

Summary
Native RNA immunoprecipitation (nRIP) coupled with high throughput sequencing (nRIP-seq) is a powerful technique that allows transcriptome-wide identification of the entire subset of coding- and noncoding- RNAs associated with a particular protein. Since this technology is carried out in a native condition without crosslinking, nRIP-seq detects RNAs that bind a protein directly or indirectly through a larger RNA-protein complex. Here, we use the interaction between RNA and chromatin modifiers, Polycomb proteins, as an example to describe this method. Using nRIP-seq, we provide a snapshot of Ezh2, a Polycomb component, and RNA interaction in mouse embryonic stem cells.
Keywords: RNA immunoprecipitation, Polycomb, Ezh2, long noncoding RNA
1. Introduction
Mounting evidence over the past decade suggests the vast majority of the genome is transcribed and most transcripts do not encode proteins (1, 2). A new frontier in biological research is to understand the functional importance of non-protein coding RNAs (ncRNAs) during development and in disease states. The small interfering non-coding RNAs (siRNAs) are known to silence gene expression in siRNA-dicer complexes (3), while long ncRNAs are believed to function as a scaffold to piece together different proteins at a specific genomic locus under a specific cellular context (4, 5). Recent studies suggest that RNA binds to a wide variety of proteins, including transcription factors (6, 7), chromatin modifiers (8–12), compartment proteins (13, 14) etc., suggesting that the dynamics of RNA-protein interaction play key roles in gene regulation. In order to capture a genome-wide pool of long transcripts associated with a specific protein, we developed a novel technology nRIP-seq (15). nRIP is a method that involves immunoprecipitation of RNA-binding proteins together with all RNAs associated with the protein in a native condition and further isolation of co-immunoprecipitated both coding and noncoding RNAs (16). Specific RNAs can be measured quantitatively using reverse transcription polymerase chain reaction (RT-PCR) or can be converted into cDNA libraries for high throughput sequencing. Using nRIP-seq, we captured Polycomb Repressive Complex 2 (PRC2), a chromatin modifier, transcriptome in mouse embryonic stem cells (15). Because PRC2 plays a central role in stem cell pluripotency and differentiation, such a genome-wide profile of regulatory RNAs provides invaluable information in regenerative medicine. Thus, nRIP-seq provides a powerful tool to study RNA-protein interaction in a given cell type and will facilitate our understanding of RNA’s function in gene regulation.
2. Materials
Prepare all solutions in RNAse- and DNAse-free Nanopure water (18Ω cm) and using only analytical grade reagents.
2.1 Nuclear lysate preparation
Cell lysis buffer is purchased from Active motif (Cat. No. 101212).
Polysome Lysis Buffer (PLB): 100 mM KCl, 5 mM MgCl2, 10 mM HEPES (pH 7.0), 0.5% NP40 (Igepal US Biological Cat. No. 3500). This buffer can be stored at 4°C. Before using, add 1mM Dithiothreitol (DTT), 200 units/ml RNase inhibitor (Roche Cat. No. 03335402001400), and 10 μL/mL of Protease inhibitor cocktail from Sigma (Cat. No. P8340).
The protease inhibitor cocktail from Sigma allows inhibition of serine, cysteine, aspartic proteases, and amidopeptidases. For larger amounts of liquid volume, Protease Inhibitor Cocktail Tablets are used (Roche Cat. No. 11836170001, 1 tablet for 10 mL solution).
RNAse inhibitors: In addition to the RNase inhibitor from Roche, Vanadyl Ribonucleoside Complex (VRC) can also be used.
A Dounce tissue grinder (Wheaton Cat. No. 357544) is used to break the cells and extract nuclei. There are two different sizes of pestles. The pestle labeled “tight” should be used.
DNAse I: For a more efficient nRIP, the nuclear lysate should be treated with 400 Units / mL recombinant DNAse I, RNAse free, (Roche Cat. No. 04716728001) to degrade DNA.
2.2 RNA immunoprecipitation
NT2 buffer: 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 0.05% NP40. Add 1mM DTT, 200 units/ml RNase inhibitor, 10 μL/mL of Protease inhibitor cocktail before use.
Magnetic beads: Protein A (Life Technologies Dynabeads Cat. No. 10002D) or protein G (Life Technologies, Cat. No. 10009D) beads are used for immunoprecipitation.
Magnetic rack for 1.5 mL Eppendorf tubes.
The quality of the antibody is key for the success of nRIP and should be determined experimentally before library construction. Ezh2 antibody (Active Motif) is used for Ezh2 nRIP-seq. Unfortunately this antibody has now been discontinued.
Trizol reagent (Life Technologies Cat. No. 15596018) is used to extract RNA.
The quantity of RNA after immunoprecipitation is determined by Quant-iTTM Ribogreen RNA Assay Kit (Life Technologies Cat. No. R11490).
2.3 cDNA library construction with template switch
The template switching method following the Clontech SMART protocol can be used to generate cDNA from very small amount of RNA (as little as 50 ng). During this process, modified MMLV reverse transcriptase add non-templated C’s at the end of a DNA template (17, 18). This 3′ overhang of Cs can be used to hybridize template switch primer with a G stretch.
SuperScript™ II Reverse Transcriptase (Life Technologies Cat. No. 18064) should be used to allow for non-templated addition of CCC at the termini of synthesized first strand cDNAs.
-
Primers: Primers should be designed to be compatible with the desired sequencing method. Since Illumina high throughput sequencing for genomic DNA was used for current study, we designed primers compatible with this method. New primers should be designed if other sequencing methods are used.
Illumina primers for sequencing genomic DNA
PCR Primers:
IG-Forward: 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′
IG-Reverse: 5′-CAAGCAGAAGACGGCATACGAGCTCTTCCGATCT-3′
Genomic DNA Sequencing Primer:
5′ ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′
Primers used for the current study:
Primer for first strand cDNA synthesis: a hybrid of partial IG-forward primer and random hexamer
5′-CTTTCCCTACACGACGCTCTTCCGATCTNNNNNN-3′
Template switch primer: IG-reverse primer with three Gs added to the 3′-end
5′-CAAGCAGAAGACGGCATACGAGCTCTTCCGATCTGGG-3′
PCR and sequencing primers are the same as the Illumina primers.
High fidelity Taq polymerase: Phusion Hot Start High-Fidelity DNA Polymerase (NEB Cat. No. M0535) is used for cDNA amplification.
2.4 cDNA library preparation for Illumina sequencing
Due to the presence of primer dimers in the lower molecular weight region of gels, only PCR products with size ranges larger than 200 bp were excised and extracted for sequencing.
3% NuSieve 3-1 agarose gel (Lonza, Cat. No. 50091) is used for size-selection.
QIAEX II Agarose Gel Extraction Kit (Qiagen Cat. No. 20021) is used to for DNA extraction.
DNA concentrations were quantitated by Quant-iT PicoGreen dsDNA assay kit (Life Technologies, Cat. No. P7589).
1. Methods
A general outline of nRIP-seq is shown in Fig. 1.
Figure 1.

Flow chart for nRIP-seq.
4.1 Preparation of nuclear lysate
Typically 1 X 107 cells are used for each IP. The cell number should be determined experimentally due to the variations of protein abundance in cells.
Cells are washed two times with ice cold 1XPBS.
Add 2 mL scrape buffer (1XPBS with 10 μL PMSF, 20 μL PIC, and 10 μL RNAse inhibitor) to the plate and get cells off the plate using a cell lifter.
Pellet the cells by centrifuge for 10 min at 720 g at 4°C and remove the supernatant.
Add 1 mL ice-cold lysis buffer with 10 μL PIC and 5 μL PMSF and 2 μL RNase inhibitor before use and incubate on ice for 30 minutes.
Transfer the cells to the ice-cold Dounce homogenizer. Dounce 10X to aid the release of nuclei.
Transfer cells to an cold 15 mL conical tubes and centrifuge at 2400xg for 10 min
Remove supernatant. Resuspend the pellet in 1mL polysome lysis buffer.
Pass through 27 1/2 gauge needle 4X to break nuclei.
Incubate with 40 μL (400 units) DNAse / 1 mL rotating in cold room for 30 minutes to digest DNA.
Centrifuge at highest speed of the cells for 10 min to collect the supernatant for IP.
4.2 Immunoprecipitation
For each reaction, add 100 μL lysate into 850 μL NT2 supplemented with 2 μL RNAse inhibitor, 2 μL RVC; 1μL DTT, and 5 μL proteinase inhibitors.
Take 9.5 μL out as 1% input.
Add ~ 5 μg antibody to the reaction mix and incubate at 4°C overnight by end-to-end rotation.
The next morning, wash protein A/G beads 3X with NT2 buffer.
Add 50 μL beads into the reaction mix and incubate at 4°C for 2 hr by end-to-end rotation.
Wash beads 6X with NT2 buffer, each time rotating for 5 minutes at 4°C.
4.3 RNA extraction
Add Trizol reagent directly to the beads and perform RNA extraction following the manufacturer’s protocol. It is important at this stage to add glycogen and glycoblue during
-
RNA precipitation in order to visualize trace amounts of RNA in the eppendorf tube.
Determine the RNA concentration using Quant-iTTM Ribogreen RNA Assay Kit.
4.4 cDNA synthesis using template switch
Typically, 100 ng RNA is used as starting material. Negative mock control without any RNA product should also be carried out to control for any trace amounts of reagent contamination and nonspecific amplification.
Dilute the primer for first strand synthesis at 2 μM.
Preheat a PCR block at 72°C.
-
Add these reagents on ice:
Reagent Sample (μL) Control (μL) RNAse-Free H2O 0 1.5 RNA (100ng/1.5μL) 1.5 0 First strand synthesis primer 2μM 0.5 0.5 Heat mix to 72°C for 10 min followed by 2 min on ice.
-
Add reserve transcriptase into the above mix:
Reagent Volume Superscript II RT buffer 1 μL 20mM DTT 0.5 μL 10mM dNTP 0.5 μL Superscript II RT 0.5 μL Total volume 4.5 μL Put samples back onto the thermocycler and incubate the samples at 20°C for 10 min, followed by 37°C for 10 min, then 42°C 45 min. Turn off the heated lid of the cycler.
Denature the template switch primer at 72°C for 5 min.
Add 0.5 μL heat denatured 10 μM template switch primer into the reaction mix and incubate at 42°C for another 15 min.
Stop the reaction at 95°C for 5 min.
4.5 PCR amplification
-
Set up the PCR reaction using using IG-Forward and IG-Reverse primers and Phusion polymerase as below:
H2O Add to 50 μL 5XPhusion HF buffer 10 μL 10mM dNTPs 1 μL IG-Forward 10μM 1 μL IG-Reverse 10μM 1 μL Template DNA 5 μL Polymerase 0.5 μL PCR amplification is carried out using the following cycle conditions: 98°C 30 sec, [98°C 10 sec, 65°C 1 min, 72°C 30 sec] X n cycles, 72°C 5 min, 4°C.
The optimal cycle numbers should be determined experimentally by qPCR analysis (Fig. 2).
Figure 2.
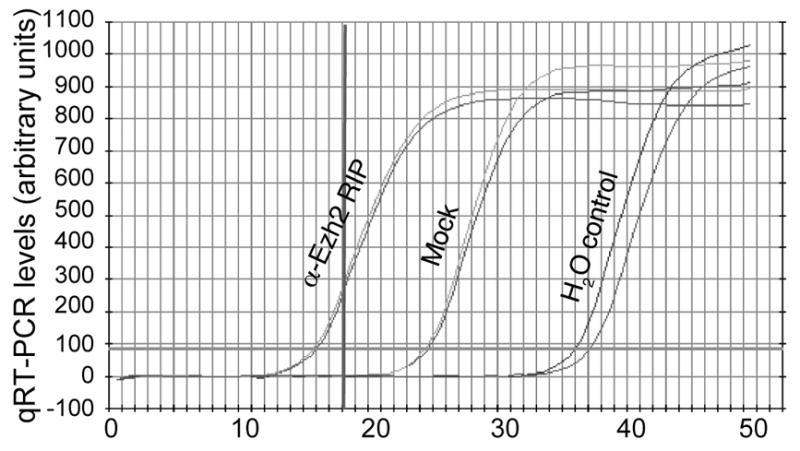
Example of determining a cycle number for library construction. An optimal cycle number should amplify the initial material in the linear phase but also provide enough product for sequencing.
4.6 Library preparation
PCR products are loaded on a 3% NuSieve agarose gel for size-selection. As shown in Fig. 3, PCR products below 200 bp are mainly primer dimers that are present both in wild type and control libraries. Little product was detected beyond 1.2 Kb, most likely due to the degradation of RNA during the RIP process.
Products ranging from 200–1,200 bp were excised and extracted by the QIAEX II Agarose Gel Extraction Kit.
DNA concentrations were quantitated by a PicoGreen Assay.
Five to ten microliters of 2 – 20 nM cDNA samples were sent for Illumina high throughput sequencing.
Figure 3.

Example of cDNA library prepared from RIP products. The box highlights the size selection of the RIP product. WT: wild type mESC: mouse embryonic stem cells.
5. Notes
A ribonuclease-free environment should be created and maintained whenever working with RNA. All the reagents should be RNAse free and RNAse inhibitor should be added into solutions to prevent RNA degradation. RNAse Zap (Life Technologies, Cat. No. AM9780) can be used to treat small equipment, such as Dounce homogenizers, and bench surfaces, followed by rinsing with DEPC-treated water and air drying.
It is recommended to process the cell pellets and carry out the RIP procedure right away. However, it is possible to snap freeze the cell pellets in liquid nitrogen and store at −80°C for future use.
Which type of magnetic beads to use depends on the immunoglobulin type and species of the primary antibody. Please consult the Life Technologies website to determine the beads that will be most suitable for your antibody. Typically, rabbit polyclonal antibodies have strong affinity for A or G beads and mouse monoclonal antibodies bind strongly to protein G beads. You can also mix A/G beads during immunoprecipitation. In general, protein A beads are less “sticky” than G beads and provide lower backgrounds.
Negative control samples or antibodies should be used during RIP. Typically, normal IgG is used during IP to control for nonspecific binding. A cell line that has protein of interest knocked down or knocked out is ideal to be used as a negative control for antibody specificity.
The wash condition is critical to decrease background and should be determined experimentally. Up to 1M salt can be used for stringent washes by adding NaCl directly into the NT2 buffer to reach the appropriate concentration. Since the experiment is carried out in native conditions without crosslinking, ionic detergents such as SDS, deoxycholate, or urea are not recommended to be included during washes.
It’s important that one changes pipette tips between different samples during all steps, to avoid cross contamination.
For some antibodies, it is recommended to coat the beads with antibody first, followed by lysate addition. For others, such as Ezh2, it is critical to add antibody into lysate first, followed by protein A/G pull down. Such conditions need to be determined experimentally.
The template switching method relies on the ability of MMLV reverse transcriptase to add a few non-templated cytosines to the 3′-ends of newly synthesized cDNAs upon reaching the 5′-ends of the RNA templates. The commercially available MMLV-derived reverse transcriptases include superscript II (Life Technologies), RevertAid H minus (Fermentas), and RevertAid Premium (Fermentas) (19). Superscript III reverse transcriptase from Life Technologies is not suitable for such an assay.
RNA fragmentation upon IP can be carried out using the fragmentation solution and protocol from Life Technologies (Cat. No. AM8740). However, this step will further degrade RNA and is not recommended for very small amounts of RNA obtained from RIP.
Since this experiment is carried out under native conditions, RNA extraction reagents, such as Trizol, can be added to beads directly for RNA extraction. No proteinase K digestion step is needed.
Since a small amount of RNA is used as the initial material, necessary controls, including no cell, no RNA, no RNA and no RT samples should be included during cDNA synthesis and PCR amplification.
To validate the library, quantitative PCR should be carried out to determine whether positive control RNA is present in the library.
References
- 1.Claverie JM. Fewer genes, more noncoding RNA. Science. 2005;309:1529–1530. doi: 10.1126/science.1116800. [DOI] [PubMed] [Google Scholar]
- 2.Carninci P, et al. The transcriptional landscape of the mammalian genome. Science. 2005;2309:1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 3.Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet. 2011;12:19–31. doi: 10.1038/nrg2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guttman M, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanz RB, et al. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. doi: 10.1016/s0092-8674(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 7.Willingham AT, et al. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309:1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 8.Rinn JL, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao J, et al. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pandey RR, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 11.Nagano T, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–1720. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 12.Bertani S, et al. The noncoding RNA Mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting MLL1 to chromatin. Mol Cell. 2011;43:1040–1046. doi: 10.1016/j.molcel.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Chen LL, Carmichael GG. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol Cell. 2009;35:467–478. doi: 10.1016/j.molcel.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clemson CM, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33:717–26. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keene JD, Komisarow JM, Friedersdorf MB. RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc. 2006;1:302–307. doi: 10.1038/nprot.2006.47. [DOI] [PubMed] [Google Scholar]
- 17.Cloonan N, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- 18.Matz M, et al. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 1999;27:1558–1560. doi: 10.1093/nar/27.6.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinto FL, Lindblad P. A guide for in-house design of template-switch-based 5′ rapid amplification of cDNA ends systems. Anal Biochem. 2010;397:227–232. doi: 10.1016/j.ab.2009.10.022. [DOI] [PubMed] [Google Scholar]