group in proximity of lysines in the active site. The side chain amino group of lysine covalently reacts with probe, releasing ATP, and covalently attaches desthiobiotin to kinase through an amide bond. (B) The DGKα inhibitor ritanserin and matching negative control inhibitor ketanserin.
Figure 3. ATP acyl phosphates enable gel-based activity-based profiling of DGKα.

(A) In vitro IC50 values for DGKα inhibition by ritanserin as measured by DAG phosphorylation substrate assay described in Figure S2. Data shown are mean +/− SEM. for triplicate measurements. Results are representative of two independent biological replicates. 95% confidence intervals for IC50 values: 20–30 μM. (B) Gel-based ATP acyl phosphate assay was used to determine in vitro IC50 values for DGKα inhibition by ritanserin (95% confidence intervals for IC50 values: 17–192 μM). Details of the assay and representative gels used to calculate potency values can be found in Figure S3. (C) DGKα-HEK293T soluble proteomes were pretreated with ritanserin (100 μM), ketanserin (100 μM), or ATP (1 mM) for 30 min prior to addition of ATP acyl phosphate probe (10 μM, 30 min) and gel-based analysis as described in Figure S3. Pretreatment with ritanserin but not ketanserin blocked probe labeling of ~80 kDa recombinant DGKα. Western blot analysis (anti-FLAG, 0.8 μg/mL) confirmed equivalent recombinant protein expression across treatment conditions.
Figure 5. Chemical proteomic profiling of the DGK superfamily.

(A) Heatmap showing SILAC ratios for probe-binding sites of respective DGK isoforms in ATP (1 mM, 30 min)- versus DMSO vehicle-treated recombinant HEK293T proteomes. DGK ATP binding sites are defined by SR > 5. (B–C) Sequence similarity of ATP binding sites of DGK isoforms measured by quantitative proteomics. Multiple sequence alignments of probe-modified peptides were performed using Clustal Omega and results analyzed by sequence logos (See STAR Methods for details) to search for common motifs within the DAGKc (B) and DAGKa (C) ATP-binding sites of DGKs. The height of each stack denotes sequence conservation at the respective position (measured in bits). The height of individual residues within the stack indicates the relative frequency of corresponding amino acids at that position. The numbers in parentheses indicate the number of DGKs that show modification at the respective probe-modified lysine. Color scheme for amino acids in sequence logos is as follows: hydrophilic, blue; neutral, green; hydrophobic, black. Probe modified peptides used for sequence alignment and logo analysis can be found in Figure S6 and Table S1.
Next, we set out to determine whether we could use desthiobiotin-tagged, ATP acyl-phosphates (Patricelli et al., 2011; Patricelli et al., 2007) as a surrogate chemical proteomic assay for measuring recombinant DGKα activity in cell proteomes (Figure 2A). ATP acyl-phosphate probes enable global profiling of kinase activities by covalent attachment of reporter tags to conserved lysine residues in the ATP binding site of a wide range of kinases as well as other ATP-binding proteins (Patricelli et al., 2011; Patricelli et al., 2007). Initially, we performed gel-based profiling experiments to allow rapid optimization of probe labeling parameters (Figure S3A). In brief, DGKα-HEK293T soluble lysates were reacted with the ATP acyl-phosphate probe, desthiobiotin-modified proteins separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probe-modified proteins detected using a fluorescently-labeled streptavidin. We observed concentration dependent labeling of a ~80 kDa fluorescent band in DGKα- but not mock-transfected HEK293T proteomes (Figure S3B). We confirmed by western blot that differences in fluorescence signals in our probe binding assay were not due to expression levels of recombinant DGKα (Bottom panel, Figure S3B).
From these studies, we identified experimental conditions where ATP acyl-phosphate labeling of DGKα was not saturating to allow competitive profiling of reversible inhibitors (Adibekian et al., 2012) (10 μM ATP probe, 30 min; Figure S3C). Using these kinetically-controlled conditions, we showed that pretreatment with ritanserin but not ketanserin resulted in concentration-dependent blockade of probe labeling (IC50 = 57 μM, Figure 3B and Figure S3D). We included analysis of the non-selective DGK inhibitors, R59949 (de Chaffoy de Courcelles et al., 1989) and R59022 (de Chaffoy de Courcelles et al., 1985), to show that our gel activity assay can be used to generally profile inhibitor activity against DGKα (Figure S3E). Finally, we showed that treatment with free ATP (1 mM) resulted in global reductions in fluorescent protein signals (Figure 3C). These results support specific detection of probe labeling events occurring in the ATP binding site of recombinant DGKα as well as other native proteins detected in HEK293T cell proteomes. In all of our probe-labeling studies, changes in fluorescent signals in compound-treated samples were not due to differences in DGKα protein levels as confirmed by western blot analysis (Bottom panels; Figure 3C, Figure S3D and E). Collectively, the comparable potency values determined using substrate- (Figure 3A) versus chemical proteomic-assays (Figure 3B) demonstrate that ATP acyl phosphates are capable of measuring authentic DGKα activity with the advantage of enabling rapid assessment of compound activity across ATP-binding sites detected in native cell proteomes (Figure 3C).
Mapping the ATP binding site of DGKα using quantitative chemical proteomics
Results from gel profiling analyses demonstrated the probe binding of DGKα is competed by ATP substrate. While suited for rapid screening, gel-based chemical proteomic assays do not provide information on site of binding of compounds. Thus, we implemented a liquid chromatography-mass spectrometry (LC-MS) assay to discover the ATP binding site(s) of DGKα. For these studies, we overexpressed DGKα in isotopically light and heavy amino acid-labeled HEK293T cells to enable quantitative LC-MS by stable isotope labeling with amino acids in cell culture (SILAC (Mann, 2006), Figure 4A). In brief, light and heavy DGKα-HEK293T lysates were treated differentially with DMSO vehicle or free ATP (1 mM) respectively, prior to addition of ATP acyl phosphate to label active site lysines. After probe labeling, light and heavy proteomes were combined, digested with trypsin protease, and desthiobiotin-modified peptides were enriched by avidin affinity chromatography and analyzed by LC-MS/MS to identify and quantify isotopically tagged active-site peptides from DGKα (Figure 4A, see STAR Methods for more details).
Figure 4. Elucidation of ATP and ligand-binding sites of DGKα by quantitative chemical proteomics.

(A) Schematic of quantitative LC-MS proteomics workflow to identify ligand binding sites of recombinant DGKs using ATP acyl phosphate probe. See STAR Methods for more details. (B) Left - MS2 spectra of probe-modified peptides corresponding to the active site of DGKα. Major b- and y-ion fragments derived from neutral losses of the precursor (M) are indicated on spectrum in red. An asterisk denotes fragments containing probe-modified lysine residues corresponding to the red-labeled lysine shown in the peptide sequence. Right - MS1 extracted ion chromatograms of probe-modified peptides with corresponding SILAC ratios quantifying vehicle-treated (light): compound-treated (heavy). (C) Schematic of DGKα showing domains where ATP probe binding is detected by quantitative chemical proteomics. Orange circles represent ATP probe binding at K237 of the C1 domain, K377 of the DAGKc domain, and K539 of the DAGKa domain.
Using our quantitative chemical proteomics assay, we identified two probe-labeled peptides that were highly competed with ATP treatment as determined by SILAC ratios (SR) of MS1 chromatographic peak areas >5 in DMSO/ATP comparisons (Figure 4B and Table S1). All peptides reported met quality control criteria and were observed in 2 biological replicates (see STAR Methods for more details). The sites of labeling for probe-modified peptides of DGKα were confirmed by identifying ions corresponding to peptide fragments that contain the modified lysine residue in MS2 spectra (red asterisk, Figure 4B). Both ATP-sensitive peptides are located within the predicted catalytic domain, albeit at different subdomain regions (K377 – DAGKc, SR = 16.3; K539 – DAGKa, SR = 16.4; Figure 4B). Comparison of these peptide sequences with ATP-binding motifs found in protein kinases (Hanks et al., 1988; Sakane et al., 1990) revealed no apparent homology, supporting previous speculation that DGKs mediate ATP binding through a non-canonical binding motif (Schaap et al., 1994). Closer inspection of the DAGKc peptide did reveal homology with ATP binding sites of DgkB from S. aureus, and placement of K377 at the homologous residue in the bacterial DGK crystal structure (threonine 12) positions this lysine in vicinity of phosphate groups of ADP (Miller et al., 2008) (Figure S4). The second ATP-sensitive peptide (K539, Figure 4B) is located in the poorly annotated DAGKa subdomain that, to the best of our knowledge, has not been implicated in ATP binding in DGKα or any other DGK isoform. Our results help explain previous findings from other groups showing that C-terminal truncations (which remove the DAGKa subdomain) result in impaired DGK catalytic activity (Los et al., 2004).
Finally, we identified a probe-modified peptide located in the first C1 domain of DGKα (K237, Figure 4B). The C1 site was competed with ATP treatments (K237, SR = 2.4, ~58% inhibition; Figure 4B and Table S1) but with lower potency compared with probe-modified peptides from DAGKc and DAGKa subdomains (~94% competition with ATP). The difference in sensitivity to ATP competition at C1 versus DAGKc/DAGKa suggests that the latter sites largely mediate ATP binding of DGKα. The partial sensitivity of C1 to ATP competition suggests the existence of a distinct binding site in the C1 domain that can bind ATP probe separate from interactions at the DAGKc/DAGKa sites (Figure 4C). In summary, our LC-MS findings provide evidence that ATP acyl phosphate probes can map important binding regions of DGKα domains to reveal ATP (DAGKc/DAGKa) and other ligand binding sites (C1) important for mediating catalytic functions.
Chemical proteomic profiling of the DGK superfamily using ATP acyl phosphates
Next, we sought to expand our chemical proteomics analysis to other DGK subtypes to identify conserved and distinguishing features of active sites in comparison with type 1 DGKα. For these studies, we chose to test a representative member from each of the DGK subtypes: kappa (DGKκ (Imai et al., 2005)), type 2; epsilon (DGKε (Tang et al., 1996)), type 3; zeta (DGKζ (Goto and Kondo, 1996)), type 4; and theta (DGKθ (Houssa et al., 1997)), type 5 (Figure 1B). Recombinant DGKs were transiently transfected in light and heavy HEK293T cells, protein overexpression confirmed by western blot (Figure S1), and recombinant lysates subjected to quantitative chemical proteomics (Figure 4A). The identified probe-modified peptides for each DGK isoform and their corresponding sensitivities to ATP competition are listed in Table S1.
Akin to DGKα, we identified probe-modified peptides in C1, DAGKc, and DAGKa binding sites of DGKζ and DGKθ (Figure 5A). Treatment with free ATP resulted in potent competition at DAGKc (K596, SR = 7.6) and DAGKa (K768, SR = 14.4) sites within the catalytic domain of DGKθ (Figure 5A and Table S1). Similar inhibition profiles were observed for DGKζ with the exception of a lower sensitivity to ATP competition at the DAGKc (K500, SR = 2.9) compared with DAGKa site (K662, SR = 16.7, Figure 5A and Table S1). Probe-modified peptides corresponding to C1 domains of both DGKζ (K323) and DGKθ (K202) showed moderate competition with ATP (SR ~3 for both isoforms, Figure 5A and Table S1). Of the remaining subtypes, we identified a single probe-modified peptide in the DAGKa subdomain of DGKκ and DGKε that were competed with ATP with high (K892, SR = 15.0) or moderate inhibition (K392, SR = 2.6; Figure 5A and Table S1), respectively. Based on ATP sensitivity, our findings position the primary ATP binding site within the DAGKa subdomain of type 2 (DGKκ), 3 (DGKε), and 4 (DGKζ) enzymes. Similar to DGKα, type 5 DGKθ likely requires both DAGKa and DAGKc regions for ATP substrate binding.
We performed multiple sequence alignments and sequence logo analysis (Crooks et al., 2004; Schneider and Stephens, 1990) to identify a potential DGK-specific ATP binding motif. We used ATP-competed peptide sequences identified in our LC-MS analyses (Table S1) to discover potential regions of sequence conservation across all 5 DGK subtypes tested. Our analyses identified clusters of amino acid conservation in regions that contained probe-modified lysines within both DAGKc (positions 7–17, Figure 5B) and DAGKa subdomains (positions 7–19, Figure 5C). We used our results to determine whether DGKs are probe-labeled at conserved lysines in the active site, which would provide preliminary evidence of a common ATP binding orientation. Closer inspection of the data revealed that the lysine showing highest conservation in the DAGKc motif was also probe modified with the highest frequency (position 9, Figure 5B). In contrast, the correlation between conserved lysines and frequency of probe modifications at these sites was less clear in the DAGKa motif. For example, probe modification of the lysine with highest conservation (position 19) was only observed in the DGKκ active site peptide (Figure 5C). The identification of probe modifications at both conserved (DAGKc) and non-conserved lysines (DAGKa) in the DGK ATP binding site is different from protein kinases, which are probe modified largely at conserved lysines in the ATP binding site (Patricelli et al., 2007). Future studies are needed to determine how these differences in DGK active sites impart substrate specificity in vivo and whether these features can be exploited for inhibitor development.
Inhibitor profiling to determine ritanserin binding mode and selectivity
We next asked whether we could use quantitative chemical proteomics to determine the binding mode and selectivity of inhibitors against DGK isoforms. Ritanserin was originally tested in the clinic as a serotonin receptor antagonist for treatment of psychiatric disorders (Barone et al., 1986) and has recently generated interest as a lead DGKα inhibitor for drug repurposing to treat cancer (Boroda et al., 2017; Purow, 2015). DGKα-HEK293T soluble proteomes were treated with ritanserin or ketanserin (100 μM compounds, Figure 2B) followed by labeling with ATP acyl phosphate and quantitative chemical proteomics analysis (Figure 4A). Ritanserin concentrations were chosen to provide ~70% blockade of DGKα activity as determined from substrate- (Figure 3A) and chemical proteomic-assays (Figure 3B). Probe-modified peptides showing high competition, as judged by SILAC ratios, were identified as ritanserin binding sites in DMSO/ritanserin comparisons (SR > 5, Figure 6A and Table S1).
Figure 6. Inhibitor profiling of the DGK superfamily.

(A) Heat map showing SILAC ratios of DGK probe-binding sites that are competed (SR > 5) with ritanserin- versus DMSO vehicle control-treated samples. Lack of competition at respective sites in ketanserin-treated samples indicates ritanserin competition was specific. (B) Representative extracted ion chromatograms (MS1) of probe modified peptides from DGKα C1 and DAGKa domains, which represent the primary sites of binding for ritanserin. Ketanserin is inactive at these same sites. For all studies, proteomes were pretreated with compounds (100 μM) for 30 min prior to labeling with ATP acyl phosphate probe (10 μM, 30 min).
We used these criteria to discover that ritanserin inhibits DGKα predominantly through binding interactions at the C1 (K237, SR = 7.0) and DAGKa sites (K539, SR = 7.0; Figure 6B and Table S1). Surprisingly, we observed minimal competition at the DAGKc domain (K377, SR = 2.0; Table S1). Ritanserin competed at common (DAGKa) as well as distinct binding sites (C1) compared with ATP substrate (Table S1). The unusual binding mode of ritanserin identified from our LC-MS studies may help explain previous kinetic assays describing a mixed competitive mechanism of inhibition for this inhibitor; ritanserin is hypothesized to bind a DGKα-ATP complex through an unidentified binding site (Boroda et al., 2017). We propose that C1 could be a potential site mediating ritanserin binding to DGKα distinct from the ATP pocket. Inhibitor profiling of other DGKs revealed that ritanserin showed minimal activity against other isoforms (SR < 2 at all binding sites detected, Figure 6A and Table S1). Ritanserin competition was specific because treatment with the negative control probe ketanserin resulted in negligible competition at all probe-binding sites with the exception of a lower SILAC ratio for DGKκ peptide, which indicates a potential activating effect for this isoform (Figure 6A and Table S1). Future studies will be required to determine whether ritanserin shows similar selectivity profiles against native DGKs.
One of the advantages of using chemical proteomics is the ability to simultaneously evaluate on- and off-target activity of inhibitors directly in cell proteomes (Chang et al., 2015; Nagano et al., 2013). Here, we measured the selectivity of ritanserin against >50 native kinases quantified in HEK293T soluble proteomes (Figure 7A). On average, we detected >200 probe-modified peptides from ~85 protein and lipid kinases per individual SILAC sample. Our kinome coverage is comparable with previous reports using ATP acyl phosphates and data-dependent MS scan modes (Patricelli et al., 2007). Native kinases reported in Figure 7A and Table S1 were quantified in at least 2 biological replicates across all treatment conditions and competed by treatment with free ATP (SR > 5, Table S1). The latter criterion (i.e. ATP competition) was important for identifying non-specific probe labeling events in our studies (see STAR Methods for more details). Kinase targets of ritanserin were defined as those active site peptides that showed SILAC ratios ≥ 5. Based on this criterion, the most potent targets of ritanserin were DGKα and the non-receptor tyrosine protein kinase FER (Greer, 2002) (SR = 7.9; Figure 7A and B). We confirmed that ritanserin was competing at ATP binding sites of FER by demonstrating potent competition at the same site (K591) with free ATP (SR = 19.3, Table S1). Collectively, our studies demonstrate the use of chemical proteomics to elucidate the binding mode and selectivity of ritanserin, resulting in discovery of the C1 domain as a novel ligand binding site and FER as an unanticipated off-target.
Figure 7. Discovery of a lead fragment inhibitor of DGKα by chemical proteomics.

(A) Heat map showing potency and selectivity of ritanserin and RF001 against recombinant DGKs and native kinases detected in HEK293T proteomes. (B) Representative extracted ion chromatograms (MS1) of probe modified peptide from FER, showing potent competition with ritanserin but not RF001. (C) Ritanserin deconstruction to identify the fragment RF001, which shows concentration-dependent blockade of recombinant DGKα as measured by substrate assay (Figure S2). Data shown are mean +/− SEM. for triplicate measurements. Results are representative of two independent biological replicates. 95% confidence intervals for IC50 values: 120–414 μM. Dotted line represents background activity detected in non-transfected HEK293T proteomes. (D) Representative extracted ion chromatograms (MS1) showing the primary sites of binding for RF001 against DGKα. Quantified SILAC ratios shown are averages of two biological replicates. (E) Bar graph comparing the total number of kinase targets (recombinant DGKs and native kinases in HEK293T proteomes) observed with potent (SR ≥ 5), moderate (SR ≥ 3), and weak competition (SR ≥ 2) at respective probe-binding sites in ritanserin- versus RF001-treated samples. For quantitative LC-MS experiments, proteomes were pretreated with compounds (100 μM) for 30 min prior to labeling with ATP acyl phosphate probe (10 μM, 30 min).
Discovery of a lead fragment inhibitor of DGKα by ritanserin deconstruction
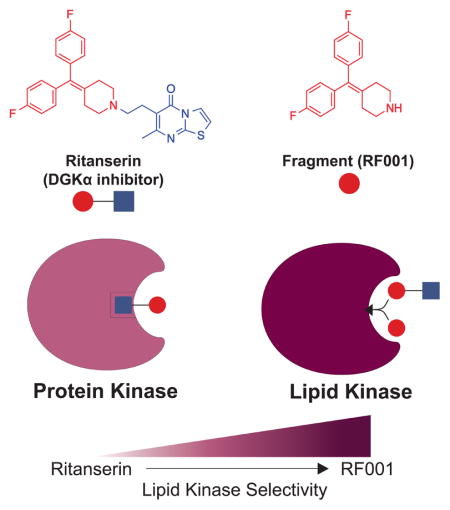
In an effort to improve selectivity of ritanserin for DGKα, we explored ligand deconstruction (Hajduk et al., 2000; Kozakov et al., 2015; Lingel et al., 2017) strategies to evaluate the contributions of representative fragments for binding affinity and selectivity. We hypothesized that the 4-substituted piperidine moiety of ritanserin (highlighted in red, Figure 7C) is a likely pharmacophore required for DGKα inhibition because of conservation of this motif across several DGKα inhibitors (Boroda et al., 2017) (Figure S3E). Here, we tested the capacity of quantitative chemical proteomics to evaluate binding mode and selectivity of a ritanserin fragment (designated RF001, Figure 7C) against recombinant DGKs and endogenous kinases directly in native cell proteomes.
We confirmed that RF001 blocked DGKα activity in a concentration-dependent manner using the DAG phosphorylation substrate assay (IC50 = 223 μM, Figure 7C). Our data show substantially lower potency of RF001 compared with ritanserin (~10-fold difference in IC50 values when comparing Figure 3A and 7C), which is expected of low molecular weight fragments (<300 Da) that typically exhibit binding affinities in the high micromolar to millimolar range (Erlanson et al., 2016). To account for differences in potency, we tested RF001 at 10-fold higher concentrations (1 mM) in our subsequent LC-MS assays. This concentration of RF001 was chosen to provide >80% inhibition of DGKα activity in our probe-binding assay (Figure S5). Akin to ritanserin, RF001 treatment resulted in potent competition at C1 (SR = 12.6) and DAGKa sites (SR = 9.1) while showing weak activity at the DAGKc site (SR = 1.7, Figure 7D and Table S1). We also confirmed that RF001 was largely inactive against other DGK subtypes as determined by low SILAC ratios at all detected DGK probe-modified sites (average SR ~1, Figure 7A and Table S1). The similar inhibition profiles of RF001 and ritanserin observed in our LC-MS analyses support that the ritanserin binding mode is conserved with the ritanserin fragment and that the 4-substituted piperidine group represents a core binding motif of DGKα inhibitors.
While ritanserin and RF001 share similar inhibition profiles within the DGK family, they differed substantially in cross-reactivity against the kinome. A striking finding from our studies is the dramatic improvement in selectivity against the kinome observed with RF001 compared with ritanserin (Figure 7A). Specifically, the potent FER off-target activity observed with ritanserin was largely eliminated using RF001 (SR = 1.2, Figure 7B). In fact, RF001 showed potent activity (SR ≥ 5) against a single kinase target, DGKα, across all detectable kinases (native and recombinant DGKs) quantified in our chemical proteomics studies (Figure 7A and Table S1). Closer inspection of the data revealed that unlike ritanserin, RF001 maintained good selectivity even for kinase targets that show moderate to weak inhibitory activity (25 versus 3 kinase targets that show SR ≥ 2 for ritanserin versus RF001, respectively; Figure 7E). Future studies are needed to explore whether synthetic elaboration of RF001, potentially using fragment-based approaches (Erlanson et al., 2016), can improve affinity for DGKα while maintaining selectivity against the kinome.
DISCUSSION
We used ATP acyl phosphates and quantitative LC-MS to map ligand-binding regions corresponding to the active site of mammalian DGKs. We defined, for the first time, the location of the ATP binding site of representative isoforms from all five principal DGK subtypes (Figure 5). Inspection of the DGK ATP binding sites reveals several important features that are unique to this lipid kinase family. First, we identified ATP-sensitive, probe-modified peptides from both DAGKc and DAGKa subdomains, supporting interactions between these regions within the catalytic domain to constitute a potential ATP binding cleft. Crystal structures of soluble bacterial lipid kinases with homology to mammalian DGKs have also been found with active sites located in an interdomain cleft (Bakali et al., 2007). Our finding that the DAGKa region is involved in substrate binding was important for assigning a catalytic role to this domain and helps explain previous reports that C-terminal truncations impair DGK enzymatic activity (Los et al., 2004). Second, conserved sequences corresponding to ATP binding sites of DGKs (Figure 5B and C) are not homologous with glycine-rich loops mediating ATP binding of protein kinases (Hanks et al., 1988; Hemmer et al., 1997). Our data provide the first experimental evidence in support of a unique DGK ATP binding motif that was postulated over 20 years ago (Schaap et al., 1994). Finally, it is tempting to speculate that detection of a single ATP binding site (as opposed to 2 sites in other DGKs) for DGKκ and DGKε is a reflection of functional differences in substrate binding of DGK subtypes (Figure 5A). In support of this hypothesis, DGKκ, along with other type 2 members, contain an unusual peptide motif that physically separates the DAGKc and DAGKa subdomains (Imai et al., 2005). DGKε, the sole type 3 member, is the only subtype that lacks regulatory domains and shows acyl chain preference in DAG substrate assays in vitro (Tang et al., 1996). We should note that DGKκ and DGKε showed lower recombinant protein expression compared with other isoforms (Figure S1) and so we cannot rule out the possibility of detection limits using our LC-MS approach. Future studies will be required to evaluate how these distinctions in active sites influence substrate (DAG) specificity and function across DGK subtypes.
We also discovered important clues to domain binding sites of DGKs and how to exploit these regions for development of DGKα-selective inhibitors. The identification of a probe-modified site at C1 domain provided the first evidence of a ligand binding site remote from the ATP binding region of DGKs. Although we cannot rule out the possibility of alternative mechanisms, e.g. probe binding due to domain- (Nordin et al., 2015) or protein-interactions (Okerberg et al., 2014), we do provide evidence that the C1 domain serves as a ligand binding site for ritanserin distinct from the ATP binding region of DGKα (Figure 5A and 6A). The overlapping (DAGKa) and distinct (C1) binding sites of ritanserin compared with ATP helps explain previous kinetic findings of a mixed competitive mechanism of inhibition whereby ritanserin prefers to bind a DGKα-ATP complex (Boroda et al., 2017). We investigated how the binding mode of ritanserin affects selectivity against other DGK isoforms as well as >50 native kinases detected in cell proteomes. While ritanserin showed good selectivity within the DGK superfamily, we discovered substantial cross-reactivity against protein kinases, including the non-receptor tyrosine kinase FER that was inactivated to a similar magnitude as DGKα (SR = 7.9, Figure 7A and B). An unexpected finding was the discovery that a ritanserin fragment (RF001) functioned as a DGKα inhibitor that retained binding at C1 and DAGKa sites (Figure 7D) and largely removed FER and other kinase off-target activity (Figure 7A and E). Conservation of fragment binding mode is characteristic of ligand-binding hotspots (Hall et al., 2015; Kozakov et al., 2015) of proteins suitable for fragment-based lead and drug discovery (Erlanson et al., 2016). In this regard, future studies are needed to investigate whether RF001 can serve as a core fragment for synthetic elaboration of high affinity ligands with selectivity for DGKα.
SIGNIFICANCE
Our studies describe the first functional proteomic map of ligand-binding regions that mediate substrate (ATP) and inhibitor binding in the poorly annotated active site of the mammalian diacylglycerol kinase (DGK) superfamily. Given the dearth of lipid kinase inhibitors available in the clinic and the emerging role of DGKs as anticancer and immunotherapy targets, we believe our findings offer exciting new prospects for development of new chemical probes to study and target lipid kinases. We define, for the first time, the location of the ATP binding site of representative isoforms from all five principal DGK subtypes (type 1 – 5). Inspection of DGK ATP binding sites identify conserved features that are distinct from protein kinases, providing the first experimental evidence in support of a DGK-specific ATP binding motif that was postulated over 20 years ago. We discovered clues to domain regions of DGKs important for inhibitor development by identifying probe-modified sites in C1 and accessory (DAGKa) domains that serve as primary binding sites for the DGKα inhibitor ritanserin. An unexpected finding was the discovery that a fragment of ritanserin (RF001) functioned as a DGKα inhibitor that retained binding at C1 and DAGKa domains and largely removed protein kinase off-target activity. While few examples have been reported, conservation of fragment binding mode is characteristic of ligand-binding hotspots of proteins suitable for fragment-based lead discovery. Thus, we believe the C1 and DAGKa sites are key binding regions of DGKs to enable development of high affinity, isoform-selective inhibitors of this lipid kinase superfamily.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ku-Lung Hsu (kenhsu@virginia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
HEK293T cells were cultured in DMEM with 10% FBS (U.S. Source, Omega Scientific) and 1% L-glutamine (Thermo Fisher Scientific) in 10cm2 plates. SILAC HEK293T cells were cultured in DMEM for SILAC (Fisher Scientific) supplemented with 10% dialyzed FBS (Omega Scientific) and either ‘Light’ 12C, 14N-labeled lysine and arginine or ‘Heavy’ 13C, 15N-labeled lysine and arginine (100ug/mL) in 10cm2 plates. Light or heavy amino acids were incorporated for at least 5 passages prior to utilizing SILAC HEK293T cells for experiments. All cells were grown to ~80% confluency in a 37°C incubator with 5% CO2.
METHOD DETAILS
Transient transfection
Recombinant DGK proteins were produced by transient transfection of HEK293T cells with recombinant DNA. pDONR223-DGKK was a gift from William Hahn & David Root (Addgene plasmid # 23487). pCSF107mT-GATEWAY-3′-FLAG was a gift from Todd Stukenberg (Addgene plasmid # 67619). pCSF107mT-DGKK-FLAG construct was generated by recombination of the Addgene plasmids using the Gateway cloning system (Invitrogen). All other vectors were gifted to Dr. Kevin Lynch (University of Virginia, School of Medicine) by Dr. Kaoru Goto (Yamagata University, School of Medicine) and Dr. Fumio Sakane (Chiba University) and were kindly shared with us: pcDNA3-FLAG-DGKA (rat), pCMV-Tag2B-FLAG-DGKQ (human), pcDNA3-DGKE-3xFlag (human), and pCMV-SPORT6-HA-DGKZ (human). HEK293T cells were plated at a concentration of 400,000 cells in complete DMEM and grown to 50–60% confluency. A polyethyleneimine (PEI) stock solution was prepared (1mg/mL, pH 7.4) and filter sterilized. Serum-free DMEM (600 μL) was mixed gently with 2.6 μg DNA and 20 μL of sterile PEI (1 mg/mL, pH 7.4) in a sterile microfuge tube. Mixtures were incubated for 30 min at 25 °C. The mixture was then added drop-wise to each 10 cm2 plate, rocked back and forth to mix, and placed back in the incubator. Cell pellets were harvested after two full days of growth, snap-frozen in liquid N2, and stored at −80°C until use. Recombinant proteins were produced by transient transfection in SILAC HEK293T cells using the procedure described above, except that cells were plated at a concentration of 1 × 106 cells per 10 cm2 plate and grown to ~70% confluency prior to introducing transfection mixture.
Western blot analysis of recombinant protein expression
Cell lysates were separated via centrifugation at 100,000 x g for 45 min at 4 °C. Proteins separated by SDS-PAGE (7.5% polyacrylamide, TGX Stain-Free Mini Gel) at 150 V for 55 min. Gel transfers were performed using the Bio-Rad Trans-Blot Turbo RTA Midi Nitrocellulose Transfer Kit with a Bio-Rad Trans-Blot Turbo Transfer System (25V, 10 min). The nitrocellulose blot was then incubated in blocking solution (30 mL, 5% Milk in TBS-T (1.5 M NaCl, 0.25 M Tris pH 7.4 in ddH2O)) for 1 h at 25 °C with gentle shaking. The blot was then transferred immediately to primary antibody solution (1:1,000 anti-FLAG or 1:10,000 anti-HA in TBS-T) and incubated overnight at 4°C with gentle shaking. The blot was then rinse d 5 times for 5 min in TBS-T, transferred immediately into secondary antibody solution (1:10,000 anti-species DyLight 550 or DyLight 650 in TBS-T), and incubated for 1 h at 25 °C with gentle shaking. The blot was then rinsed 5 times for 5 min in TBS-T, transferred into ddH2O, and imaged by in-blot fluorescence scanning on a ChemiDoc MP Imaging System.
Preparation of cell lysates for gel-based chemical proteomics
Cell pellets were resuspended in kinase buffer (Dulbecco’s PBS (DPBS, Hyclone), 20 mM MgCl2, EDTA-free protease inhibitors (Pierce)) and then lysed by sonication (3 × 1 sec pulse, 20% amplitude). The cell lysates were then subjected to centrifugation (100,000 x g, 45 min at 4 °C) to isolate the cytosolic fraction in the supernatant and the membrane fraction as a pellet. The membrane pellet was resuspended in kinase buffer by sonication. For all further analyses, only the soluble (cytosolic) fraction was used to prevent the need for detergents, which have been shown to interfere with DGK activity (Yada et al., 1990). The only exception was experiments involving DGKE; recombinant DGKE protein was most prominently expressed in the membrane fraction and so this fraction was utilized to study DGKE enzyme (see Figure S1). Protein concentrations were measured using the Bio-Rad DC protein assay. Samples were stored at −80 °C until use.
Gel-based chemical proteomic assay
Proteome concentration was adjusted to 2 mg/mL in kinase buffer. Proteomes were first pre-treated with compound (0.6 μL, 50X stock in DMSO) mixed with gentle flicking, and incubated for 30 min at 25 °C in a microfuge tube (30 μL reaction volume). Desthiobiotin ATP acyl phosphate nucleotide probe (0.5 mM in ddH2O) was then added to each sample (0.6 μL, 10 μM final) and incubated for 30 min at 25 °C. Reactions were then quenched with 10 μL of 4X SDS-PAGE loading buffer. Protein samples (15 μL) were loaded onto 4–20% TGX Stain-Free Protein Midi Gel and resolved by SDS-PAGE at 150V for 55 min. Proteins were then transferred to a nitrocellulose blot by Bio-Rad Trans-Blot Turbo Transfer System (25V, 10 min) to enhance sensitivity. The nitrocellulose blot was then incubated in blocking solution (30 mL, 3% BSA in TBS-T) for 1 h at 25 °C with gentle shaking. The blot was then transferred immediately to antibody solution (30 mL, 5% BSA in ddH2O with 0.1% Tween20 and 1:3000 Streptavidin DyLight 550) and incubated for 2 h at 25 °C with gentle shaking. The blot was then rinsed 5 times for 5 min in TBS-T, and then transferred into ddH2O. The blot was then imaged by in-blot fluorescence scanning on a ChemiDoc MP Imaging System. Fluorescence intensity signals were normalized to total lane protein using the Bio-rad Stain Free imaging (Posch et al., 2013).
Preparation of cell lysates for ADP-glo assay
The ADP-Glo DAG phosphorylation substrate assay was adapted from Sato et al (Sato et al., 2013). Transfected HEK cells expressing recombinant FLAG-DGKA were harvested in DPBS and centrifuged at 1400 x g for 3 min. Supernatant was removed and 1 mL Lysis Buffer (50 mM HEPES (pH 7.2), 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1 mM phenylmethysulfonyl chloride, Phosphatase Inhibitor Cocktail 2 (Sigma-Aldrich), and EDTA-Free Protease Inhibitor Mini Tablets (Pierce)) was added and cells re-suspended. Solutions were sonicated (3 × 1 sec pulse, 20% amplitude) and then centrifuged at 400 x g for 5 min. Supernatant was separated and protein concentrations were measured using the Bio-Rad DC protein assay and diluted in Lysis Buffer as appropriate. Samples were stored at −80°C until use.
ADP-glo DAG phosphorylation substrate assay
Micelles were prepared from lipid stocks as follows: Reaction Buffer (50 mM MOPS (pH 7.4), 1 mM DTT, 100 mM NaCl, 20 mM NaF, and 1 μM CaCl2) was prepared in ddH2O. From this stock, a solution of Reaction Buffer with 50 mM MgCl2 and 1 mM ATP (‘Reaction Initiator’) and a solution of 0.3% Triton-X100 in Reaction Buffer (‘Triton Buffer’) were separately prepared. 1, 2-dioleoyl-sn-glycerol (DG) and 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) in chloroform were mixed and then dried under nitrogen. Triton Buffer was added to the dried lipids to a final concentration of 10 mM DG and 8 mM DOPS. This solution was incubated at room temperature for 5 min with gentle shaking, followed by sonication (3 × 1 sec pulse, 20% amplitude). The micelles were then diluted 4-fold in reaction buffer to yield the final micelle buffer. 1 mg of lysate was aliquoted into each well of a 96 well plate, followed by micelle buffer to a final volume of 20 μL. 19 μL of this mix was added to 1 μL of DMSO or inhibitor solution and incubated at 30 °C for 30 min. After incubation, 5 μL of reaction initiator was added to each well and mixed thoroughly, followed by aliquoting 5 μL of each reaction mixture to a 96-well half area black polystyrene plate and incubated at 30 °C for 30 min. At this point the procedure for the ADP-Glo™ assay (Promega) was performed. 5 μL of ‘ADP-Glo Reagent’ was added to each well, mixed thoroughly, and allowed to incubate at 25 °C for 40 min. Then 10 μL of the ‘Kinase Detection Reagent’ was added to each well, mixed thoroughly, and allowed to incubate at 25 °C for 40 min. Luminescence was measured with no filter and an integration time of 1 sec per well on a BMG Labtech CLARIOstar plate reader.
Sample preparation for quantitative LC-MS analysis using ATP acyl phosphates
Proteomes were diluted to 2 mg/mL in kinase buffer. The light and heavy proteomes (0.5 mg, 250 μL total reaction volume) were pre-treated with vehicle or compound, respectively (5 μL, DMSO (light) or 50X stock in DMSO (heavy)), mixed gently, and incubated at 25 °C for 30 min. Desthiobiotin ATP acyl phosphate nucleotide probe (0.5 mM in ddH2O) was then added to each sample (5 μL, 10 μM final), mixed gently, and allowed to incubate at 25 °C for 30 min. After incubation, matched light and heavy proteomes were transferred and mixed in a 1:1 ratio in a two-dram vial containing 4:1:3 MeOH/CHCl3/H2O (2 mL MeOH, 500 μL CHCl3, 1.5 mL H2O) for extraction of proteins to remove excess probe, quickly vortexed, and centrifuged at 1,400 x g for 3 min to pellet protein. Organic and aqueous layers were removed using a Pasteur pipette, and the protein pellet was transferred to a screw-top tube in 600 μL MeOH. A second extraction was performed by adding CHCl3 (150 μL) and H2O (600 μL) to each sample, vortexed, and centrifuged at 1,400 x g for 3 min to pellet protein. Organic and aqueous layers were removed by pipetting, MeOH added to pellet (600 μL) and pellets were re-suspended by sonication (3 × 1 sec pulse, 20% amplitude) for a final extraction. Samples were then centrifuged at 17,000 x g for 5 min to pellet protein and MeOH was removed by pipetting. The pellets were re-suspended in 10 M urea/25 mM ammonium bicarbonate (500 mL), brought to a final volume of 1 mL with 25 mM ammonium bicarbonate, reduced with 10 mM DTT for 15 min at 65 °C, allowed to cool, and then alkylated with 40 mM iodoacetamide for 30 min at 25°C in the dark. To de salt the samples, each was transferred to a two-dram glass vial, and to the vial 4:1:2 MeOH/CHCl3/H2O (2 mL MeOH, 500 μL CHCl3, 1 mL H2O) was added. The vials were vortexed quickly, spun at 1,400 x g for 3 min to pellet protein, and aqueous and organic layers were removed using a Pasteur pipette. The resulting protein pellet was transferred to a screw-top tube in 600 μL MeOH, and then CHCl3 (150 μL) and H2O (600 μL) were added to extract protein a second time. The samples were vortexed quickly, centrifuged at 1,400 x g to pellet protein, and the aqueous and organic layers were removed by pipetting. Resulting protein pellet was suspended in MeOH (600 μL) via sonication (3 × 1sec pulse, 20% amplitude), centrifuged at 17,000 x g for 5 min to pellet protein, and MeOH removed by pipetting. Protein pellets were then re-suspended in 25 mM ammonium bicarbonate (500 μL) and digested with 7.5 μg Trypsin/Lys-C (Promega, 15 μL, 0.5 μg/μL) for 3 h at 37 °C. Avidin-agarose beads (Thermo Scientific Pierce, 100 μL aliquot per sample) were washed three times by adding 10 mL DPBS, centrifuged at 1,400 x g for 1 min, and decanting. This wash step was repeated for a total of 3 times. Digested protein samples were mixed with washed avidin beads (100 μL) and brought to a volume of 5.5 mL with DPBS in a 15 mL conical and rotated for 1 h to enrich samples for the covalent desthiobiotin modification. The beads were washed with 25 mM ammonium bicarbonate (3X with 10 mL, centrifuge at 1,400 x g for 3 min, decant) and then H2O (3X with 10 mL, centrifuge at 1,400 x g for 3 min, decant). Washed beads were then transferred to a low-bind microfuge tube, centrifuged at 1,400 x g for 3 min, allowed to rest for 1 min to settle beads, and then excess H2O was removed carefully using a gel-loading pipette tip. To elute peptides, 100 μL of elution buffer (50% acetonitrile, ACN; 0.1% formic acid) was added to each sample and incubated for 3 min. Beads were spun down at 1,400 x g for 3 min, allowed to rest for 1 min to settle beads, and then 75 μL of peptide-containing supernatant was removed carefully using a gel-loading pipette tip and transferred to a new low bind centrifuge tube. This step was repeated two more times with 75 μL of elution buffer and all eluent were collected into the same centrifuge tube (~225 μL total). Peptides were dried on a speed vacuum, resulting peptide samples acidified in 5% (v/v) formic acid, and stored at −80 °C until analysis.
LC-MS/MS analysis of SILAC samples
The peptide samples were analyzed by liquid chromatography-mass spectrometry. An integrated autosampler-LC (Ultimate 3000 RSLC nanoSystem, Dionex) was used to load the peptides onto a trap column (Nano-Trap, Thermo Scientific, 2 cm, 5 μm C18) and washed for 2 minutes with 1% B (80% ACN, 1% formic acid). The peptides were eluted from the trap column and through a homemade nanocapillary analytical column (20 cm, 5 μm C18 packed in 360 μm o.d. x 75 μm i.d. fused silica), with an integrated electrospray tip, using a 180 min 1–95% reverse-phase LC gradient (A: 0.1% formic acid; B: 80% ACN, 0.1% formic acid) with the following parameters: 0–2 min 1% B, 400 nL/min; 2–144 min to 95% B, 300 nL/min; 144.1–180 min 1% B, 400 nL/min. The eluting peptides were electrosprayed into an Orbitrap Q Exactive Plus mass spectrometer (Thermo Scientific), which was operated with a top 10 data-dependent acquisition method that consisted of one full MS1 scan (375 – 1,500 m/z) followed by 10 MS2 scans of the most abundant ions recorded in the MS1 scan. For recombinant DGKE samples, a data-independent parallel reaction monitoring (PRM) method was used to detect DGKE peptides. One full MS1 scan (375 – 1,500 m/z) was followed by MS2 scans of targeted parent ions from a curated inclusion list (DGKE: EKAPSLFSSR, +2 charge state, 659.3617 m/z (light), 668.3729 m/z (heavy), 103.00–110.00 min). Data analysis was accomplished using the IP2 (Integrated Proteomics Applications) software package, in which RawConverter was used to generate searchable MS1 and MS2 data from the .raw file followed by using the ProLuCID algorithm to search the data against a modified human protein database (UniProt human protein database with rat DGKs, angiotensin I and vasoactive intestinal peptide standards; 40,660 proteins) with the following parameters: static carbamidomethyl modification of cysteine (+57.0142 Da), differential modifications of oxidized methionine (+15.9949 Da) and desthiobiotin-labeled lysine residues (+196.1212 Da), added masses of the SILAC “heavy”-labeled amino acids (+10.0083 Da for R, +8.0142 Da for K), and trypsin enzyme specificity with 2 missed cleavages. The resulting MS2 spectra matches were assembled into protein identifications and filtered using DTA Select 2.0 using the --mass, --modstat, and --trypstat options with a 1% peptide FDR. mzIdent files corresponding to searches were generated in IP2-Integrated Proteomics Pipeline, mzXML spectra data was extracted from the raw file using RawConverter, and uploaded into Skyline-daily (Schilling et al., 2012) to determine SILAC ratios (SR) of light/heavy (vehicle/compound treated) peptides. Peptides used for analysis were assessed for quality in Skyline by the following criteria: isotope dot-product (iDOTP) ≥ 0.8, ratio dot-product (rDOTP) ≥ 0.8, and singletons defined by L/H ratios > 20 were set to 20. Dot-product values are measures of similarity between the precursor peak area and expected isotope distribution (iDOTP) and between the light and heavy peak area (rDOTP) as calculated in Skyline and described by Schilling et al (Schilling et al., 2012). Probe-modified peptides that met these criteria were manually inspected and integrated. Peptide ratios reported were normalized to DMSO/DMSO peptide ratios to account for potential variations in mixing and sample preparations. Additionally, reported DGK and FER peptides were verified by manual inspection of the raw data (MS1 and MS2).
Sequence alignments and generation of sequence logos
Lipid kinase sequences were obtained from Uniprot (http://www.uniprot.org/) and aligned using Clustal Omega (Goujon et al., 2010; Sievers et al., 2011). Sequence logos shown in Figure 5 were generated with WebLogo (Crooks et al., 2004; Schneider and Stephens, 1990) (http://weblogo.threeplusone.com).
DgkB monomer molecular model and alignment
PDB model 2QV7 visualized and colored using PyMol software. Partial Structure-Aided Sequence Alignment completed as described previously (Miller et al., 2008) and added to Figure S4 using GIMP software package.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis and determination of IC50 values
The percentage of enzyme activity remaining was determined by comparing integrated band intensities or luminescence of inhibitor- with DMSO-treated samples for gel-based chemical proteomic or ADP-glo assays, respectively. For both chemical proteomic and ADP-glo methods, nonlinear regression analysis was used to determine the IC50 values from a dose-response curve generated using GraphPad Prism. Data are shown as mean ± s.e.m. Determination of significance was performed by one-way ANOVA. All statistical analyses were performed using GraphPad Prism.
Supplementary Material
Table S1. Related to Figure 4 – 7. SILAC ratios for ATP, Ritanserin, Ketanserin, and RF001 treatment of recombinant DGK-HEK293T soluble lysates. Multiple sequence alignments for sequence logo analysis.
HIGHLIGHTS.
First functional proteomic annotation of diacylglycerol kinase (DGK) active sites
Elucidation of a ATP binding motif unique for lipid kinases
Identification of ligand binding sites for developing DGKα-selective inhibitors
Acknowledgments
We thank Mark M. Ross and Adam Borne for assistance with experiments and manuscript preparation. We thank all members of the Hsu Lab and colleagues for review of this manuscript. This work was supported by the LaunchPad for Diabetes Program funded by the Manning Family Foundation at the University of Virginia (K.-L.H.), University of Virginia Start-up Funds (K.-L.H.), and National Institutes of Health (DA035864 to K.-L.H.; T32 GM007055 to C.E.F.; T32 CA009109 to S.T.C.; DK101946 to T.E.H; CA180699 and CA189524 to B.W.P.).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, C.E.F., B.W.P., T.E.H., and K.-L.H.; Methodology, C.E.F. and K.-L.H.; Investigation, C.E.F. and S.T.C.; Validation, C.E.F., S.T.C., and K.-L.H., Writing – Original Draft, C.E.F., S.T.C., and K.-L.H.; Writing – Review and Editing, C.E.F. and K.-L.H.; Resources, T.E.H.; Funding Acquisition, C.E.F., S.T.C., B.W.P., T.E.H., and K.-L.H.; Supervision, K.-L.H.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe T, Lu X, Jiang Y, Boccone CE, Qian S, Vattem KM, Wek RC, Walsh JP. Site-directed mutagenesis of the active site of diacylglycerol kinase alpha: calcium and phosphatidylserine stimulate enzyme activity via distinct mechanisms. Biochem J. 2003;375:673–680. doi: 10.1042/BJ20031052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams DR, Pyne S, Pyne NJ. Sphingosine Kinases: Emerging Structure-Function Insights. Trends Biochem Sci. 2016;41:395–409. doi: 10.1016/j.tibs.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Adibekian A, Martin BR, Chang JW, Hsu KL, Tsuboi K, Bachovchin DA, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, et al. Confirming target engagement for reversible inhibitors in vivo by kinetically tuned activity-based probes. J Am Chem Soc. 2012;134:10345–10348. doi: 10.1021/ja303400u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakali HM, Herman MD, Johnson KA, Kelly AA, Wieslander A, Hallberg BM, Nordlund P. Crystal structure of YegS, a homologue to the mammalian diacylglycerol kinases, reveals a novel regulatory metal binding site. J Biol Chem. 2007;282:19644–19652. doi: 10.1074/jbc.M604852200. [DOI] [PubMed] [Google Scholar]
- Barone JA, Bierman RH, Cornish JW, Hsuan A, Drake ND, Colaizzi JL. Safety evaluation of ritanserin--an investigational serotonin antagonist. Drug Intell Clin Pharm. 1986;20:770–775. doi: 10.1177/106002808602001006. [DOI] [PubMed] [Google Scholar]
- Boroda S, Niccum M, Raje V, Purow BW, Harris TE. Dual activities of ritanserin and R59022 as DGKalpha inhibitors and serotonin receptor antagonists. Biochem Pharmacol. 2017;123:29–39. doi: 10.1016/j.bcp.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HA, Thomas PG, Lindsley CW. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat Rev Drug Discov. 2017;16:351–367. doi: 10.1038/nrd.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco S, Merida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. 2007;32:27–36. doi: 10.1016/j.tibs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Chang JW, Zuhl AM, Speers AE, Niessen S, Brown SJ, Mulvihill MM, Fan YC, Spicer TP, Southern M, Scampavia L, et al. Selective inhibitor of platelet-activating factor acetylhydrolases 1b2 and 1b3 that impairs cancer cell survival. ACS Chem Biol. 2015;10:925–932. doi: 10.1021/cb500893q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty T, Cai J, Sakane F, Taketomi A, Prescott SM, Topham MK. Diacylglycerol kinase delta regulates protein kinase C and epidermal growth factor receptor signaling. Proc Natl Acad Sci U S A. 2006;103:15485–15490. doi: 10.1073/pnas.0604104103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chaffoy de Courcelles D, Roevens P, Van Belle H, Kennis L, Somers Y, De Clerck F. The role of endogenously formed diacylglycerol in the propagation and termination of platelet activation. A biochemical and functional analysis using the novel diacylglycerol kinase inhibitor, R 59 949. J Biol Chem. 1989;264:3274–3285. [PubMed] [Google Scholar]
- de Chaffoy de Courcelles DC, Roevens P, Van Belle H. R 59 022, a diacylglycerol kinase inhibitor. Its effect on diacylglycerol and thrombin-induced C kinase activation in the intact platelet. J Biol Chem. 1985;260:15762–15770. [PubMed] [Google Scholar]
- Dominguez CL, Floyd DH, Xiao A, Mullins GR, Kefas BA, Xin W, Yacur MN, Abounader R, Lee JK, Wilson GM, et al. Diacylglycerol kinase alpha is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 2013;3:782–797. doi: 10.1158/2159-8290.CD-12-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov. 2016;15:605–619. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- Goto K, Kondo H. A 104-kDa diacylglycerol kinase containing ankyrin-like repeats localizes in the cell nucleus. Proc Natl Acad Sci U S A. 1996;93:11196–11201. doi: 10.1073/pnas.93.20.11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer P. Closing in on the biological functions of Fps/Fes and Fer. Nat Rev Mol Cell Biol. 2002;3:278–289. doi: 10.1038/nrm783. [DOI] [PubMed] [Google Scholar]
- Hajduk PJ, Gomtsyan A, Didomenico S, Cowart M, Bayburt EK, Solomon L, Severin J, Smith R, Walter K, Holzman TF, et al. Design of adenosine kinase inhibitors from the NMR-based screening of fragments. J Med Chem. 2000;43:4781–4786. doi: 10.1021/jm000373a. [DOI] [PubMed] [Google Scholar]
- Hall DR, Kozakov D, Whitty A, Vajda S. Lessons from Hot Spot Analysis for Fragment-Based Drug Discovery. Trends Pharmacol Sci. 2015;36:724–736. doi: 10.1016/j.tips.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Hemmer W, McGlone M, Tsigelny I, Taylor SS. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J Biol Chem. 1997;272:16946–16954. doi: 10.1074/jbc.272.27.16946. [DOI] [PubMed] [Google Scholar]
- Houssa B, Schaap D, van der Wal J, Goto K, Kondo H, Yamakawa A, Shibata M, Takenawa T, van Blitterswijk WJ. Cloning of a novel human diacylglycerol kinase (DGKtheta) containing three cysteine-rich domains, a proline-rich region, and a pleckstrin homology domain with an overlapping Ras-associating domain. J Biol Chem. 1997;272:10422–10428. doi: 10.1074/jbc.272.16.10422. [DOI] [PubMed] [Google Scholar]
- Hsu KL, Tsuboi K, Adibekian A, Pugh H, Masuda K, Cravatt BF. DAGLbeta inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol. 2012;8:999–1007. doi: 10.1038/nchembio.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Misra S. Signaling and subcellular targeting by membrane-binding domains. Annu Rev Biophys Biomol Struct. 2000;29:49–79. doi: 10.1146/annurev.biophys.29.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Kai M, Yasuda S, Kanoh H, Sakane F. Identification and characterization of a novel human type II diacylglycerol kinase, DGK kappa. J Biol Chem. 2005;280:39870–39881. doi: 10.1074/jbc.M500669200. [DOI] [PubMed] [Google Scholar]
- Kozakov D, Hall DR, Jehle S, Luo L, Ochiana SO, Jones EV, Pollastri M, Allen KN, Whitty A, Vajda S. Ligand deconstruction: Why some fragment binding positions are conserved and others are not. Proc Natl Acad Sci U S A. 2015;112:E2585–2594. doi: 10.1073/pnas.1501567112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingel A, Sendzik M, Huang Y, Shultz MD, Cantwell J, Dillon MP, Fu X, Fuller J, Gabriel T, Gu J, et al. Structure-Guided Design of EED Binders Allosterically Inhibiting the Epigenetic Polycomb Repressive Complex 2 (PRC2) Methyltransferase. J Med Chem. 2017;60:415–427. doi: 10.1021/acs.jmedchem.6b01473. [DOI] [PubMed] [Google Scholar]
- Liu K, Kunii N, Sakuma M, Yamaki A, Mizuno S, Sato M, Sakai H, Kado S, Kumagai K, Kojima H, et al. A novel diacylglycerol kinase alpha-selective inhibitor, CU-3, induces cancer cell apoptosis and enhances immune response. J Lipid Res. 2016;57:368–379. doi: 10.1194/jlr.M062794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los AP, van Baal J, de Widt J, Divecha N, van Blitterswijk WJ. Structure-activity relationship of diacylglycerol kinase theta. Biochim Biophys Acta. 2004;1636:169–174. doi: 10.1016/j.bbalip.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- Merida I, Torres-Ayuso P, Avila-Flores A, Arranz-Nicolas J, Andrada E, Tello-Lafoz M, Liebana R, Arcos R. Diacylglycerol kinases in cancer. Adv Biol Regul. 2017;63:22–31. doi: 10.1016/j.jbior.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Merino E, Sanjuan MA, Moraga I, Cipres A, Merida I. Role of the diacylglycerol kinase alpha-conserved domains in membrane targeting in intact T cells. J Biol Chem. 2007;282:35396–35404. doi: 10.1074/jbc.M702085200. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Jerga A, Rock CO, White SW. Analysis of the Staphylococcus aureus DgkB structure reveals a common catalytic mechanism for the soluble diacylglycerol kinases. Structure. 2008;16:1036–1046. doi: 10.1016/j.str.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano JM, Hsu KL, Whitby LR, Niphakis MJ, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Hodder P, et al. Selective inhibitors and tailored activity probes for lipoprotein-associated phospholipase A(2) Bioorg Med Chem Lett. 2013;23:839–843. doi: 10.1016/j.bmcl.2012.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC, Koshland DE., Jr High cooperativity, specificity, and multiplicity in the protein kinase C-lipid interaction. J Biol Chem. 1989;264:14909–14915. [PubMed] [Google Scholar]
- Nordin BE, Liu Y, Aban A, Brown HE, Wu J, Hainley AK, Rosenblum JS, Nomanbhoy TK, Kozarich JW. ATP Acyl Phosphate Reactivity Reveals Native Conformations of Hsp90 Paralogs and Inhibitor Target Engagement. Biochemistry. 2015;54:3024–3036. doi: 10.1021/acs.biochem.5b00148. [DOI] [PubMed] [Google Scholar]
- Okerberg ES, Brown HE, Minimo L, Alemayehu S, Rosenblum J, Patricelli M, Nomanbhoy T, Kozarich JW. Monitoring native p38alpha:MK2/3 complexes via trans delivery of an ATP acyl phosphate probe. J Am Chem Soc. 2014;136:4664–4669. doi: 10.1021/ja4129907. [DOI] [PubMed] [Google Scholar]
- Patricelli Matthew P, Nomanbhoy Tyzoon K, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, et al. In Situ Kinase Profiling Reveals Functionally Relevant Properties of Native Kinases. Chemistry & Biology. 2011;18:699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry. 2007;46:350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- Posch A, Kohn J, Oh K, Hammond M, Liu N. V3 stain-free workflow for a practical, convenient, and reliable total protein loading control in western blotting. J Vis Exp. 2013:50948. doi: 10.3791/50948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz PU, Mendler AN, Masouris I, Durner L, Oberneder R, Noessner E. High DGK-alpha and disabled MAPK pathways cause dysfunction of human tumor-infiltrating CD8+ T cells that is reversible by pharmacologic intervention. J Immunol. 2012;188:5990–6000. doi: 10.4049/jimmunol.1103028. [DOI] [PubMed] [Google Scholar]
- Purow B. Molecular Pathways: Targeting Diacylglycerol Kinase Alpha in Cancer. Clinical Cancer Research. 2015;21:5008–5012. doi: 10.1158/1078-0432.CCR-15-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane F, Kai M, Wada I, Imai S, Kanoh H. The C-terminal part of diacylglycerol kinase alpha lacking zinc fingers serves as a catalytic domain. Biochem J. 1996;318(Pt 2):583–590. doi: 10.1042/bj3180583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane F, Mizuno S, Komenoi S. Diacylglycerol Kinases as Emerging Potential Drug Targets for a Variety of Diseases: An Update. Front Cell Dev Biol. 2016;4:82. doi: 10.3389/fcell.2016.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane F, Yamada K, Kanoh H, Yokoyama C, Tanabe T. Porcine diacylglycerol kinase sequence has zinc finger and E–F hand motifs. Nature. 1990;344:345–348. doi: 10.1038/344345a0. [DOI] [PubMed] [Google Scholar]
- Santos T, Carrasco S, Jones DR, Merida I, Eguinoa A. Dynamics of diacylglycerol kinase zeta translocation in living T-cells. Study of the structural domain requirements for translocation and activity. J Biol Chem. 2002;277:30300–30309. doi: 10.1074/jbc.M200999200. [DOI] [PubMed] [Google Scholar]
- Sato M, Liu K, Sasaki S, Kunii N, Sakai H, Mizuno H, Saga H, Sakane F. Evaluations of the selectivities of the diacylglycerol kinase inhibitors R59022 and R59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology. 2013;92:99–107. doi: 10.1159/000351849. [DOI] [PubMed] [Google Scholar]
- Schaap D, van der Wal J, van Blitterswijk WJ. Consensus sequences for ATP-binding sites in protein kinases do not apply to diacylglycerol kinases. Biochem J. 1994;304(Pt 2):661–662. doi: 10.1042/bj3040661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling B, Rardin MJ, MacLean BX, Zawadzka AM, Frewen BE, Cusack MP, Sorensen DJ, Bereman MS, Jing E, Wu CC, et al. Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Mol Cell Proteomics. 2012;11:202–214. doi: 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: Identification of signaling domains. Proceedings of the National Academy of Sciences. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo M, Irie K, Masuda A, Ohigashi H, Shirai Y, Miyasaka K, Saito N. Synthesis and phorbol ester binding of the cysteine-rich domains of diacylglycerol kinase (DGK) isozymes. DGKgamma and DGKbeta are new targets of tumor-promoting phorbol esters. J Biol Chem. 2003;278:18448–18454. doi: 10.1074/jbc.M300400200. [DOI] [PubMed] [Google Scholar]
- Shulga YV, Topham MK, Epand RM. Regulation and Functions of Diacylglycerol Kinases. Chemical Reviews. 2011;111:6186–6208. doi: 10.1021/cr1004106. [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Kishimoto A, Kikkawa U, Mori T, Nishizuka Y. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochem Biophys Res Commun. 1979;91:1218–1224. doi: 10.1016/0006-291x(79)91197-5. [DOI] [PubMed] [Google Scholar]
- Tang W, Bunting M, Zimmerman GA, McIntyre TM, Prescott SM. Molecular cloning of a novel human diacylglycerol kinase highly selective for arachidonate-containing substrates. J Biol Chem. 1996;271:10237–10241. [PubMed] [Google Scholar]
- Yada Y, Ozeki T, Kanoh H, Nozawa Y. Purification and characterization of cytosolic diacylglycerol kinases of human platelets. J Biol Chem. 1990;265:19237–19243. [PubMed] [Google Scholar]
- Yetukuri L, Ekroos K, Vidal-Puig A, Oresic M. Informatics and computational strategies for the study of lipids. Mol Biosyst. 2008;4:121–127. doi: 10.1039/b715468b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 4 – 7. SILAC ratios for ATP, Ritanserin, Ketanserin, and RF001 treatment of recombinant DGK-HEK293T soluble lysates. Multiple sequence alignments for sequence logo analysis.