

Abstract
Opioid receptor antagonist naltrexone reduces alcohol consumption and relapse in both humans and rodents. The present study investigated whether hypothalamic proopiomelanocortin (POMC) neurons (producing beta-endorphin and melanocortins) play a role in alcohol drinking behaviors. Both male and female mice with targeted deletion of two neuronal Pomc enhancers nPE1 and nPE2 (nPE−/−), resulting in hypothalamic-specific POMC deficiency, were studied in short-access (4-h/day) drinking-in-the-dark (DID, alcohol in one bottle), intermittent access (IA, 24-hour cycles of alcohol access every other day, alcohol vs. water in a two-bottle choice), and alcohol deprivation effect (ADE) models. Wild-type nPE+/+ exposed to 1-week DID rapidly established stable alcohol drinking behavior with more intake in females, whereas the nPE−/− of both sexes had less intake and less preference. Though nPE−/− showed less saccharin intake and preference than nPE+/+, there was no genotype difference in sucrose intake or preference in the DID paradigm. After 3-week IA, nPE+/+ gradually escalated to high alcohol intake and preference, with more intake in females, whereas nPE−/− showed less escalation. Pharmacological blockade of mu-opioid receptors with naltrexone dose-dependently reduced intake in nPE+/+, but had blunted effects in nPE−/− of both sexes. When alcohol was presented again after 1-week abstinence from IA, nPE+/+ of both sexes displayed significant increases in alcohol intake (ADE or relapse-like drinking), with more pronounced ADE in females, whereas nPE−/− did not show ADE in either sex. Our results suggest that neuronal POMC is involved in modulation of alcohol “binge” drinking, escalation and “relapse”, probably via hypothalamic-mediated mechanisms, with sex differences.
Keywords: Hypothalamic-specific POMC-deficient mice, intermittent access drinking, alcohol deprivation effect, drinking-in-the-dark, naltrexone, sex differences
Introduction
Alcohol alters the activity of the endogenous opioid peptide systems, especially the proopiomelanocortin (POMC) system. POMC is a large peptide precursor that is cleaved to form several biologically active neuropeptides, including the potent opioid peptide beta-endorphin, adrenocorticotropic hormone (ACTH), beta-lipotropin and alpha-melanocyte-stimulating hormone. In the brain, a specialized population of neurons concentrated in the hypothalamic arcuate nucleus expresses POMC [Rubinstein et al. 1996; Cowley et al. 2001; de Souza et al. 2005; Zhou et al. 2013]. In the rat hypothalamus, alcohol increased POMC mRNA levels after 15 days of an alcohol-containing liquid diet [Angelogianni & Gianoulakis 1993]. Compared with Sardinian alcohol non-preferring (sNP) rats, there were higher basal POMC mRNA levels in the hypothalamus of Sardinian alcohol-preferring (sP) rats, and chronic alcohol exposure further increased hypothalamic POMC mRNA levels in sP rats [Zhou et al. 2013]. Taking into account the well-established role of beta-endorphin in reward-related and feeding behaviours [Koch et al. 2015], the genetically-determined POMC gene expression at basal levels and in response to alcohol exposure may contribute to the high alcohol preference and consumption observed in alcohol-preferring rats [Zhou et al. 2013] and mice [Jamensky & Gianoulakis 1999]. Decreased POMC mRNA and peptide levels were also reported in rat hypothalamus after chronic alcohol exposure [Rasmussen et al. 2002; Navarro et al. 2013].
Acute alcohol administration stimulated the release of beta-endorphin in the nucleus accumbens [Marinelli et al. 2003]. Since activation of the mu-opioid receptor (MOP-r) by beta-endorphin (the main endogenous ligand of MOP-r) is rewarding and increases dopamine release [Spanagel et al. 1991], beta-endorphin is involved in the reinforcing effects and motivational behaviors of alcohol [Koob & Kreek 2007; Roth-Deri et al. 2008]. Indeed, central beta-endorphin administration was found to induce conditioned place preference in rats [Amalric et al. 1987]. Numerous pharmacological studies provide strong evidence that opioid antagonists decrease alcohol reward, consumption, cue-induced reinstatement of alcohol seeking, and relapse-like drinking in rodents and primates [Altschuler et al. 1980; Volpicelli et al. 1986; Heyser et al. 1999], as well as alcohol drinking, craving and relapse episodes in human alcoholics [O’Malley et al. 1992, 2002; Volpicelli et al. 1992]. Moreover, alcohol consumption is reduced in MOP-r knockout mice [Roberts et al. 2000; Hall et al. 2001; Becker et al. 2002], further suggesting that the MOP-r is involved in the regulation of alcohol drinking.
Previous studies on the role of beta-endorphin in modulation of alcohol drinking using beta-endorphin deficient mice have been inconclusive. For example, earlier publications reported no effect of the global knockout of pituitary and brain beta-endorphin on alcohol intake in mice [Grisel et al. 1999; Grahame et al. 2000]. However, in another study, beta-endorphin knockout mice were reported to show decreased alcohol consumption [Racz et al. 2008]. A limitation of this global beta-endorphin knockout mouse model is that it does not allow for clarification of which specific regions of POMC cells (e.g., hypothalamus) are involved in alcohol drinking behaviors. Recently, two neuronal Pomc enhancers (nPE1 and nPE2), that are necessary and sufficient for POMC expression specifically in hypothalamic arcuate neurons, have been identified [de Souza et al. 2005]. Simultaneous transcriptional interference of nPE1 and nPE2 enhancer function by insertion of a neomycin selection cassette in the enhancer vicinity abolishes POMC gene expression in the arcuate nucleus of transgenic mice, while leaving POMC expression in pituitary cells at normal levels [Bumaschny et al. 2012]. Therefore, to determine the role of hypothalamic POMC neurons in alcohol drinking behaviors, in the present study, we used transgenic mice with region-specific POMC deficiency resulting from selective deletion of both nPE1 and nPE2 in combination with the neomycin insertion [Lam et al. 2015]. Specifically, we determined the effect of the tissue-specific Pomc gene manipulation on: (a) binge-like drinking in a 1-week drinking-in-the-dark model; (b) acquisition and escalation in a chronic (3-week) intermittent access drinking model of dependence-like behavior, and (c) relapse-like drinking in an alcohol deprivation effect model in both male and female mice.
MATERIALS AND METHODS
ANIMALS
[1] Pomc neuronal enhancers nPE1 and nPE2 knockout mice
The present study used intact, male and female, single-housed mice with targeted deletion of the POMC neuronal enhancers nPE1 and nPE2 (nPE−/−) [Lam et al. 2015]. The gene mutations were generated by homologous recombination in 129S6/SvEvTac Taffy ES cells to produce the chimeric founder mice, followed by 7–10 generations of backcrossing onto the C57BL/6J strain for the mice used in these studies. Specifically, in these transgenic mice, simultaneous deletion of nPE1 and nPE2 and insertion of a neomycin selection cassette in the enhancer vicinity in the context of the intact Pomc pituitary enhancer region and proximal promoter abolishes POMC gene expression in the arcuate nucleus, without altering POMC expression in pituitary cells. Consistent with this previous report, the nPE−/− mice with the lack of hypothalamic POMC expression had greater daily food intake in 3 measures at 8–9 weeks of age (5.3g in males and 5.4g in females, respectively) than the nPE+/+ or nP+/− mice (~3.3g in males and 3.2g in females, respectively, for both genotypes). At the time the experiments started (age 8–10 weeks), the nPE−/− mice had greater body weight (~ 40g and 35g in males and females, respectively) than the nPE+/+ or nP+/− mice (~ 27g and 23g in males and females, respectively, for both genotypes). Such phenotypes are not shown until the age of 7–8 weeks.
[2] Beta-endorphin deficient (B-END KO) mice
The mice were generated and described previously [Rubinstein et al. 1996]. A premature STOP codon was inserted into the coding region of exon 3 of Pomc, to prevent the translation of the N -terminal beta-endorphin peptide by homologous recombination in 129/Sv-derived D3 ES cells. The original chimeric founder was subsequently backcrossed onto the C57BL/6N strain for 5 generations and then onto the C57BL/6J strain for a minimum of 5 additional generations to produce the mice used in these studies. These mice have a global deletion of beta-endorphin peptides, but intact melanocortins and ACTH (including in the pituitary and hypothalamus).
EXPERIMENTS
I. Effects of genotype and sex on alcohol (7.5%, 15% or 30%) drinking in a 4-day drinking-in-the-dark (DID) model in nPE and B-END mice
Based on the publication by Rhodes et al. (2005), the basic paradigm with our modifications was as follows: Starting at 3 hours after lights off, water bottles were replaced with 10-ml alcohol tubes filled with alcohol solution, kept for 4 hours and then replaced with the water bottles [Zhou et al. 2016]. For 4 days, alcohol intake values were recorded daily after 4 hours of alcohol access. These data were used to calculate alcohol intake (i.e., g ⁄ kg). Genotype (nPE+/+, nPE+/− vs. nPE−/−) and sex (male vs. female) differences were then tested in this 4-hour short access model (Table 1A). To further assess the genotype differences in alcohol preference, nPE+/+ and nPE−/− mice of both sexes were exposed to the 2-bottle “alcohol vs. water” free choice regimen on day 5, after 4 days of DID. In control experiments, sucrose (8% or 16%) or saccharin (0.1%, 0.2% or 0.4%) was used in place of alcohol, with an otherwise identical procedure. For each concentration of alcohol, saccharin or sucrose, separate groups of transgenic mice were used to test the following behaviors: [1.1] Alcohol drinking in nPE; [1.2] Sucrose drinking in nPE; and [1.3] Saccharin drinking in nPE; as well as [1.4] Alcohol drinking in B-END knockout.
Table 1.
Experimental timelines
| A. The 4-day alcohol drinking-in-the-dark (DID) model and alcohol preference (alcohol vs. water) on day 5 | ||||
|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Day 4 | Day 5 |
| 4-h alcohol | 4-h alcohol | 4-h alcohol | 4-h alcohol | 4-h alcohol vs water |
| B. The 3-week chronic intermittent access (IA) drinking model (15% alcohol vs. water) every other day with mu- (naltrexone) or kappa- (nor-BNI) opioid antagonist or morphine. | |||
|---|---|---|---|
| Week 1 | Week 2 | Week 3 | Week 4 |
| 24-h alcohol vs water | 24-h alcohol vs water | 24-h alcohol vs water | Naltrexone, nor-BNI, morphine |
| C. The alcohol deprivation effect (ADE) model after 1-week abstinence from a 3-week chronic intermittent access (IA) drinking (15% alcohol vs. water) | ||||
|---|---|---|---|---|
| Week 1 | Week 2 | Week 3 | Week 4 | Week 5 |
| 24-h alcohol vs water | 24-h alcohol vs water | 24-h alcohol vs water Baseline (BL) |
Alcohol abstinence | ADE, alcohol vs water Post-abstinence (P) |
II. Effects of genotype and sex on alcohol (15%) drinking in a 3-week intermittent access (IA) drinking model with acute administration of naltrexone, morphine or nor-binaltorphimine (nor-BNI) in nPE mice
The primary objective of the following experiments was to determine whether intermittent 24-hour long access to alcohol for 3 weeks led to elevated alcohol intake and preference in each sex and genotype. The IA model was similar to an earlier protocol [Hwa et al. 2011], with some modifications: mice had 24 hours of access to 15% alcohol every other day for 3 weeks with food available at all times in a two-bottle free choice paradigm. For IA alcohol drinking [Experiment 2.1], the procedures were identical to those of the above DID model with the following exceptions: starting at 3 hours after lights off, both the water and 15% alcohol tubes were placed on home cages for 24 hours. The left ⁄right position of the tubes was set up randomly. Alcohol and water intake values were recorded after 4, 8 and 24 hours of alcohol access on the drinking days. These data were used to calculate alcohol intake (i.e., g ⁄ kg) and relative preference for alcohol (i.e., alcohol intake ⁄ total fluid intake) (Table 1B).
After 3 weeks of IA, the next objective was to determine whether there were potential genotype or sex differences in drinking behavior in the IA mice following mu- and kappa-opioid receptor activation or blockade (morphine, naltrexone or nor-BNI). Separate groups of mice were used for each dose of each compound in the following experiments: [2.2] IA with acute naltrexone (1 or 2 mg/kg); [2.3] IA with acute morphine (0.1 or 0.3 mg/kg); and [2.4] IA with acute nor-BNI (5 mg/kg). On the test day, alcohol (15% concentration) was presented after a single injection of the compound or vehicle, and then alcohol and water intake values were recorded at 4-, 8- and 24-hour time points.
III. Effects of genotype and sex on alcohol “relapse” drinking in an alcohol deprivation effect (ADE) model in nPE mice
In the baseline session, alcohol and water intake was recorded at 4, 8 and 24 hours after 3 weeks of IA. The alcohol bottles were then removed and presented again after 7 days of abstinence, at which point the alcohol and water intake values were recorded in 2 post-abstinence sessions (on 2 consecutive days). We determined whether the IA to alcohol for 3 weeks led to the ADE with potential genotype and sex differences (Table 1C).
IV. Effects of genotype and sex on basal corticosterone levels, analgesic threshold and locomotor activity in nPE mice
To determine genotype and sex differences in basal levels of the stress hormone corticosterone, blood samples from nPE+/+ and nPE−/− mice were collected 3 hours after lights off (the time of the drinking sessions), and plasma corticosterone levels were assayed by radioimmunoassays. The thermal analgesic threshold was measured 3 hours after lights off in another set of nPE mice, and the time to response (rear paw flutter, rear paw licking or jumping) at 52° C on the hot plate was measured. Spontaneous locomotor activity was evaluated 3 hours after lights off in another set of nPE mice of both sexes in a place conditioning apparatus for 30 min. In the first of two tests, half the mice for each genotype were placed in the white chamber and half in the black one. In the second test, the chambers were switched. The average values of both tests were used as the locomotor activity score.
Data analysis
In most experiments, group differences in alcohol (sucrose or saccharin) intake and preference ratios were analyzed using 2-, 3- or 4-way ANOVA for genotype, sex, time (hour, day or session) and treatments followed by Newman-Keuls post-hoc multiple comparison tests. The statistical analysis results for all the figures and tables are provided in detail in the Supplemental Information.
RESULTS
I. Effects of genotype and sex on alcohol drinking in a DID model in nPE and B-END mice
1.1. Genotype and sex differences in alcohol intake and preference in the nPE mice
Between the genotypes, the nPE−/− mice of both sexes drank less alcohol at every concentration tested than the nPE+/+ ones over the 4-day DID experiment (Figure 1) [p<0.000001]. Over time, wild-type nPE+/+ mice of both sexes escalated their intake; in contrast, only the male nPE−/− mice escalated their intake. Though 3-way ANOVA revealed no significant main effect of sex at 15% alcohol, previous studies in mice have found sex differences in alcohol drinking behaviors [Racz et al. 2008; Hall et al. 2001; Hwa et al. 2011; Becker & Koob 2016], Using these findings as an a priori hypothesis, post-hoc result showed that the nPE+/+ females had more 15% alcohol intake on day 3 than the nPE+/+ males [p<0.05] (Figure 1B). Between the sexes, the nPE+/+ females consumed more 30% alcohol than the nPE+/+ males during the first 2 days [p<0.05] (Figure 1C). When alcohol preference was tested on day 5 using a 4-h, 2-bottle (alcohol vs. water) choice paradigm, the nPE−/− mice of both sexes had less preference than the nPE+/+ ones at 7.5% and 15% concentrations [p<0.05] (Table 2A, 2B). However, the heterozygous nPE+/− mice did not show any differences from the nPE+/+ in either sex after exposure to 15% alcohol over the 4-day DID experiment (Figure 1 Additional Information in Supplementary Information).
Figure 1. Genotype and sex differences in alcohol (7.5, 15 and 30%) intake in a 4-day drinking-in-the-dark (DID) model in male (left) and female (right) nPE mice.
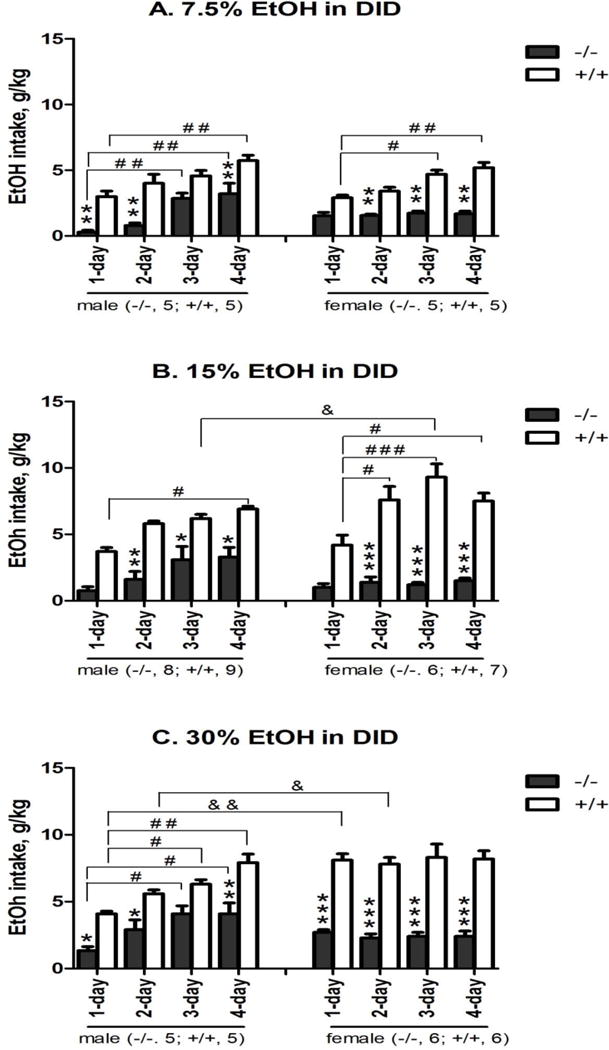
Alcohol at 7.5% (A), 15% (B) or 30% (C) concentration was presented 3 hours after the beginning of dark cycle, and alcohol intake values (EtOH intake, g/kg) were recorded after 4 hours of alcohol access for 4 days in the nPE+/+ and nPE−/− mice. Genotype difference: *p<0.05, **p<0.01 or ***p<0.005 vs. nPE+/+ at the same day; Day difference: #p<0.05, ##p<0.01 or ###p<0.005 vs. the same genotype at day 1 session; and Sex difference: &p<0.05 or &p<0.01 vs. the same genotype on the same day by 3-way ANOVA with Newman-Keuls post-hoc tests (n=5–8 in nPE−/−; n=5–9 in nPE+/+). (A) 7.5% alcohol: 3-way ANOVA revealed significant effects of genotype [F (1, 64) = 181, p<0.000001] and day [F (3, 64) = 25, p<0.00001], with no interaction between genotype and sex. Post hoc analysis showed that: (1) between genotypes, nPE−/− males had less intake than nPE+/+ ones on day 1, day 2 and day 4 [p<0.01 for all]; nPE−/− females had less intake than nPE+/+ ones on day 2, day 3 and day 4 [p<0.01 for all]; and (2) between the days, nPE+/+ males had more intake on day 4 than on day 1 [p<0.01]; nPE−/− males had more intake on day 3 and day 4 than on day 1 [p<0.01 for both]; and nPE+/+ females had more intake on day 3 and day 4 than on day 1 [p<0.05 and p<0.01, respectively]. (B) 15% alcohol: 3-way ANOVA revealed significant effects of genotype [F (1, 104) = 156, p<0.000001], day [F (3, 104) = 9.7, p<0.00005] and interaction between genotype and sex [F (1, 104) = 9.9, p<0.005]. Post hoc analysis showed that: (1) between genotypes, nPE−/− males had less intake than nPE+/+ ones on day 2, day 3 and day 4 [p<0.05 for all]; nPE−/− females had less intake than nPE+/+ ones on day 2, day 3 and day 4 [p<0.001 for all]; and (2) between the days, nPE+/+ males had more intake on day 4 than on day 1 [p<0.05]; nPE+/+ females had more intake on day 2, day 3 and day 4 than on day 1 [p<0.05 for all]. Though overall sex difference was not significant, the post-hoc showed that nPE+/+ females had more intake on day 3 than nPE+/+ males [p<0.05]. (C) 30% alcohol: 3-way ANOVA revealed significant effects of genotype [F (1, 72) = 196, p<0.000001], day [F (3, 72) = 5.1, p<0.005], sex [F (1, 72) = 5.3, p<0.05], interaction between genotype and sex [F (1, 72) = 21, p<0.0001] and interaction between day and sex [F (3, 72) = 5.7, p<0.005]. Post hoc analysis showed that: (1) between genotypes, nPE−/− males had less intake than nPE+/+ ones on day 1, day 2 and day 4 [p<0.05 for all]; nPE−/− females had less intake than the nPE+/+ ones from day 1 to day 4 [p<0.005 for all]; (2) between the days, nPE+/+ males had more intake on day 3 and day 4 than on day 1 [p<0.05 and p<0.01, respectively]; nPE−/− males had more intake on day 3 and day 4 than on day 1 [p<0.05 for both]; and (3) between sex, nPE+/+ females had more intake on day 1 and day 2 than nPE+/+ males [p<0.01 and p<0.05, respectively].
Table 2. Genotype differences in alcohol (7.5, 15 or 30%) preference after 4 days of drinking-in-the-dark (DID) in male (A) and female (B) nPE mice.
On the test day, both alcohol (7.5%, 15% or 30% concentration) and water were presented 3 hours after the beginning of dark cycle, and alcohol and water intake values were recorded after 4 hours of alcohol access in the nPE+/+ and nPE−/− mice. Genotype difference: *p<0.05 vs. nPE+/+ by Student’s t-tests.
| A. Males | ||||||
|---|---|---|---|---|---|---|
| Genotype | 7.5% alcohol | 15% alcohol | 30% alcohol | |||
| nPE+/+ (n=5) |
nPE−/− (n=5) |
nPE+/+ (n=9) |
nPE−/− (n=8) |
nPE+/+ (n=5) |
nPE−/− (n=5) |
|
| Alcohol Preference | 0.83±0.04 | 0.60±0.03* | 0.67±0.08 | 0.42±0.04* | 0.50±0.04 | 0.51±0.04 |
| B. Females | ||||||
|---|---|---|---|---|---|---|
| Genotype | 7.5% alcohol | 15% alcohol | 30% alcohol | |||
| nPE+/+ (n=5) |
nPE−/− (n=5) |
nPE+/+ (n=6) |
nPE−/− (n=7) |
nPE+/+ (n=6) |
nPE−/− (n=6) |
|
| Alcohol Preference | 0.85±0.03 | 0.54±0.02* | 0.61±0.06 | 0.24±0.07* | 0.55±0.07 | 0.59±0.02 |
1.2. No genotype or sex differences in sucrose drinking or preference
The specificity of the genotype differences on alcohol intake was tested using sucrose, another caloric reinforcer, with the same DID paradigm. During 4 days of 8% or 16% sucrose drinking, there was no genotype difference in either sex (Table S2), though there was a slight, but not significant, decrease in nPE−/− females at 16% concentration.
1.3. Genotype and sex differences in saccharin drinking and preference
The specificity of the genotype differences in alcohol intake was further tested by examining intake of non-caloric reinforcer saccharin. The nPE−/− mice of both sexes drank less 0.4% saccharin than the nPE+/+ ones over the 4-day DID experiment [p<0.01], with apparent, but not significant, less intakes at 0.1% and 0.2% saccharin (Table 3A, 3B). When saccharin preference was tested on day 5 using a 4-h, 2-bottle choice paradigm, the nPE−/− mice of both sexes had less 0.2% saccharin preference than the nPE+/+ ones [p<0.05] (Table 3C, 3D), with no genotype differences in 0.1% or 0.4% saccharin preference. Between the sexes, the nPE+/+ females had more saccharin intake than the nPE+/+ males at 0.4% over the 4-day DID experiment [p<0.01] (Table 3A, 3B).
Table 3. Genotype and sex differences in saccharin (0.1, 0.2 and 0.4%) intake in a 4-day drinking-in-the-dark (DID) model in male (A) and female (B) nPE mice, and in saccharin preference after the 4-day DID model in male (C) and female (D) nPE mice.
In A and B, saccharin was presented 3 hours after the beginning of the dark cycle, and saccharin intake values were recorded after 4 hours of saccharin access for 4 days in the nPE+/+ and nPE−/− mice. In C and D, on the test day (day 5), both saccharin (0.1%, 0.2% or 0.4% concentration) and water were presented, and saccharin and water intake were recorded after 4 hours of saccharin access. Genotype difference: *p<0.05, **p<0.01 or ***p<0.005 vs. nPE+/+ on the same day; Sex difference: &p<0.01 vs. the same genotype on the same day.
At 0.1% or 0.2% saccharin, 3-way ANOVA did not show any differences between the genotypes, sexes or any effects of their interaction. At 0.4% saccharin, 3-way ANOVA revealed significant effects of genotype [F (1, 64) = 53, p<0.000001], sex [F (1, 64) = 56, p<0.000001], and interaction between genotype and sex [F (1, 64) = 43, p<0.00005]. Post hoc analysis showed that: (1) between genotypes, both nPE−/− males and females had less intake from day 1 to day 4 than nPE+/+ ones [p<0.01 for all]; nPE−/− females had less intake at 8 and 24 hours than nPE+/+ ones [p<0.005 for both], and (2) between sexes, nPE+/+ males had less intake from day 1 to day 4 than nPE+/+ females [p<0.01 for all].
| A. Saccharin intake in males (n=5) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | Day 1 | Day 2 | Day 3 | Day 4 | ||||
| nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | |
| 0.1%, g/kg | 0.16± 0.02 | 0.10± 0.03 | 0.19± 0.03 | 0.08± 0.02 | 0.25± 0.02 | 0.07± 0.04 | 0.28± 0.03 | 0.09± 0.03 |
| 0.2%, g/kg | 0.29± 0.03 | 0.18± 0.03 | 0.31± 0.02 | 0.13± 0.02 | 0.48± 0.08 | 0.19± 0.03 | 0.53± 0.07 | 0.20± 0.04 |
| 0.4%, g/kg | 0.65± 0.08 && | 0.32± 0.07* | 0.69± 0.10 && | 0.38± 0.08* | 0.78± 0.13 && | 0.32± 0.08* | 0.81± 0.04 && | 0.33± 0.08** |
| B. Saccharin intake in females (n=5) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | Day 1 | Day 2 | Day 3 | Day 4 | ||||
| nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | |
| 0.1%, g/kg | 0.19± 0.03 | 0.04± 0.01 | 0.22± 0.04 | 0.12± 0.02 | 0.25± 0.01 | 0.11± 0.01 | 0.23± 0.02 | 0.07± 0.02 |
| 0.2%, g/kg | 0.29± 0.08 | 0.24± 0.05 | 0.54± 0.05 | 0.22± 0.05 | 0.49± 0.04 | 0.23± 0.04 | 0.53± 0.02 | 0.12± 0.04 |
| 0.4%, g/kg | 1.03± 0.05 | 0.38± 0.08*** | 1.03± 0.08 | 0.39± 0.02*** | 1.16± 0.04 | 0.36± 0.07*** | 1.07± 0.06 | 0.31± 0.08*** |
| C. Saccharin preference in males (n=5). Genotype difference: *p<0.05 vs. nPE+/+ on the same day by Student’s t-tests. | ||||||
|---|---|---|---|---|---|---|
| Genotype | 0.1% | 0.2% | 0.4% | |||
| nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | |
| Preference | 0.94±0.02 | 0.90±0.01 | 0.91±0.04 | 0.67±0.07* | 0.95±0.01 | 0.92±0.02 |
| D. Saccharin preference in females (n=5). Genotype difference: *p<0.05 vs. nPE+/+ on the same day by Student’s t-tests. | ||||||
|---|---|---|---|---|---|---|
| Genotype | 0.1% | 0.2% | 0.4% | |||
| nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | nPE+/+ | nPE−/− | |
| Preference | 0.92±0.02 | 0.90±0.01 | 0.93±0.02 | 0.71±0.01* | 0.94±0.02 | 0.92±0.02 |
1.4. Sex, but no genotype, differences in alcohol intake in B-END+/+ and B-END−/− mice
In the following experiments, the alcohol exposure procedure was identical to the above experiment. During 4 days of DID experiment, there was no genotype difference in alcohol intake in either sex (Table S4), though the B-END+/+ females showed more alcohol intake than the B-END+/+ males in all alcohol concentrations tested [p<0.000001]. When 15% alcohol preference was tested on day 5, there was no genotype difference (data not shown).
II. Effects of genotype and sex on alcohol (15%) drinking in an IA model with acute administration of naltrexone, morphine or nor-BNI in nPE mice
2.1 Genotype and sex differences in alcohol intake and preference in an IA model
The nPE+/+ males exposed to the 2-bottle “alcohol (15%) vs. water” free choice regimen every other day for 3 weeks had more alcohol intake and higher preference ratios in session 10 than in session 1 [p<0.00001] (Figure 2A, left). To test our a priori hypothesis if there were sex differences, we included the post-hoc result showing that nPE+/+ females had more intake than the nPE+/+ males in sessions 1 and 10 [p<0.01], even though 4-way ANOVA showed no significant main effect of sex and session interaction (Figure 2A, right). In contrast, neither the male nor the female nPE−/− mice escalated their intake significantly over 3 weeks, with lower consumption [p<0.000001] and preference [p<0.000001] than the nPE+/+ ones.
Figure 2. Genotype and sex differences in alcohol intake (A) and alcohol preference (B) in male (left) and female (right) nPE mice in a 3-week intermittent access (IA) drinking model.
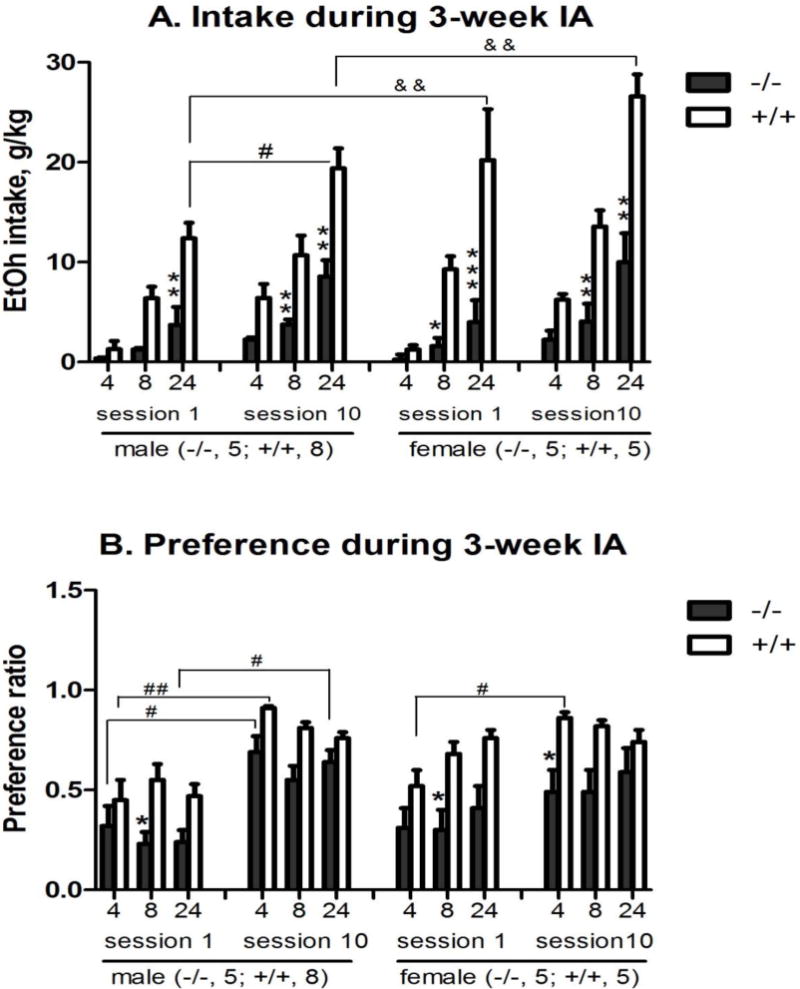
The mice were exposed to the 2-bottle “alcohol (15%) vs. water” choice regimen every other day for 3 weeks. Data are presented after 4, 8 and 24 hours of alcohol access in session 1 on the first day and in session 10 after 3 weeks of IA. Genotype difference: *p<0.05, **p<0.01 or *** p<0.005 vs. nPE+/+ at the same hour; Session difference: #p<0.05, ##p<0.01 vs. the same genotype at the same hour in session 1; and Sex difference: &p<0.01 vs. the same genotype in the same session by 4-way ANOVA with Newman-Keuls post-hoc tests (n=5 in nPE−/−; n=5–8 in nPE+/+). (A) Alcohol intake: 4-way ANOVA revealed significant effects of genotype [F (1, 114) = 146, p<0.000001], session [F (1, 114) = 44, p<0.00001], sex [F (1, 114) = 13, p<0.0005], and interaction between genotype and sex [F (1, 114) = 6.9, p<0.01]. Post hoc analysis showed that: (1) in Session 1 between genotypes, nPE−/− males had less intake at 24 hours than nPE+/+ males [p<0.01]; nPE−/− females had less intake at 8 and 24 hours than nPE+/+ females [p<0.05 and p<0.001, respectively]; (2) in Session 10 between genotypes, both nPE−/− males and females had less intake at 8 and 24 hours than nPE+/+ ones [p<0.01 for all]; and (3) between the Sessions, nPE+/+ males had more intake at 24 hours in Session 10 than in Session 1 [p<0.05]. Though overall interaction between sex and session was not significant, nPE+/+ females had more intake at 24 hours than nPE+/+ males [p<0.01] in both Sessions 1 and 10. (B) Alcohol preference: 4-way ANOVA revealed significant effects of genotype [F (1, 114) = 70, p<0.000001] and session [F (1, 114) = 63, p<0.000001]. Post hoc analysis showed that: (1) in Session 1 between genotypes, both nPE−/− males and females had less preference at 8 hours than nPE+/+ ones [p<0.05]; (2) in Session 10 between genotypes, nPE−/− females had less preference at 4 hours than nPE+/+ ones [p<0.05]; and (3) between the Sessions, both nPE+/+ males and females had more preference at 4 hours in Session 10 than in Session 1 [p<0.01 and p<0.05, respectively]; and nPE−/− males had more preference at 4 and 24 hours in Session 10 than in Session 1 [p<0.05 for both].
To test if there were genotype or sex effects on consumption at other alcohol concentrations, after 3 weeks of IA, the alcohol concentration was switched to 7.5% and then 30% alcohol. In both sexes, the nPE−/− mice had significantly less alcohol intake at both concentrations than the nPE+/+ ones [p<0.000001] (Table S5A, S5B). Additionally, the nPE+/+ females had more intake than the nPE+/+ males at both concentrations [p<0.05].
2.2. Genotype differences in the effect of mu-opioid receptor antagonist naltrexone on alcohol drinking after IA
Naltrexone at 1 mg/kg had no significant effect on alcohol intake or preference ratio in nPE+/+ or nPE−/− mice of either sex (Table S6). At 2 mg/kg, naltrexone reduced alcohol intake in the nPE+/+ mice of both sexes, with a significant effect at 24 hours [p<0.01] (Table 4). In contrast, naltrexone did not reduce alcohol intake in the nPE−/− mice of either sex.
Table 4. Genotype differences in effects of acute naltrexone (NTN, 2 mg/kg) on alcohol (15%) intake and its preference in male (A) and female (B) nPE mice after 3 weeks of intermittent access (IA) drinking.
On the test day, 15% alcohol was presented 10 min after a single i.p. injection of NTN in saline or vehicle, and then alcohol and water intake values were recorded after 4, 8 and 24 hours of alcohol access. In the following experiments, mice in each sex were assigned to one of four treatment groups: (1) nPE+/+ with vehicle as control; (2) nPE+/+ with 2 mg/kg NTN; (3) nPE−/− with vehicle as control; and (4) nPE−/− with 2 mg/kg NTN. Data are presented after 4, 8 and 24 hours of alcohol access. Genotype difference: *p<0.05 or *** p<0.005 vs. nPE+/+ mice at the same hour after the same treatment; Naltrexone treatment difference: ##p<0.01or ###p<0.005 vs. vehicle control in the same genotype at the same hour by 4-way ANOVA with Newman-Keuls post-hoc tests.
| A. Males | |||||
|---|---|---|---|---|---|
| Genotype | nPE+/+ (n = 6) | nPE−/− (n = 5) | |||
| Treatment | vehicle | 2 mg/kg NTN | vehicle | 2 mg/kg NTN | |
| Intake g/kg | 4h | 7.6 ± 1.6 | 2.0 ± 0.7 | 2.9 ± 0.51 | 1.0 ± 0.08 |
| 8h | 10.9 ± 2.2 | 5.8 ± 1.1 | 4.5 ± 0.8* | 2.8 ± 0.10 | |
| 24h | 21.4 ± 2.1 | 15.5 ± 1.3## | 9.8 ± 2.2*** | 7.4 ± 0.7 | |
| Preference | 4h | 0.86 ± 0.02 | 0.40 ± 0.13 | 0.68 ± 0.11 | 0.51 ± 0.10 |
| 8h | 0.71 ± 0.04 | 0.54 ± 0.08 | 0.66 ± 0.12 | 0.53 ± 0.12 | |
| 24h | 0.67 ± 0.04 | 0.66 ± 0.06 | 0.58 ± 0.11 | 0.58 ± 0.13 | |
| B. Females | |||||
|---|---|---|---|---|---|
| Genotype | nPE+/+ (n = 5) | nPE−/− (n = 5) | |||
| Treatment | vehicle | 2 mg/kg NTN | vehicle | 2 mg/kg NTN | |
| Intake g/kg | 4h | 5.6 ± 0.79 | 4.1 ± 0.37 | 1.4 ± 0.57 | 1.2 ± 0.46 |
| 8h | 12.0 ± 0.89 | 8.7 ± 0.64 | 2.9 ± 1.03*** | 3.3 ± 0.88 | |
| 24h | 24.4 ± 1.85 | 17.2 ± 1.3### | 6.2 ± 1.9*** | 8.4 ± 1.8 | |
| Preference | 4h | 0.81 ± 0.06 | 0.72 ± 0.08 | 0.51 ± 0.14 | 0.59 ± 0.08 |
| 8h | 0.74 ± 0.07 | 0.68 ± 0.09 | 0.42 ± 0.14 | 0.53 ± 0.12 | |
| 24h | 0.65 ± 0.04 | 0.63 ± 0.07 | 0.42 ± 0.12 | 0.50 ± 0.11 | |
Alcohol intake: 4-way ANOVA revealed significant effects of genotype [F (1, 102) = 181, p<0.000001], NTN treatment [F (1, 102) = 28, p<0.00005], interaction between genotype and sex [F (1, 102) = 4.2, p<0.05], interaction between genotype and NTN treatment [F (1, 102) = 15, p<0.0005], and interaction between sex and NTN treatment [F (1, 102) = 4.9, p<0.05]. Post hoc analysis showed that: (1) between genotypes, nPE−/− males and females had less intake at 8 and 24 hours than nPE+/+ ones [p<0.05 and p<0.005, respectively]; and (2) NTN treatment at 2 mg/kg reduced intake at 24 hours in nPE+/+ males and females [p<0.01 and p<0.005, respectively], but not in the nPE−/− ones.
Alcohol preference: 4-way ANOVA revealed significant effects of genotype [F (1, 102) = 12, p<0.001], NTN treatment [F (1, 102) = 4.3, p<0.05], interaction between genotype and sex [F (1, 102) = 4.5, p<0.05], and interaction between sex and NTN treatment [F (1, 102) = 5.2, p<0.05].
2.3. No genotype differences in the effect of mu-opioid receptor agonist morphine on alcohol drinking after IA
Morphine at 0.1 mg/kg (Table S7) and 0.3 mg/kg doses (Table S8) had no effect on alcohol intake or preference in nPE+/+ or nPE−/− mice of either sex.
2.4. Sex, but no genotype, differences in the effect of selective kappa-opioid receptor antagonist nor-BNI on alcohol drinking after IA
At 5 mg/kg, nor-BNI significantly reduced alcohol intake at 24 hours in male mice of both the nPE+/+ and nPE−/− genotypes [p<0.05] (Table S9A). In females, however, there was no effect of acute nor-BNI treatment on either genotype (Table S9B).
III. Effects of genotype and sex on alcohol relapse-like drinking in an ADE model
The nPE+/+ mice of both sexes had significantly more alcohol intake at the 4-hour time point in the post-abstinence session 1 (P1) than the ones in the baseline session (BL) [p<0.05] (Figure 3A). The nPE−/− mice did not show this ADE. To test our a priori hypothesis if there were sex differences, we included the post-hoc result showing that nPE+/+ females had more intake than the nPE+/+ males in session P1 [p<0.05], though 3-way ANOVA showed no significant main effect of sex (Figure 3A). There was no change in alcohol preference in either genotype at 4 hours (Figure 3B). There was no ADE observed at 8 or 24 hours in either genotype (Table S10).
Figure 3. Genotype and sex differences in 15% alcohol intake (A) and preference (B) in male (left) and female (right) nPE mice in an alcohol deprivation effect (ADE) model at the 4-hour time point.
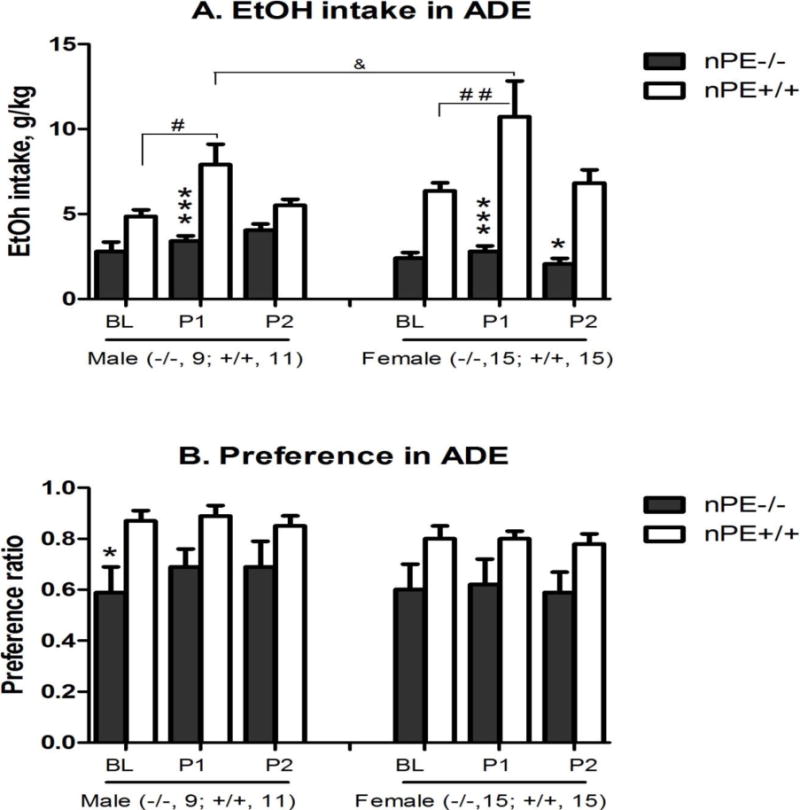
In the baseline session (BL) after 3 weeks of intermittent access (IA) drinking, alcohol and water intake values (EtOH intake, g/kg) were recorded after 4, 8 and 24 hours of alcohol access. After 7 days of abstinence, alcohol bottles were presented again and the alcohol and water intake were recorded for the 1st post-abstinence session (P1). The 2nd post-abstinence session (P2) was 1 day later. Data in this figure is at the 4-hour time point. Genotype difference: *p<0.05 or ***p<0.005 vs. nPE+/+ in the same session; Session difference, #p<0.05 or ##p<0.01 vs. the same genotype at the baseline session BL; and Sex difference: &p<0.05 vs. the same genotype in the same session by 3-way ANOVA with Newman-Keuls post-hoc tests (n=9–15 in nPE−/−; n=11–15 in nPE+/+). (A) Alcohol intake: 3-way ANOVA revealed significant effects of genotype [F (1, 138) = 58, p<0.000001], session [F (2, 138) = 8.8, p<0.005], interaction between genotype and sex [F (1, 138) = 6.6, p<0.05] and interaction between genotype and session [F (2, 138) = 3.9, p<0.05]. Post hoc analysis showed that: (1) In comparison with baseline session BL, nPE+/+ males and females had more intake in session P1 [p<0.05 and p<0.01, respectively]; and (2) between genotypes, nPE−/− males and females had less intake in session P1 than nPE+/+ ones [p<0.001 for both]; nPE−/− females had less intake in session P2 than nPE+/+ ones [p<0.05]. Though overall sex difference was not significant, nPE+/+ females had more intake in session P1 than nPE+/+ males [p<0.05]. (B) Alcohol preference: 3-way ANOVA revealed a significant effect of genotype [F (1, 138) = 19.9, p<0.00005]. Post hoc analysis showed that in session BL, nPE−/− males had less alcohol preference than nPE+/+ [p<0.05].
IV. Effects of genotype and sex on basal corticosterone levels and analgesic threshold, but not locomotor activity in nPE mice
As shown in Table 5A, there were higher basal corticosterone levels in the female nPE+/+ than the male nPE+/+ [p=0.05]. Between the genotypes, the nPE−/− had significantly higher basal corticosterone levels than the nPE+/+ in each sex [p<0.005]. In the nPE+/+, there was a longer basal hot plate response in females than males [p<0.01], and the nPE−/− had significantly shorter basal hot plate responses than the nPE+/+ in each sex [p<0.05] (Table 5B). There were no sex or genotype differences in locomotor activity (Table 5C).
Table 5. Genotype and sex differences in basal plasma corticosterone levels (A), thermal analgesic thresholds (B), and spontaneous locomotor activity (C) in male and female alcohol naive nPE mice.
(A) Blood was collected 3 hours after lights off and plasma corticosterone levels were assayed by radioimmunoassays; (B) The thermal analgesic threshold test was carried out on a hot plate at 52° C 3 hours after lights off and the time to response (rear paw flutter, rear paw licking or jumping) was measured; and (C) 3 hours after lights off, mice were placed into the appropriate chamber of conditioned place preference apparatus for 30 min, and locomotor activity was assessed as the number of “crossovers”. Genotype difference: *p<0.05 or ***p<0.005 vs. nPE+/+ mice in the same sex; and Sex difference: &p=0.05 or &p<0.01 v. nPE+/+ mice in the same genotype by 2-way ANOVA with Newman-Keuls post-hoc tests.
| A. Plasma corticosterone: Significant effects of genotype [F (1, 13) = 57.6, p<0.000005] and interaction between genotype and sex [F (2, 13) = 5.51, p<0.05]. Post hoc analysis showed that between genotypes, nPE−/− of both sex had higher plasma corticosterone levels than nPE+/+ [p<0.005 for both]. Though overall sex difference was not significant, nPE+/+ females nPE+/+ females had higher plasma corticosterone levels than nPE+/+ males [p=0.05]. | ||||
|---|---|---|---|---|
| 5A | males | females | ||
| Genotype | nPE+/+ (n=4) | nPE−/− (n=5) | nPE+/+ (n=5) | nPE−/− (n=4) |
| Corticosterone, ng/ml | 41 ± 5 | 187 ± 10*** | 84 ± 25 & | 162 ± 13*** |
| B. Hot plate responses: Significant effects of genotype [F (1, 29) = 18.8, p<0.0005] and sex [F (2, 29) = 17.5, p<0.0005]. Post hoc analysis showed that: (1) between genotypes, nPE−/− of both sex had shorter responses than nPE+/+ [p<0.05 for both]; and (2) between sexes, nPE+/+ females had longer hot plate responses than nPE+/+ males [p<0.01]. | ||||
|---|---|---|---|---|
| 5B | males | females | ||
| Genotype | nPE+/+ (n=9) | nPE−/− (n=8) | nPE+/+ (n=9) | nPE−/− (n=7) |
| second | 27 ± 4.1 | 14 ± 1.0* | 38 ± 2.3 && | ± 2.0* |
| C. Locomotor activity | ||||
|---|---|---|---|---|
| 5C | males | females | ||
| Genotype | nPE+/+ (n=8) | nPE−/− (n=8) | nPE+/+ (n=5) | nPE−/− (n=5) |
| Crossover (30 min) | 273 ± 45 | 255 ± 27 | 280 ± 62 | 231 ± 59 |
DISCUSSION
The main objective in this study was to investigate the potential role of hypothalamic-specific POMC deficiency in alcohol drinking behaviors. Activation of POMC neurons affects food intake (which is increased and decreased by endorphin and melanocortins, respectively) especially at the onset of the dark cycle [Mercer et al. 2013], so we purposely monitored the drinking activity at the beginning of the dark cycle using the DID model. We found that the nPE−/− mice of both sexes displayed lower alcohol intake than the wild-type nPE+/+ mice during the 4-hour DID “binge” drinking (Figure 1), suggesting a contribution of basal hypothalamic POMC expression to the genetically determined tendency of nPE−/− mice towards reduced voluntary alcohol consumption. Consistently, the nPE−/− mice displayed less alcohol preference at 7.5% and 15% (but not 30%) alcohol concentrations than the nPE+/+ mice in both sexes (Table 2). When the blood alcohol concentration was measured after DID, the nPE−/− males had significantly lower blood alcohol levels than the nPE+/+ males (Table S1), which correlated with their reduced alcohol consumption levels, suggesting that the differences in alcohol intake between two genotypes resulted in altered alcohol levels in the blood. This result is consistent with earlier studies showing that there are lower basal POMC mRNA levels in the hypothalamus of DBA/2 mice or sNP and ANA rats, which have low consumption and preference, than those in the hypothalamus of C57BL/6 mice or sP and AA rats, which have high consumption and preference [Jamensky & Gianoulakis 1999; Marinelli et al. 2000; Zhou et al. 2013].
An issue with standard methods of measuring alcohol intake in terms of grams consumed/kilograms of body weight emerges when comparing groups with different body weights (e.g., genotype or sex). The nPE−/− mice had greater body weights (~40%) than the nPE+/+ mice, resulting in an exaggerated reduction in alcohol intake (g/kg). Similarly, males in general have greater body weight than females, with relatively less intake (g/kg). Of importance, the nPE−/− mice showed less alcohol preference than the nPE+/+ ones in both the DID and IA models, which did not take into account the body weight differences between the genotypes.
It is unlikely that the genotype differences between the nPE−/− and nPE+/+ mice on alcohol intake were secondary to a general suppression of consumption behaviors. In fact, the nPE−/− mice displayed higher food consumption, replicating results from a previous report [Bumaschny et al. 2012]. In the DID paradigm, the nPE deletion did not alter sucrose preference and only slightly reduced sucrose consumption without reaching statistical significance (Table S2). When both nPE−/− and nPE+/+ females were exposed to 15% alcohol in 0.1% saccharin, nPE−/− had less alcohol intake of and preference for the palatable alcohol/saccharin solution than the wild-type, indicating that the nPE deletion may not alter “alcohol-tasting” (Table S3). Further, the reduction of alcohol intake found in the nPE−/− mice in the 2-bottle, free choice IA model was coupled with a compensatory increase in water intake, clearly indicating the absence of nonspecific sedative behaviors, with normal locomotor activity (Table 5C).
Consistent with a previous report using a 2-bottle choice paradigm [Grisel et al. 1999], the present study did not find any genotype differences in alcohol intake in either male or female B-END knockout mice using the DID paradigm (Table S4). Because more (~10–20 fold) beta-endorphin is synthesized in the pituitary than in the hypothalamus [Zhou et al. 2013], global beta-endorphin deletion may affect pituitary functions more dramatically than hypothalamic ones. It is very possible that pituitary POMC may not be involved in certain alcohol-related behaviors. In our unpublished study, using Tpit knockout mice provided by Dr. Drouin (resulting in pituitary-specific POMC knock-out with intact hypothalamic POMC), we did not observe any differences in DID (15%) drinking between Tpit−/− and wild-type in either males or females (n=7–8 for each genotype). Though our data were preliminary, they suggest that the pituitary POMC is not involved in regulation of DID drinking behavior.
The gradual increase in alcohol intake which results from long access to voluntary alcohol drinking is analogous to the loss of control over drug-taking found in human alcoholics [Koob 2008]. In the nPE+/+ mice in the IA paradigm (intermittent 24-hour cycles of alcohol access) [Hwa et al. 2011], alcohol intake rapidly escalated over the course of weeks to high levels of consumption, with a parallel progressive elevation of alcohol preference (Figure 2). Therefore, we further tested the effect of hypothalamic POMC deletion on alcohol drinking during chronic escalation using an IA model, and found the nPE−/− mice showed less alcohol intake of and preference for 15% alcohol in both sexes than the wild-type (Figure 2). Our results suggest that the activation of hypothalamic POMC is involved in modulation of alcohol escalation drinking behavior.
The phenomenon of a transient increase in alcohol consumption and/or preference after a period of imposed abstinence has been termed the alcohol deprivation effect (ADE) and has been observed in several species, including humans [Burish et al. 1981], rats and mice [Holter & Spanagel 1999; Sajja & Rahman 2013]. Since the ADE in a long-term alcohol drinking model persists through very long abstinence phases (up to one year) and is hardly modified by external stimuli [Vengeliene et al. 2014], it can be regarded as an animal model of relapse behavior and craving with excellent predictive validity. In the present study, the nPE+/+ mice of both sexes displayed the ADE with increased alcohol intake after 1 week of abstinence (Figure 3A). In contrast, the nPE−/− mice had a blunted ADE in both sexes, suggesting the involvement of hypothalamic POMC in relapse-like drinking behavior. Consistently, in several different rodent studies [Holter & Spanagel 1999; Heyser et al. 2003], the ADE can be suppressed by naltrexone, a compound that is also effective in reducing relapse behavior in human alcoholics [O’Malley et al. 2002]. Because high baselines of the preference ratio in the nPE+/+ mice were developed after 3 weeks of IA, it is possible that the ADE could not increase the preference ratio further when tested during post-abstinence sessions (Figure 3B).
In rats, chronic naltrexone (0.06 mg/kg) for 5 days was able to suppress the ADE only on the first post-abstinence day. Twenty-four hours after the last naltrexone injection, however, significant increases in alcohol consumption were observed [Heyser et al. 2003]. Similar results were reported after administering naloxone (1 mg/kg/h) in rats: reduced alcohol intake on post-abstinence day 1, with an increase on day 2 (1 day after the last naloxone) [Holter & Spanagel 1999]. These published results show a rebound increase in alcohol consumption in the post-abstinence period after MOP-r blockade was discontinued. One possible explanation is that the MOP-r antagonists may enhance hypothalamic POMC function, as we recently reported increased POMC mRNA levels in the rat hypothalamus 24 hours after naloxone at 1 mg/kg [Zhou & Kreek 2015]. Based on these reasons, we purposely examined drinking activity on the 2nd post-abstinence day in the present study, and found that the nPE−/− mice did not show a rebound effect on the 2nd ADE test (Figure 3). Therefore, the observed reduction of ADE behaviours by hypothalamic POMC deletion is of interest, as it would constitute a clear piece of experimental evidence that hypothalamic POMC is responsible for relapse-like alcohol drinking.
Using the IA model, our current study confirmed that acute naltrexone administration dose-dependently (1–2 mg/kg) reduced alcohol intake and preference in the wild-type mice of both sexes [Hwa et al. 2011] (Table 4). These findings raise the possibility that alcohol may cause the release of endogenous beta-endorphin [Marinelli et al. 2003], which plays a functional role in alcohol drinking. Indeed, our study found that the nPE−/− mice of both sexes had lower alcohol preference for 15% alcohol in both the DID (Table 2) and IA models (Figure 2), suggesting a reduced rewarding effect of alcohol when central POMC and beta-endorphin are reduced. In parallel, the nPE−/− had significantly less saccharin (natural non-caloric reinforcer) intake and preference (Table 3) than the wild-type, suggesting that hypothalamic POMC deletion may produce an anhedonic state in the nPE−/− mice.
In line with the result on drinking behavior, the observation of decreased analgesic thresholds and increased corticosterone levels in the nPE−/− mice (Table 5A, B) further indicates less beta-endorphin/MOP-r tone resulting from neuronal POMC deletion, as it is known that the beta-endorphin/MOP-r system plays modulatory roles in pain and hypothalamic-pituitary-adrenal activity in both humans and rodents [Rubinstein et al. 1996; O’Malley et al. 2002]. Because enkephalins can also bind and activate MOP-r, we purposely investigated whether naltrexone could affect alcohol drinking in the nPE−/− mice lacking beta-endorphin, the main peptide ligand of MOP-r. We observed no effect of acute naltrexone treatment at 2 mg/kg on alcohol drinking in the IA paradigm in either nPE−/− males or females (Table 4), though the same naltrexone treatment significantly reduced alcohol drinking in the nPE+/+. Therefore, it is unlikely that enkephalins contribute much to the MOP-r mediated modulation of alcohol drinking. Our results agree with one earlier study showing that voluntary alcohol intake was not affected in enkephalin knockout mice [Racz et al. 2008]. For comparison with naltrexone, we tested the selective kappa-opioid receptor antagonist nor-BNI in both genotypes, and found that both nPE+/+ and nPE−/− males displayed similar reductions in intake after the blockade of kappa-opioid receptors (Table S9), indicating no disruption of kappa function after the neuronal POMC deletion. This also suggests that the lack of significant effect of naltrexone in the nPE−/− males was not due to the lowered basal alcohol intake in the nPE−/−, as nor-BNI still significantly reduced alcohol intake in the nPE−/− males. Together, these findings suggest that POMC/beta-endorphin acting on MOP-r plays a critical role in alcohol drinking.
Consistent with previous studies in mice [Racz et al. 2008; Hall et al. 2001; Hwa et al. 2011] and in rats (see a recent review by Becker & Koob in 2016), we confirmed sex differences in alcohol drinking with higher alcohol intake in females and further observed that the genotype differences in alcohol consumption between the nPE+/+ and nPE−/− mice was much greater in females than in males in the DID, IA and ADE models. The POMC deficiency affected female mice more strongly than males, suggesting that POMC may influence alcohol consumption in a sex-specific manner. Our findings are in line with previous studies showing decreased voluntary alcohol intake in beta-endorphin and MOP-r knockout mice with more notable differences in females [Racz et al. 2008; Hall et al. 2001]. Sex differences have also been observed for alcoholism in a human genetic study: the Pomc two-marker haplotype was associated with alcoholism in women, but not in men [Racz et al. 2008]. Our results further contribute to the idea that opioid regulation of alcohol dependence differs between sexes [Becker & Koob in 2016]. Additionally, there may be many physiological differences between sexes, such as metabolism [Burke et al. 2016], pain regulation and endocrine systems (presented in the current study), that could underlie sex differences in alcohol-related behaviors.
The role of endogenous melanocortins (encoded by the Pomc gene) in regulation of alcohol-related behavior is not clear. Pharmacological studies have found that activation of melanocortin 4 receptors (MC4) by specific agonists significantly reduced alcohol “binge”-like drinking in a DID model, with reduced appetitive and consumption behaviors [Sprow et al. 2016]. A very recent study, however, found that MC4 receptor blockade in the ventral tegmental area reduced alcohol self-administration in rats [Shelkar et al. 2015], indicating that endogenous melanocortins mediated alcohol reinforcing effect via the MC4 activation, which is consistent with reports on other drugs of abuse [Hsu et al. 2005]. The present study also suggests that melanocortins and MC4 receptors may be involved in reduced alcohol drinking in hypothalamic POMC/melanocortin deficient mice. Further investigation of this is needed [Olney et al. 2014].
Summary
Use of a transgenic mouse line with tissue-specific deletion of POMC in hypothalamic neurons uniquely allowed us to explore the potential roles of hypothalamic POMC neurons in the context of multiple behavioral models of alcohol addiction (including DID, IA and ADE). Our studies clearly indicate an essential role of the POMC system in the regulation of alcohol-related behaviors in both male and female mice. This corroborates earlier studies showing that voluntary alcohol intake is decreased in MOP-r knockout mice lacking the receptor for the endogenous ligand beta-endorphin produced in POMC neurons, with more pronounced effects in females.
Supplementary Material
Acknowledgments
NIAAA grant AA021970 (YZ), NIH DK068400 (MJL & MR) and the Adelson Medical Research Foundation (MJK). Special thanks to Dr. R. Schaefer and Konrad Ben for providing their comments and corrections on the manuscript; to Dr. J. Correa Da Rosa for providing his comments on statistical analyses. Select data were presented in the 38th Annual Research Society on Alcoholism Scientific Meeting in 2015.
Footnotes
Financial Discloses: No conflict of interest.
References
- Altschuler HL, Phillips PE, Feinhandler DA. Alteration of ethanol self-administration by naltrexone. Life Sci. 1980;26:679–688. doi: 10.1016/0024-3205(80)90257-x. [DOI] [PubMed] [Google Scholar]
- Amalric M, Cline EJ, Martinez JL, Jr, Bloom FE, Koob GF. Rewarding properties of beta-endorphin as measured by conditioned place preference. Psychopharmacology (Berl) 1987;91:14–19. doi: 10.1007/BF00690919. [DOI] [PubMed] [Google Scholar]
- Angelogianni P, Gianoulakis C. Chronic ethanol increases pro-opiomelanocortin gene expression in the rat hypothalamus. Neuroendocrinology. 1993;57:106–114. doi: 10.1159/000126348. [DOI] [PubMed] [Google Scholar]
- Becker A, Grecksch G, Kraus J, Loh HH, Schroeder H, Hollt V. Rewarding effects of ethanol and cocaine in mu opioid receptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:296–302. doi: 10.1007/s00210-002-0533-2. [DOI] [PubMed] [Google Scholar]
- Becker JB, Koob GF. Sex differences in animal models: focus on addiction. Pharmacol Rev. 2016;68:242–263. doi: 10.1124/pr.115.011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumaschny VF, Yamashita M, Casas-Cordero R, Otero-Corchón V, de Souza FS, Rubinstein M, Low MJ. Obesity-programmed mice are rescued by early genetic intervention. J Clin Invest. 2012;122:4203–4212. doi: 10.1172/JCI62543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burish TG, Maisto SA, Cooper AM, Sobell MB. Effects of voluntary short-term abstinence from alcohol on subsequent drinking patterns of college students. J Stud Alcohol. 1981;42:1013–1020. doi: 10.15288/jsa.1981.42.1013. [DOI] [PubMed] [Google Scholar]
- Burke LK, Doslikova B, D’Agostino G, Greenwald-Yarnell M, Georgescu T, Chianese R, Martinez de Morentin PB, Ogunnowo-Bada E, Cansell C, Valencia-Torres L, Garfield AS, Apergis-Schoute J, Lam DD, Speakman JR, Rubinstein M, Low MJ, Rochford JJ, Myers MG, Evans ML, Heisler LK. Sex difference in physical activity, energy expenditure and obesity driven by a subpopulation of hypothalamic POMC neurons. Mol Metab. 2016;5:245–252. doi: 10.1016/j.molmet.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- de Souza FS, Santangelo AM, Bumaschny V, Avale ME, Smart JL, Low MJ, Rubinstein M. Identification of neuronal enhancers of the proopiomelanocortin gene by transgenic mouse analysis and phylogenetic footprinting. Mol Cell Biol. 2005;25:3076–3086. doi: 10.1128/MCB.25.8.3076-3086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahame NJ, Mosemiller AK, Low MJ, Froehlich JC. Naltrexone and alcohol drinking in mice lacking beta-endorphin by site-directed mutagenesis. Pharmacol Biochem Behav. 2000;67:759–766. doi: 10.1016/s0091-3057(00)00411-1. [DOI] [PubMed] [Google Scholar]
- Grisel JE, Mogil JS, Grahame NJ, Rubinstein M, Belknap JK, Crabbe JC, Low MJ. Ethanol oral self-administration is increased in mutant mice with decreased beta-endorphin expression. Brain Res. 1999;835:62–67. doi: 10.1016/s0006-8993(99)01384-0. [DOI] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- Heyser C, Roberts A, Schulteis G, Koob GF. Central administration of an opiate antagonist decreases oral ethanol self-administration in rats. Alcohol Clin Exp Res. 1999;23:468–1476. [PubMed] [Google Scholar]
- Heyser CJ, Moc K, Koob GF. Effects of naltrexone alone and in combination with acamprosate on the alcohol deprivation effect in rats. Neuropsychopharmacology. 2003;28:1463–1471. doi: 10.1038/sj.npp.1300175. [DOI] [PubMed] [Google Scholar]
- Holter SM, Spanagel R. Effects of opiate antagonist treatment on the alcohol deprivation effect in long-term ethanol-experienced rats. Psychopharmacology (Berl) 1999;145:360–369. doi: 10.1007/s002130051069. [DOI] [PubMed] [Google Scholar]
- Hsu R, Taylor JR, Newton SS, Alvaro JD, Haile C, Han G, Hruby VJ, Nestler EJ, Duman RS. Blockade of melanocortin transmission inhibits cocaine reward. Eur J Neurosci. 2005;21:2233–2242. doi: 10.1111/j.1460-9568.2005.04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa LS, Chu A, Levinson SA, Kayyali TM, DeBold JF, Miczek KA. Persistent escalation of alcohol drinking in C57BL/6J mice with intermittent access to 20% alcohol. Alcohol Clin Exp Res. 2011;35:1938–1947. doi: 10.1111/j.1530-0277.2011.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamensky NT, Gianoulakis C. Comparison of the proopiomelanocortin and proenkephalin opioid peptide systems in brain regions of the alcohol-preferring C57BL/6 and alcohol-avoiding DBA/2 mice. Alcohol. 1999;18:177–187. doi: 10.1016/s0741-8329(99)00002-6. [DOI] [PubMed] [Google Scholar]
- Koch M, Varela L, Kim JG, Kim JD, Hernández-Nuño F, Simonds SE, Castorena CM, Vianna CR, Elmquist JK, Morozov YM, Rakic P, Bechmann I, Cowley MA, Szigeti-Buck K, Dietrich MO, Gao XB, Diano S, Horvath TL. Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature. 2015;519:45–50. doi: 10.1038/nature14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DD, de Souza FSJ, Nasif S, Yamashita M, López-Leal R, Otero-Corchon V, Meece K, Sampath H, Mercer AJ, Wardlaw SL, Rubinstein M, Low MJ. Partially redundant enhancers cooperatively maintain Mammalian Pomc expression above a critical functional threshold. PLoS Genetics. 2015;11(2):e1004935. doi: 10.1371/journal.pgen.1004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli PW, Kiianmaa K, Gianoulakis C. Opioid propeptide mRNA content and receptor density in the brains of AA and ANA rats. Life Sci. 2000;66:1915–1927. doi: 10.1016/s0024-3205(00)00517-8. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Quirion R, Gianoulakis C. A microdialysis profile of beta-endorphin and catecholamines in the rat nucleus accumbens following alcohol administration. Psychopharmacology (Berl) 2003;169:60–67. doi: 10.1007/s00213-003-1490-2. [DOI] [PubMed] [Google Scholar]
- Mercer AJ, Hentges ST, Meshul CK, Low MJ. Unraveling the central proopiomelanocortin neural circuits. Front Neurosci. 2013;7 doi: 10.3389/fnins.2013.00019. Article 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro M, Cubero I, Thiele TE. Decreased immunoreactivity of the polypeptide precursor pro-opiomelanocortin (POMC) and the prohormone convertase pc1/3 after chronic ethanol exposure in Sprague-Dawley rats. Alcohol Clin Exp Res. 2013;37:399–406. doi: 10.1111/j.1530-0277.2012.01951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JJ, Navarro M, Thiele TE. Targeting central melanocortin receptors: a promising novel approach for treating alcohol abuse disorders. Front Neurosci. 2014;8:128. doi: 10.3389/fnins.2014.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley SS, Jaffe A, Chang G, Schottenfeld RS, Meyer RE, Rounsaville BJ. Naltrexone and coping skills therapy for alcohol dependence: a controlled study. Arch Gen Psychiatry. 1992;49:881–887. doi: 10.1001/archpsyc.1992.01820110045007. [DOI] [PubMed] [Google Scholar]
- O’Malley S, Krishnan-Sarin S, Farren C, Sinha R, Kreek MJ. Naltrexone decreases craving and alcohol self-administration in alcohol-dependent subjects and activates the hypothalamic-pituitary-adrenocortical axis. Psychopharmacology. 2002;160:19–29. doi: 10.1007/s002130100919. [DOI] [PubMed] [Google Scholar]
- Racz I, Schurmann B, Karpushova A, Reuter M, Cichon S, Montag C, Furst R, Schutz C, Franke PE, Strohmaier J, Wienker TF, Terenius L, Osby U, Gunnar A, Maier W, Bilkei-Gorzo A, Nothen M, Zimmer A. The opioid peptides enkephalin and beta-endorphin in alcohol dependence. Biol Psychiatry. 2008;64:989–997. doi: 10.1016/j.biopsych.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen DD, Boldt BM, Wilkinson CW, Mitton DR. Chronic daily ethanol and withdrawal: 3. Forebrain pro-opiomelanocortin gene expression and implications for dependence, relapse, and deprivation effect. Alcohol Clin Exp Res. 2002;26:535–546. [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of alcohol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Roberts A, McDonald J, Heyser C, Kieffer B, Matthes H, Koob GF, Gold L. μ-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–1008. [PubMed] [Google Scholar]
- Roth-Deri I, Green-Sadan T, Yadid G. Beta-endorphin and drug-induced reward and reinforcement. Prog Neurobiol. 2008;86:1–21. doi: 10.1016/j.pneurobio.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Mogil JS, Japon M, Chan EC, Allen RG, Low MJ. Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc Natl Acad Sci USA. 1996;93:3995–4000. doi: 10.1073/pnas.93.9.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajja RK, Rahman S. Nicotinic receptor partial agonists modulate alcohol deprivation effect in C57BL/6J mice. Pharmacol Biochem Behav. 2013;110:161–167. doi: 10.1016/j.pbb.2013.07.009. [DOI] [PubMed] [Google Scholar]
- Shelkar GP, Kale AD, Singh U, Singru PS, Subhedar NK, Kokare DM. Alpha-melanocyte stimulating hormone modulates ethanol self-administration in posterior ventral tegmental area through melanocortin-4 receptors. Addict Biol. 2015;20:302–315. doi: 10.1111/adb.12126. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Bals-Kubik R, Shippenberg TS. Beta-endorphin-induced locomotor stimulation and reinforcement are associated with an increase in dopamine release in the nucleus accumbens. Psychopharmacology (Berl) 1991;104:51–56. doi: 10.1007/BF02244553. [DOI] [PubMed] [Google Scholar]
- Sprow GM, Rinker JA, Lowery-Gointa EG, Sparrow AM, Navarro M, Thiele TE. Lateral hypothalamic melanocortin receptor signaling modulates binge-like ethanol drinking in C57BL/6J mice. Addict Biol. 2016;21:835–846. doi: 10.1111/adb.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengeliene V, Bilbao A, Spanagel R. The alcohol deprivation effect model for studying relapse behavior: a comparison between rats and mice. Alcohol. 2014;48:313–320. doi: 10.1016/j.alcohol.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Volpicelli JR, Davis MA, Olgin JE. Naltrexone blocks the post-shock increase of ethanol consumption. Life Sci. 1986;38:841–847. doi: 10.1016/0024-3205(86)90601-6. [DOI] [PubMed] [Google Scholar]
- Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Colombo G, Niikura K, Carai MAM, Femenía T, García-Gutiérrez MS, Manzanares J, Ho A, Gessa GL, Kreek MJ. Voluntary alcohol drinking enhances proopiomelanocortin (POMC) gene expression in nucleus accumbens shell and hypothalamus of Sardinian alcohol-preferring rats. Alcohol Clin Exp Res. 2013;37:E131–140. doi: 10.1111/j.1530-0277.2012.01867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Kreek MJ. Persistent increases in rat hypothalamic POMC gene expression following chronic withdrawal from chronic “binge” pattern escalating-dose, but not steady-dose, cocaine. Neuroscience. 2015;289:63–70. doi: 10.1016/j.neuroscience.2014.12.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Huang T, Lee F, Kreek MJ. Involvement of Endocannabinoids in Alcohol “Binge” Drinking: Studies of Mice with Human Fatty Acid Amide Hydrolase Genetic Variation and After CB1 Receptor Antagonists. Alcohol Clin Exp Res. 2016;40:467–473. doi: 10.1111/acer.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.