

Abstract
Background and Purpose
Glucagon‐like peptide‐1 (GLP‐1) is an important target for diabetes therapy based on its key role in maintaining glucose and lipid homeostasis. This study was designed to investigate antidiabetic and hepatoprotective effects of a novel oleanolic acid derivative DKS26 in diabetic mice and elucidate its underlying GLP‐1 related antidiabetic mechanisms in vitro and in vivo.
Experimental Approach
The therapeutic effects of DKS26 were investigated in streptozotocin (STZ)‐induced and db/db diabetic mouse models. Levels of plasma glucose, glycosylated serum protein (GSP), lipid profiles, insulin, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), oral glucose tolerance (OGT), pancreatic islets and hepatic histopathological morphology, liver lipid levels and expression of pro‐inflammatory cytokines were assessed. Intestinal NCI‐H716 cells and diabetic models were used to further validate its potential GLP‐1‐related antidiabetic mechanisms.
Key Results
DKS26 treatment (100 mg·kg−1·day−1 ) decreased plasma levels of glucose, GSP, ALT and AST; ameliorated OGT and plasma lipid profiles; augmented plasma insulin levels; alleviated islets and hepatic pathological morphology; and reduced liver lipid accumulation, inflammation and necrosis in vivo. Furthermore, DKS26 enhanced GLP‐1 release and expression, accompanied by elevated levels of cAMP and phosphorylated PKA in vitro and in vivo.
Conclusion and Implications
DKS26 exerted hypoglycaemic, hypolipidaemic and islets protective effects, which were associated with an enhanced release and expression of GLP‐1 mediated by the activation of the cAMP/PKA signalling pathway, and alleviated hepatic damage by reducing liver lipid levels and inflammation. These findings firmly identified DKS26 as a new viable therapeutic option for diabetes control.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CMC‐Na
sodium carboxymethyl cellulose
- CPT1
carnitine palmitoyltransferase‐1
- DPP4
dipeptidylpeptidase 4
- FAS
fat acid synthase
- FBG
fasting blood glucose
- gcg
proglucagon
- GLP‐1
glucagon‐like peptide‐1
- GSP
glycosylated serum protein
- HDL‐C
high‐density lipoprotein cholesterol
- HE
haematoxylin‐eosin
- LDL‐C
low‐density lipoprotein cholesterol
- OA
oleanolic acid
- OGT
oral glucose tolerance
- PBG
postprandial blood glucose
- PC3
prohormone converting enzyme 3
- SREPB‐1c
sterol regulatory element binding protein‐1
- STZ
streptozotocin
- TC
total cholesterol
- TG
triglyceride
Introduction
Diabetes mellitus has been a health threat for decades for a population that has reached 382 million worldwide and will rise to 592 million by 2035 (Guariguata et al., 2014). It is a chronic metabolic disorder characterized by hyperglycaemia due to insulin secretion deficiency or/and insulin resistance (Schwartz et al., 2016). Moreover, a series of complications, such as abnormal lipid levels , hepatic damage, cardiovascular disease, ocular lesion and renal failure, subsequently appear in diabetic patients as a result of prolonged hyperglycaemia (Ghosh et al., 2015). Because of the adverse effects or/and poor long‐term efficacy of the current antidiabetic drugs (McCall, 2012; Stein et al., 2013), it is imperative to develop safer and more effective medicines for diabetes control.
Glucagon‐like peptide‐1 (GLP‐1) is an incretin hormone mainly produced in intestinal L cells in response to the activation of GPCRs [such as GPR119, GPR120, GPR40 and TGR5 (also known as GPBA receptor)] and the formation of intracellular cAMP by its secretagogues (Baggio and Drucker, 2007). GLP‐1 regulates glucolipid metabolism mainly through increased insulin release and reduced glucagon secretion; it also promotes islets beta cell proliferation, inhibits islet beta cell apoptosis and ameliorates the effect of insulin and, thereby, has become an important therapeutic target for diabetes (Campbell and Drucker, 2013). Since GLP‐1 is easily degraded by dipeptidylpeptidase 4 (DPP4), the GLP‐1 related therapeutics are composed of GLP‐1 receptor agonists and DPP4 inhibitors (Drucker and Nauck, 2006). However, GLP‐1 receptor agonists are not available for oral administration and may induce pancreatitis, and DPP4 inhibitors can cause severe polyarthritis in the course of medication (Crickx et al., 2014; Azoulay, 2015). Thus, GLP‐1 secretagogues are good potential candidates for the development of antidiabetic agents (Bruzzone et al., 2015).
Oleanolic acid (OA), a natural pentacyclic triterpenoid widely exists in many kinds of plants, has various biological activities chiefly involving hypoglycaemic, hypolipidaemic, hepatoprotective and anti‐inflammatory effects (Castellano et al., 2013). Importantly, the anti‐hyperglycaemic effect of OA is associated with the increased release of GLP‐1, mediated by activation of intestinal TGR5 (Sato et al., 2007; Bala et al., 2014). Nevertheless, relatively weak pharmacological effects and poor oral bioavailability of OA limit its application as a diabetes therapy (Ramirez‐Espinosa et al., 2014; Jiang et al., 2016). Our preliminary study found that DKS26, or 12, 13‐dihydrooleanolic acid methyl ester (Figure 1), a novel compound derived from OA, significantly augmented glucose consumption in human hepatic HepG2 cells, similar to metformin, indicating that DKS26 might exert glucose‐lowering effects in vivo (Supporting Information Figure S1). Furthermore, DKS26 enhanced GLP‐1 release and expression in human enteroendocrine NCI‐H716 L cells. However, whether DKS26 is a potential GLP‐1‐related therapeutic option for diabetes has not yet been evaluated.
Figure 1.
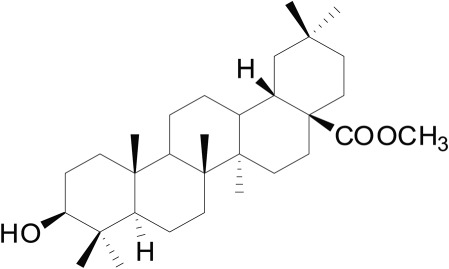
The structure of DKS26.
Accordingly, this study was undertaken to investigate the effects of DKS26 on blood glucose, lipid profiles and pancreatic islet morphology in two animal models, namely, streptozotocin (STZ)‐induced diabetic mice and genetically obese db/db diabetic mice. In addition, the effects of DKS26 on liver function and hepatic lipid levels and pro‐inflammatory cytokine expression were assessed. To further elucidate the underlying anti‐diabetic mechanism of DKS26, GLP‐1 release and expression mediated through the cAMP/PKA signalling pathway were explored in vitro and in vivo.
Methods
Animals
All the animal care and experimental studies were conducted in accordance with the guidelines of the Animal Ethical Committee of Zhejiang University and the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. All the animal studies complied with the principle for replacement, refinement or reduction (the 3Rs). Male ICR mice (20 ± 2 g) were purchased from Zhejiang Provincial Experimental Animal Centre (Hangzhou, China), and male C57BL/6 BKS.Cg‐+Leprdb/+Leprdb/J (db/db) mice (9–10 weeks old) were obtained from SLAC Laboratory Animal Co., Ltd (Shanghai, China). All the mice were fed with standard food and water under constant environment with 20 ± 2°C and 12 h light/dark cycle and adaptively raised for a week before experiments. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Cell culture
The human enteroendocrine L cell line NCI‐H716 (Cell Bank of the China Science Academy, Shanghai, China) was cultured in low‐glucose RPMI‐1640 (Gibco, Grand Island, NY, USA) including 10% FBS (Gibco) at 37°C and in the presence of 5% CO2. Moreover, the cells were maintained in matrigel (BD Biosciences, Bedford, MA, USA) with high‐glucose DMEM (Gibco) and 10% FBS for 2 days to become mature endocrine cells; 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin were added to the culture media at a ratio of 1:1000.
Blood glucose and oral glucose tolerance tests
The STZ‐induced diabetic mouse model was induced by a single i.p. injection of STZ (Sigma‐Aldrich, St. Louis, MO, USA) at a dose of 130 mg·kg−1 in ICR mice after overnight fasting. Then the mice (5 h fasted) with blood glucose levels ≥11.1 mmol·L−1 on the seventh day of STZ injection were considered as diabetic mice. These diabetic mice were randomly divided into four groups according to fasting blood glucose (FBG) levels, which were STZ model control (n = 7), metformin positive control (n = 8), OA control (n = 8) and DKS26 test group (n = 8). All drugs were administered intragastrically (i.g.) at a dose of 100 mg·kg−1·day−1. In addition, the normal (n = 5) and model control mice were administered an equal volume of 0.5% sodium carboxymethyl cellulose (CMC‐Na; vehicle). Likewise, db/db genetic obese diabetic mice with blood glucose levels ≥11.1 mmol·L−1 were randomly divided into four groups in accordance with the above criterion, namely, db/db model control (n = 7, 0.5% CMC‐Na), metformin positive control (n = 7, 100 mg·kg−1·day−1, i.g.), OA control (n = 8, 100 mg·kg−1·day−1, i.g.) and DKS26 test group (n = 8, 100 mg·kg−1·day−1, i.g.). The numbers of animals used were in line with the requirements on experimental design and analysis in pharmacology (Curtis et al., 2015).
During the 33 days of drug administration, the FBG levels in STZ‐induced diabetic mice were monitored by detecting tail blood with fast glucometers (Bayer HealthCare LLC, Mishawaka, IN, USA). And the blood glucose in db/db diabetic mice was measured in the fasting or feeding state at specific time during the 30 days of treatment. Since DKS26 exerted significantly glucose‐lowing effects at 2 h (better than 3 h) after a single administration in STZ‐induced diabetic mice (see Supporting Information Table S1), we measured blood glucose pretreatment and 2 h after dosing during the daily treatment process. The oral glucose tolerance (OGT) test was performed by glucose gavage (2 g·kg−1) at 30 min after the administration of the drug on the 24th day in STZ‐induced diabetic mice fasted for 16 h or on the 27th day in db/db diabetic mice fasted for 5 h. The blood glucose levels were determined at 0.5, 1 or 2 h after glucose loading, and OGT was expressed by the AUC.
Determination of plasma biochemical indexes
Two hours after the last treatment, blood samples were collected into the anticoagulant tubes containing 15% EDTA‐2K (Sinopharm, Shanghai, China) and 10−5 M DPP4 inhibitor MK‐0431 (Sigma‐Aldrich), and then mice were sacrificed. Blood samples were centrifuged at 4°C, 1006 × g for 5 min to obtain the upper plasma and preserved at −80°C. Next, plasma glycosylated serum protein (GSP), triglyceride (TG), total cholesterol (TC), high‐density lipoprotein cholesterol (HDL‐C), low‐density lipoprotein cholesterol (LDL‐C), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured by using commercially available kits and following the manufacturers’ instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) respectively.
Histopathological analysis of liver and pancreas
Mice were killed after a 5 h fast and 2 h after the last treatment, and their liver and pancreas were removed. The tissue samples were then fixed in 10% neutral formalin, embedded in paraffin and 5‐μm‐thick sections prepared. These histological sections were stained with haematoxylin‐eosin (HE; Beyotime, Haimeng, China), which was carried out as described previously (Liu et al., 2015). The tissue sections were observed and photographed under a microscope (100× and 200×, Leica, Germany), and the region containing islets was analysed by the software Image J (National Institutes of Health, Maryland, USA).
ELISA determination of GLP‐1, insulin and cAMP
The mature enteroendocrine NCI‐H716 cells were cultured in high‐glucose DMEM including 0.2% BSA (Sigma‐Aldrich). Experimental groups were established as solvent (0.1% ethanol) control, OA (10 μM) control and different concentration of DKS26 (0.1, 1 and 10 μM). After being treated for 2 h, the cells were collected and lysed. The cAMP levels in cell lysate were determined by using a cAMP ELISA kit (Sigma‐Aldrich) and rectified with cell protein concentration. In order to investigate the effects of DKS26 on GLP‐1 secretion, mature NCI‐H716 cells were incubated in KRB buffer (Yu et al., 2015) (128.8 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, 5 mM NaHCO3 and 10 mM HEPES, pH 7.4) and 0.2% BSA and treated with 0.1% ethanol, 10 μM OA or 10 μM DKS26 for 2 h. Then, GLP‐1 levels in cellular supernatant supplemented with 50 μg·mL−1 PMSF (Sigma‐Aldrich) were measured by the GLP‐1 (7–36) ELISA assay (Phoenix, Burlingame, CA, USA) and corrected with cell protein concentration.
The plasma GLP‐1 and insulin levels in db/db diabetic mice were detected by GLP‐1 (7–36) and insulin (Uscn life, Wuhan, China) ELISA assays according to kit instructions respectively. In addition, the ileal and colon parts of the small intestine in db/db diabetic mice were harvested 2 h after the last drug treatment and stored at −80°C for subsequent analysis of cAMP content.
Measurement of liver TG and TC
The hepatic tissue samples were homogenized in 150 μL IP buffer, which contained 150 mM NaCl, 25 mM NaF, 50 mM Tris–HCl (pH 7.5), 2 mM EDTA, 2 mM EGTA, 25 mM β‐glycerol phosphate, 5 μg·mL−1 leupeptin, 0.1% supersaturated sodium vanadate, 1 mM PMSF, 0.5% NP40 and 1% Triton X‐100. Then, tissue lysis fluids were centrifuged at 4°C, 19 480 × g for 30 min, and the supernatant lipid was mixed to determine hepatic TG and TC contents in accordance with kit instructions. The levels of liver TG and TC were corrected with tissue protein concentration.
Hepatic Oil Red O staining
Frozen liver sections (10 μm) from all groups of both STZ‐induced and db/db diabetic mice were fixed with 4% paraformaldehyde (Sigma‐Aldrich) at 4°C for 30 min. After being washed twice with PBS, sections were washed with 60% isopropyl alcohol (Sinopharm) once and stained with Oil Red O (Sigma‐Aldrich) for 1 h at room temperature. The Oil Red O was prepared with 5 mg·mL−1 solution filtered and diluted with distilled water at a ratio of 3:2. Then the dyed slices were washed with 60% isopropyl alcohol once and PBS twice. The tissue sections were photographed under a microscope (100× and 200×).
Real‐time fluorescent quantitative PCR analyses
Mature NCI‐H716 cells with 3 × 105 maintained in 2 ml high‐glucose DMEM containing 0.2% BSA and different treatments (0.1% ethanol, 10 μM OA or 10 μM DKS26) for 24 h were subjected to PCR analyses. The mRNA of liver and intestine was also extracted according to manufacturer's instructions above was also analysed. After determining the content and purity of the samples, 2 μg mRNA was considered as the quantitative standard and reversely transcribed to cDNA using a kit and following the manufacturer's instructions (TransGen Biotech, Beijing, China). Following that, PCR was performed using SYBR Premix Ex Taq™ (Takara, Shiga, Japan). The PCR programme was repeated for 40 cycles, including 94°C for 30 s, 50–60°C for 30 s, and 68°C for 30 s after 95°C degeneration for 15 min. The software Mastercycler EP Realplex (Eppendorf, Wesseling‐Berzdorf, Germany) was used to analyse the amplification curves to obtain the Ct values of target genes and reference genes (GAPDH or β‐actin). The primer sequences are listed in Supporting Information Table S2.
Immunofluorescence assay of GLP‐1 expression
The NCI‐H716 cells with 24 h treatment of 0.1% ethanol, 10 μM OA or 10 μM DKS26 were fixed with 4% paraformaldehyde at 4°C for 45 min and then washed with PBS three times. The cells were blocked with 5% BSA containing 0.1% Triton X‐100 for 1 h at room temperature and then incubated with GLP‐1 antibody (1:50; Santa Cruz, CA, USA) overnight at 4°C and subsequently goat fluorescent antibody (1:500, Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature. Next, the cell nucleus was stained with 1 μg·mL−1 DAPI (Roche, Indianapolis, IN, USA) for 2 min (avoiding light). Finally, the cells were treated with an anti‐fluorescence quenching agent and photographed under a fluorescence microscope (100×, Leica).
The intestinal tissues were directly embedded and then sliced into 5‐μm‐thick frozen sections preserved at −20°C. The frozen sections were analysed using the above immunofluorescence protocols after being placed for 30 min at room temperature.
Western blotting
The 20–40 μg protein lysis fluids from cells or tissues were run in the 10% SDS‐PAGE and then transferred to a PVDF membrane (Millipore, Billerica, MA, USA). The bands of target proteins were incubated in the corresponding antibody solution containing β‐actin, p‐PKA, PKA (1:1000; Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C or for 4 h at room temperature. Next, the protein bands were reacted with ECL liquids (Perkin Elmer, Waltham, MA, USA) and imaged after combining with the HRP‐labelled secondary antibody (1:5000, LiankeBio, Hangzhou, China). The relative quantity of protein expressed was analysed by the software Image J; the reference protein was β‐actin.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). In all studies, experiments were done by operators, who were blinded to the sample identity for treatment and detection, and data and statistical analysis were conducted by the investigator. All the experimental data are presented as mean ± SEM, and Student's t‐test was used to compare the statistical differences between two groups by use of the SPSS 18.0 programme (SPSS, Chicago, IL, USA). P < 0.05 was taken to indicate statistical significance.
Materials
OA and metformin were purchased from Zhengzhou lion Biological Technology Co., Ltd (Zhengzhou, China). The OA derivative DKS26 was provided by Prof. Lei Tang (Guizhou Medical University, Guizhou, China).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Effects of DKS26 on blood glucose, oral glucose tolerance, feeding and body weight in STZ‐induced diabetic mice
Seven days after injection of STZ, the blood glucose levels of model mice were significantly increased compared with normal control (P < 0.05), and there were no differences among the four diabetic mice groups (P > 0.05) (Figure 2A). DKS26, 100 mg·kg−1·day−1 i.g. for 33 days, obviously lowered the plasma glucose and GSP levels of diabetic mice in comparison with STZ control (P < 0.05). Metformin, positive control, had similar effects to DKS26, but the inhibitory effects of OA on plasma glucose and GSP levels in these mice were much weaker (Figure 2A, B). Moreover, it showed that the OGT in STZ control group mice was severely impaired compared with normal control, which could be effectively reversed by DKS26 (Figure 2C, D). The amelioration of OGT mediated by DKS26 was also similar to that of metformin and tended to be better than that of OA, but this latter difference was not statistically significant. Also, consistent with the hypoglycaemic effects of DKS26, the water and food intakes in diabetic mice treated with DKS26 were significantly decreased compared with STZ control; similar effects were seen with metformin, but the effects of OA on these variables were less than those of DKS26 (Figure 2E, F). However, there were no obvious changes in the body weights of mice during administration of DKS26, metformin or OA compared with STZ control (Figure 2G).
Figure 2.
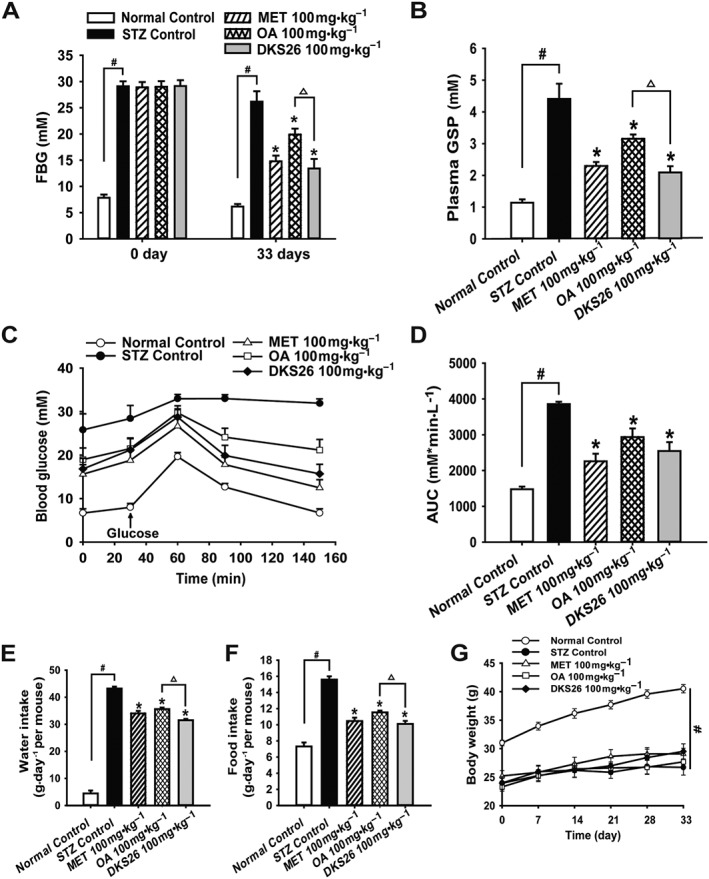
Effects of DKS26 on blood glucose, oral glucose tolerance, feeding and body weight in STZ‐induced diabetic mice after 33 days of administration. (A) The FBG on the first day and on the 33th day. (B) Plasma GSP levels. (C) and (D) Oral glucose tolerance test and the AUC. (E) Water intake expressed as the average of daily water consumption during the last week in each group of mice. (F) Food intake expressed as the average of daily food consumption during the last week in each group of mice. (G) Body weight. OA represents oleanolic acid; MET represents metformin. Values are expressed as mean ± SEM with normal control group n = 5, STZ control group n = 7 and other groups n = 8. Compared with normal control group, # P < 0.05; compared with STZ control group, * P < 0.05; compared with OA group, △ P < 0.05.
Effects of DKS26 on blood glucose, oral glucose tolerance, feeding and body weight in db/db diabetic mice
The hypoglycaemic effects of DKS26 evaluated in db/db mice are shown in Table 1 and Figure 3A. Compared with the db/db model control, DKS26 at a single dose of 100 mg·kg−1 (i.g.) significantly lowered the FBG; the same dose of metformin had a similar effect but OA did not. With continuous daily treatment for 15 days, levels of FBG in the three groups were effectively decreased as compared with model control. FBG and plasma GSP levels of db/db mice were evidently reduced after 30 days of administration of DKS26, and similar effects were observed after the metformin treatment. Notably, the glucose‐lowering effects of DKS26 were evidently better than those of OA, which was in accord with the results obtained in the STZ‐induced diabetic mice. In addition, the postprandial blood glucose (PBG) levels in mice treated with DKS26 and metformin for 20 days were lower than those in db/db control, but there was no influence of OA administration on PBG levels. Furthermore, the improved OGT was observed after both DKS26 and metformin treatment for 27 days, while it was not seen in OA‐treated group, as depicted in Figure 3B, C. Although there were no distinct differences in food intake and body weight of the three administration groups in comparison with db/db control, polydipsia, one of the typical diabetic symptoms, was effectively alleviated by the administration of DKS26 and metformin, but not OA (Figure 3D–F).
Table 1.
Effects of DKS26 on blood glucose during 30 days of treatment in db/db diabetic mice
| Groups | Blood glucose (mmol·L−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 (fasting) | Day 7 (feeding) | Day 15 (fasting) | Day 20 (feeding) | Day 30 (fasting) | ||||||
| Before | 2 h after | Before | 2 h after | Before | 2 h after | Before | 2 h after | Before | 2 h after | |
| db/db Control | 22.1 ± 1.5 | 19.8 ± 0.7 | 22.0 ± 1.9 | 20.4 ± 1.9 | 21.5 ± 0.9 | 25.4 ± 1.2 | 27.8 ± 1.2 | 24.3 ± 1.1 | 20.8 ± 1.5 | 26.5 ± 1.6 |
| MET 100 mg·kg−1 | 22.3 ± 1.7 | 12.1 ± 0.9a | 24.6 ± 1.7 | 17.6 ± 1.5 | 16.7 ± 1.8 | 12.8 ± 1.0a | 22.0 ± 1.9 | 15.7 ± 1.5a | 17.3 ± 1.2 | 13.5 ± 1.7a |
| OA 100 mg·kg−1 | 22.3 ± 1.7 | 16.0 ± 1.6 | 25.2 ± 1.6 | 23.0 ± 1.3 | 17.6 ± 1.3 | 17.8 ± 1.8a | 25.3 ± 1.5 | 23.9 ± 1.7 | 20.0 ± 1.6 | 19.8 ± 1.6a |
| DKS26 100 mg·kg−1 | 22.2 ± 1.7 | 14.7 ± 0.9a | 25.1 ± 1.1 | 21.5 ± 1.4 | 14.9 ± 1.0a | 13.9 ± 0.9a | 21.6 ± 1.4a | 17.6 ± 1.0a | 14.8 ± 1.2a | 11.8 ±1.2a, b |
Values are expressed as mean ± SEM with db/db and metformin control n = 7 and other groups n = 8. OA represented oleanolic acid; MET represented metformin.
P < 0.05, compared with db/db control group;
P < 0.05, compared with OA group.
Figure 3.
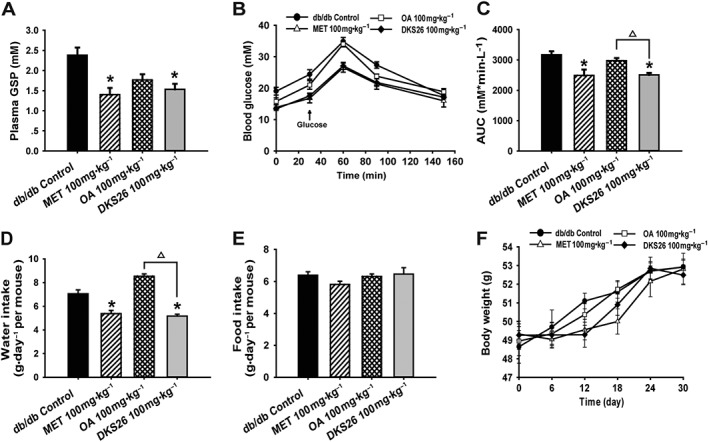
Effects of DKS26 on blood glucose, oral glucose tolerance, feeding and body weight in db/db diabetic mice after 30 days of administration. (A) Plasma GSP levels. (B) and (C) oral glucose tolerance test and the AUC. (D) Water intake expressed as the average of the last week of daily water consumption in mice of each group. (E) Food intake expressed as the average of the last week of daily food consumption in mice of each group. (F) Body weight. OA represents oleanolic acid; MET represents metformin. Values are expressed as mean ± SEM with db/db control and metformin n = 7 and OA control and DKS26 group n = 8. Compared with db/db control group, * P < 0.05; compared with OA group, △ P < 0.05.
DKS26 increased plasma insulin levels and improved the pathological morphology of pancreatic islets beta cells in vivo
To determine the impact of DKS26 on pancreatic islets beta cells, plasma insulin levels and islets area were measured in both STZ‐induced and db/db diabetic mice. As shown in Figure 4A–C, the STZ treatment markedly damaged the islets beta cells in diabetic mice compared with normal control; this was manifested as shrunken islets and a decreased number of islet beta cells accompanied by a sharp reduction in plasma insulin levels. DSK26 ameliorated the pathological morphology of pancreatic islets and elevated plasma insulin levels in comparison with STZ control (P < 0.05). However, there were no evident influences on plasma insulin levels with metformin or OA treatment despite enlarged islets. In the db/db model, plasma insulin levels were obviously augmented by the three treatments compared with db/db control (P < 0.05) (Figure 4D). Notably, hypertrophic pancreatic islets were prevented by DKS26 treatment, based on the analysis of islet area (P < 0.05) as seen in Figure 4E, F, but were not distinctly changed by metformin or OA.
Figure 4.
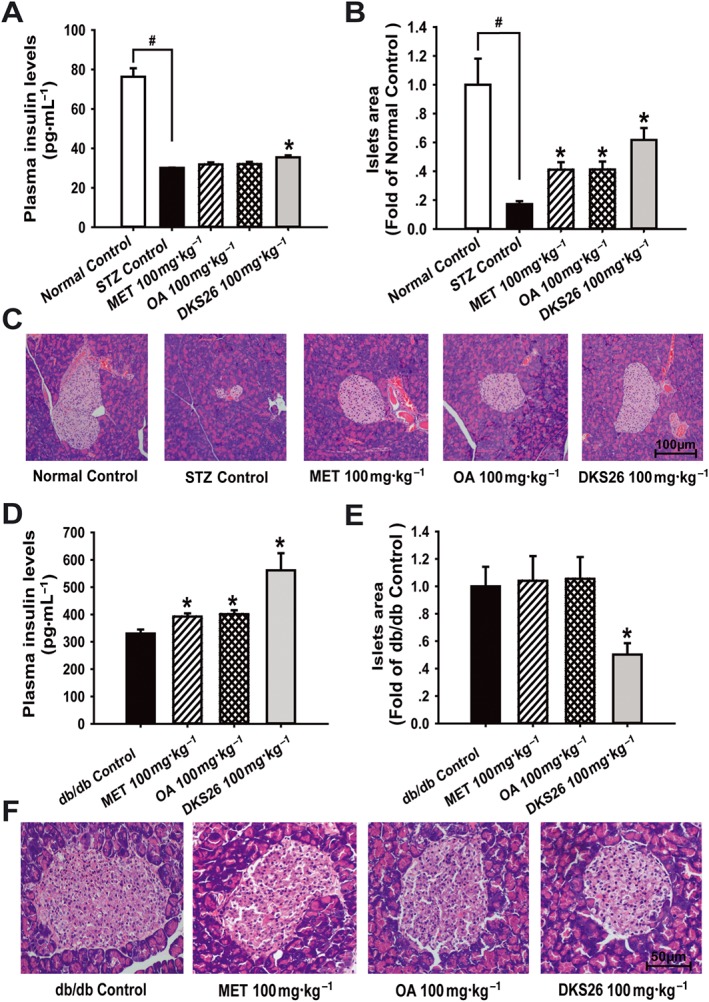
Effects of DKS26 on plasma insulin levels and pancreatic islets beta cells histopathological morphology in STZ‐induced and db/db diabetic mice. STZ‐induced diabetic mice (normal control n = 5, STZ control n = 7 and other groups n = 8): (A) plasma insulin levels, (B) islets area analysed by the software Image J (vs. normal control group) and (C) pancreas HE staining (100×). Db/db diabetic mice (db/db and metformin n = 7 and other groups n = 8): (D) plasma insulin levels, (E) islets area and (F) pancreas HE staining (200×). OA represented oleanolic acid; MET represented metformin. Values are expressed as mean ± SEM. Compared with normal control group, # P < 0.05; compared with model control group (STZ control or db/db control), * P < 0.05.
DKS26 ameliorated lipid profiles in both STZ‐induced and db/db diabetic mouse models
Lipid profiles were investigated in the present study to evaluate whether DKS26 had an anti‐hyperlipidaemic effect in diabetic models. As depicted in Table 2, the levels of plasma TG, TC, LDL‐C and ratio of LDL‐C/HDL‐C in the STZ‐induced diabetic model were significantly higher than normal control along with a reduction in plasma HDL‐C levels. In both diabetic models, the treatment with DKS26 decreased the levels of plasma TG, TC, LDL‐C and ratio of LDL‐C/HDL‐C despite no changes in plasma HDL‐C levels compared with model control. Comparatively, similar hypolipidaemic effects were displayed in the metformin‐ and OA‐treated mice on TG and HDL‐C, but there were some differences in the altered lipid profiles; the plasma TC levels in db/db mice treated with metformin and in STZ‐induced model treated with OA (P < 0.05 compared with DKS26) were not changed compared with model control (P > 0.05), and the high levels of plasma LDL‐C in both diabetic models were not lowered by OA treatment.
Table 2.
Effects of DKS26 on plasma lipid profiles in STZ‐induced and db/db diabetic mice
| Animals | Groups | TG (mM) | TC (mM) | HDL‐C (mM) | LDL‐C (mM) | LDL‐C/HDL‐C |
|---|---|---|---|---|---|---|
| STZ‐induced diabetic mice | Normal Control | 1.04 ± 0.06 | 2.34 ± 0.06 | 2.40 ± 0.10 | 0.60 ± 0.05 | 0.25 ± 0.01 |
| STZ Control | 1.66 ± 0.10a | 3.00 ± 0.20a | 1.87 ± 0.11a | 1.37 ± 0.09a | 0.78 ± 0.09a | |
| MET 100 mg·kg−1 | 1.08 ± 0.07b | 2.43 ± 0.07b | 1.93 ± 0.12 | 0.92 ± 0.04b | 0.50 ± 0.03b | |
| OA 100 mg·kg−1 | 0.90 ± 0.04b | 2.96 ± 0.11 | 1.97 ± 0.10 | 1.36 ± 0.09 | 0.63 ± 0.05 | |
| DKS26 100 mg·kg−1 | 0.96 ± 0.06b | 2.36 ± 0.12b , c | 1.98 ± 0.16 | 0.91 ± 0.07b , c | 0.47 ± 0.04b | |
| Db/db diabetic mice | db/db Control | 2.02 ± 0.22 | 5.19 ± 0.24 | 2.99 ± 0.15 | 1.18 ± 0.11 | 0.39 ± 0.02 |
| MET 100 mg·kg−1 | 1.28 ± 0.12b | 4.71 ± 0.19 | 2.61 ± 0.22 | 0.59 ± 0.10b | 0.24 ± 0.04b | |
| OA 100 mg·kg−1 | 1.34 ± 0.11b | 4.34 ± 0.17b | 2.93 ± 0.09 | 1.15 ± 0.09 | 0.39 ± 0.03 | |
| DKS26 100 mg·kg−1 | 1.17 ± 0.07b | 4.30 ± 0.09b | 2.62 ± 0.12 | 0.75 ± 0.06b | 0.28 ± 0.03b |
Values are expressed as mean ± SEM in STZ‐induced diabetic mice (normal control n = 5, STZ control n = 7 and other groups n = 8) and db/db diabetic mice (db/db and metformin control n = 7 and other groups n = 8). OA represented oleanolic acid; MET represented metformin.
P < 0.05, compared with normal control group;
P < 0.05, compared with model control group (STZ control or db/db control);
P < 0.05, compared with OA group.
DKS26 prevented diabetes‐induced hepatic damage in both STZ‐induced and db/db diabetic mouse models
To further assess the beneficial effects of DKS26 treatment, the plasma levels of ALT and AST were detected, and the expression of pro‐inflammatory cytokines, histopathology and lipid of liver in both diabetic models were assessed. Plasma ALT and AST levels in STZ‐induced mice were significantly increased compared with normal control, and these effects were prevented by DKS26 administration (Figure 5A, B). Also, DKS26 evidently reduced hepatic mRNA levels of TNF‐α, IL‐6 and IL‐1β in comparison with STZ control (Figure 5C). Furthermore, liver HE staining illustrated that the impaired hepatocellular morphology and augmented necrotic hepatocytes in STZ‐induced model were ameliorated by DKS26 treatment (Figure 5D, E). Similarly, DKS26 evidently alleviated high levels of plasma ALT and AST in db/db mice (Figure 5F, G) and inhibited the expression of liver TNF‐α, IL‐6, and IL‐1β compared with db/db control (Figure 5H). In addition, DKS26 exerted a hepatoprotective effect in db/db mice exhibiting numerous fatty degenerative hepatocytes (Figure 5I).
Figure 5.
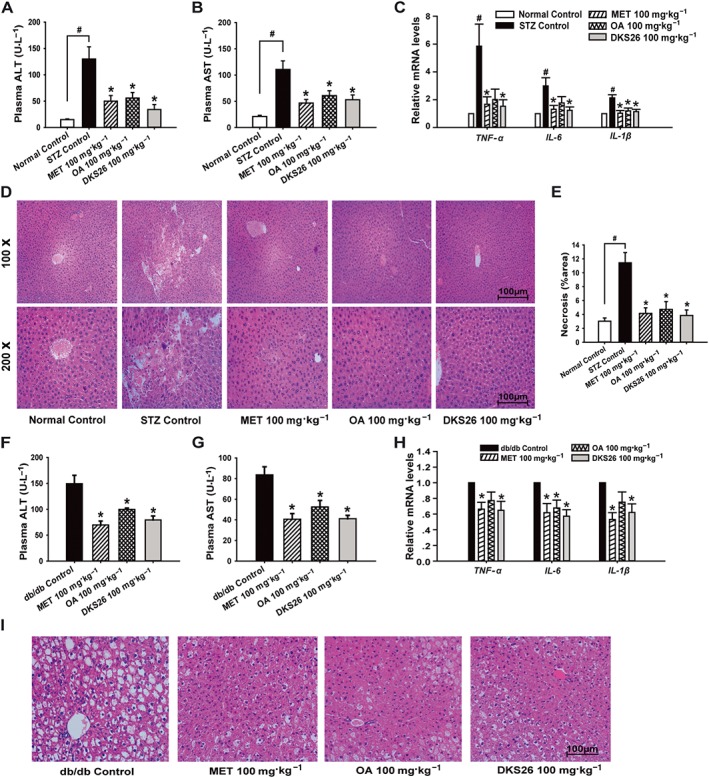
Effects of DKS26 on liver function, inflammation‐related cytokine genes and histopathology in STZ‐induced and db/db diabetic mice. STZ‐induced diabetic mice: (A) and (B) plasma ALT and AST levels (normal control n = 5, STZ control n = 7 and other groups n = 8), (C) hepatic inflammation related TNF‐α, IL‐6 and IL‐1β mRNA levels (vs. β‐actin, normal control n = 5 and other groups n = 6), (D) liver histopathology by HE staining (normal control n = 5, STZ control n = 7 and other groups n = 8, 100× and 200×), (E) hepatic necrosis area (%) analysed by 5–6 HE staining photos of each liver samples (normal control n = 5, STZ control n = 7 and other groups n = 8, 100×). Db/db diabetic mice: (F) and (G) plasma ALT and AST levels (db/db and metformin control n = 7 and other groups n = 8), (H) hepatic inflammation‐related TNF‐α, IL‐6 and IL‐1β mRNA levels (vs. β‐actin, n = 6), (I) liver histopathology by HE staining (200×). OA represents oleanolic acid; MET represented metformin. Values are expressed as mean ± SEM. Compared with normal control group, # P < 0.05; compared with model control group (STZ control or db/db control), * P < 0.05.
Furthermore, hepatic TG contents in both diabetic models were markedly decreased by DKS26 treatment, and a reduction in liver TC contents was also measured in the STZ model despite no significant change in db/db model (Figure 6A, B, E, F). Moreover, Oil Red O staining of frozen liver sections further showed that hepatic lipid accumulation was decreased in both models, manifested as relatively lighter red by DKS26 treatment compared with model control showing many lipid droplets in deep red (Figure 6C, G). In line with the hepatic lipid accumulation in the STZ model, elevated levels of sterol regulatory element binding protein‐1 (SREPB‐1c) and fat acid synthase (FAS) mRNA and reduced levels of PPAR‐α and carnitine palmitoyltransferase‐1 (CPT1) were observed in STZ control compared with normal control, and these changes were reversed by DKS26 administration as shown in Figure 6D. Similarly, DKS26 suppressed the expression of SREPB‐1c and FAS and promoted the expression of PPAR‐α and CPT1 as compared with db/db control (Figure 6H). No obvious differences in hepatic function, inflammation, histopathology or the levels of lipid and metabolism‐related gene expression were observed among the treatment groups in both diabetic models.
Figure 6.
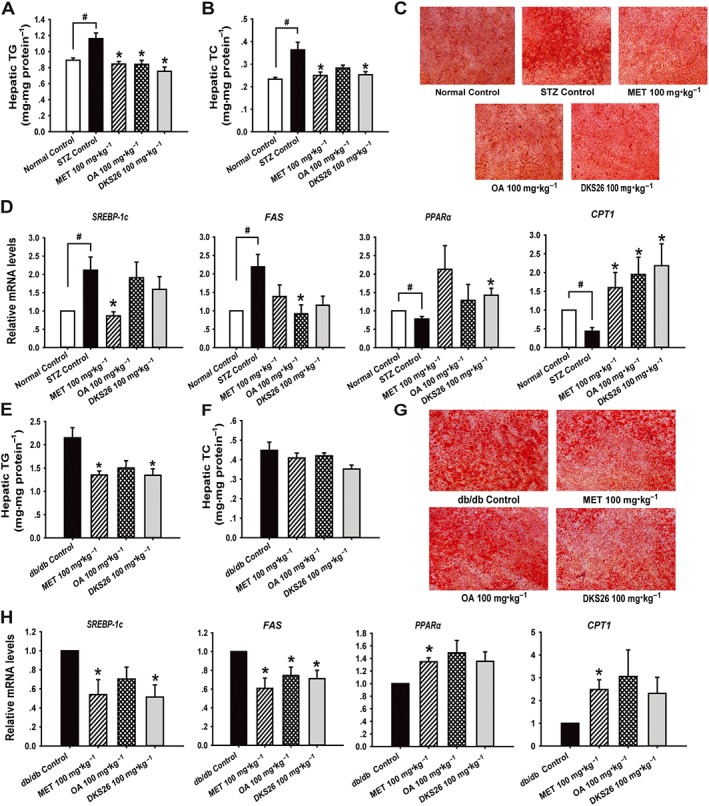
Effects of DKS26 on hepatic lipid accumulation and metabolism related genes in STZ‐induced and db/db diabetic mice. STZ‐induced diabetic mice: (A) and (B) hepatic TG and TC contents (normal control n = 5, STZ control n = 7 and other groups n = 8), (C) liver sections by Oil Red O staining (200×, scale bar 100 μm), (D) hepatic lipid metabolism related SREPB‐1c, FAS, PPAR‐α and CPT1 mRNA levels (vs. β‐actin, normal control n = 5 and other groups n = 6). Db/db diabetic mice: (E) and (F) hepatic TG and TC contents (db/db and metformin control n = 7 and other groups n = 8), (G) liver sections by Oil Red O staining (100×, scale bar 100 μm), (H) hepatic lipid metabolism related SREPB‐1c, FAS, PPAR‐α and CPT1 mRNA levels (vs. β‐actin, n = 6). OA represents oleanolic acid; MET represents metformin. Values are expressed as mean ± SEM. Compared with normal control group, # P < 0.05,; compared with model control group (STZ control or db/db control), * P < 0.05.
DKS26 augmented GLP‐1 secretion and expression via the cAMP/PKA signalling pathway in NCI‐H716 cells
The effects of DKS26 on GLP‐1 release and expression were explored in endocrine‐differentiated NCI‐H716 cells, characteristic of intestinal L cells. The contents of intracellular cAMP, a pivotal GPCR downstream target, were significantly increased by 10 μM DKS26 treatment in a concentration‐dependent manner (Figure 7A). Also, supernatant GLP‐1 levels were elevated in NCI‐H716 cells treated with 10 μM DKS26, which was similar to that of 10 μM OA (Figure 7B). Furthermore, the expression levels of the GLP‐1 related gene proglucagon (gcg) and prohormone converting enzyme 3 (PC3) in NCI‐H716 cells treated with DKS26 were obviously augmented, and this enhancing effect was stronger than that of OA at the same concentration. Immunofluorescence staining revealed that DKS26 increased the in situ expression of GLP‐1 indicating that it is capable of enhancing GLP‐1 biosynthesis (Figure 7C, D). Furthermore, up‐regulated PKA phosphorylation levels induced by DKS26 indicated that the enhanced GLP‐1 secretion and expression were related to increased activity of the cAMP/PKA signalling pathway (Figure 7E, F).
Figure 7.
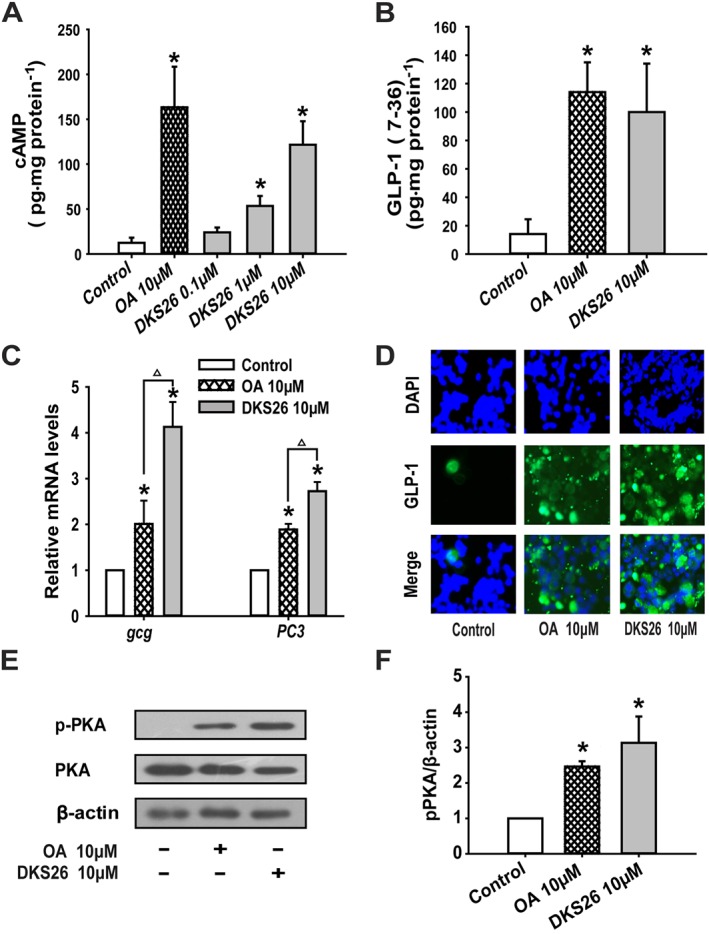
Effects of DKS26 on GLP‐1 secretion and expression in NCI‐H716 cells. (A) Intracellular cAMP levels. (B) Supernatant GLP‐1 (7–36) levels. (C) GLP‐1 related gene gcg and PC3 mRNA levels (vs. GAPDH). (D) Immunofluorescence staining for in situ expression of GLP‐1 (green); blue represents the nuclear dye DAPI and scale bar is 100 μm (100×). (E) Western blotting for expression of PKA and phosphorylated PKA. (F) Levels of PKA and phosphorylated PKA expressed relative to β‐actin levels. OA represents oleanolic acid; MET represents metformin. Values are expressed as mean ± SEM (n = 3). Compared with control group, * P < 0.05; compared with OA group, △ P < 0.05.
Effects of DKS26 on GLP‐1 release, biosynthesis and intestinal cAMP/PKA signalling pathway in vivo
Plasma GLP‐1 levels in db/db mice after 30 days treatment of DKS26 were obviously increased compared to the control group, and this enhancement was 1.55 times as much as that of OA (Figure 8A).Furthermore, the elevated intestinal levels of gcg and PC3 mRNA and enhanced GLP‐1 protein in DKS26‐treated mice demonstrated that the in situ levels of GLP‐1 were increased by DKS26 administration in db/db mice (Figure 8C, D). In contrast, there was no significant up‐regulated PC3 gene expression in OA treated mice despite a moderately enhanced gcg gene expression. The production of intestinal cAMP in db/db diabetic mice was elevated by DKS26, and this effect was more marked than that of OA (3.67 times, P < 0.05) in line with its GLP‐1 secretory effect (Figure 8B). Consistent with the above findings, intestinal PKA phosphorylation levels were distinctly up‐regulated by DKS26 treatment, which was significantly stronger than that of OA (Figure 8E, F).
Figure 8.
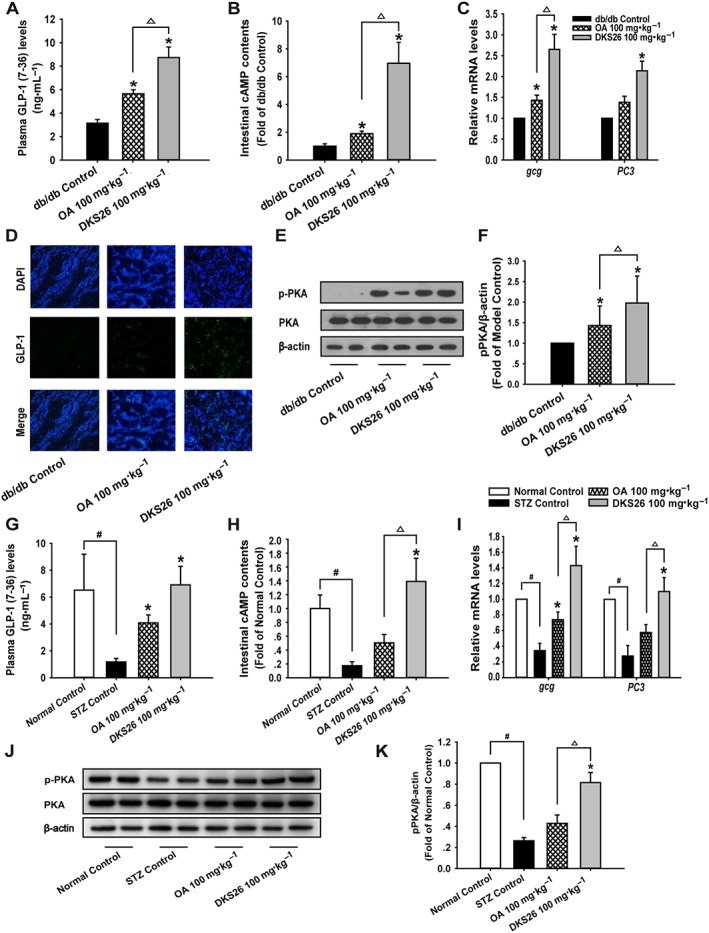
Effects of DKS26 on GLP‐1 release and biosynthesis in db/db and STZ‐induced diabetic mice. Db/db diabetic mice: (A) plasma GLP‐1 (7–36) levels (db/db and metformin control n = 7 and other groups n = 8), (B) intestinal cAMP levels (n = 6), (C) intestinal GLP‐1 biosynthesis relative gene gcg and PC3 mRNA levels (n = 5, vs. β‐actin), (D) immunofluorescence staining for intestinal GLP‐1 (green), blue represents the nuclear dye DAPI and scale bar is 100 μm (100×), (E) and (F) Western blotting for detecting PKA phosphorylation and expression levels relative to β‐actin levels (n = 6). STZ‐induced diabetic mice: (G) plasma GLP‐1 (7–36) levels (normal control n = 5, STZ control n = 7 and other groups n = 8), (H) intestinal cAMP levels (normal control n = 5 and other groups n = 6), (I) intestinal GLP‐1 biosynthesis and gcg and PC3 mRNA levels (normal control n = 5 and other groups n = 6, vs. β‐actin). (J) and (K) Western blotting for detecting PKA phosphorylation and expression levels relative to β‐actin (normal control n = 5 and other groups n = 6). OA represents oleanolic acid; MET represents metformin. Values are expressed as mean ± SEM. Compared with normal control group, # P < 0.05; compared with model control group (STZ control or db/db control), * P < 0.05; compared with OA group, △ P < 0.05.
Similarly, the significantly reduced plasma GLP‐1 levels in STZ‐induced diabetic model were ameliorated by both DKS26 and OA administration (P < 0.05; Figure 8G), and the effect of DKS26 on increased plasma GLP‐1 levels also tended to be stronger than that of OA (1.70‐fold), but this was not statistically significant. As illustrated in Figure 8I, decreased mRNA levels of intestinal gcg and PC3 were effectively enhanced by DKS26 treatment, and these effects were more marked than those of OA. DKS26 markedly enhanced intestinal cAMP contents and phosphorylation of PKA in comparison with STZ control, which exhibited low levels of cAMP and phosphorylated PKA in intestine compared with normal control, as shown in Figure 8H, J, K. In contrast, OA could not significantly restore the cAMP/PKA signalling pathway in STZ model, although slightly elevated levels of intestinal cAMP and phosphorylated PKA were detected.
Discussion and conclusions
It was initially confirmed in our study that the OA derivative DKS26 has anti‐hyperglycaemic effects equivalent to metformin and stronger than those of OA. The plasma GSP, which reflects the average blood glucose levels over 2 to 3 weeks (Nelson et al., 1985), was evidently decreased by DKS26 in both STZ‐induced and db/db diabetic mice, suggesting that DKS26 has potential as a therapeutic for controlling hyperglycaemia. In addition, DKS26 decreased feeding glucose levels and the AUC of OGTT in db/db mice, whereas OA did not, indicating that it is better at controlling postprandial blood sugar levels than OA.
The amelioration of pancreatic islet dysfunction is an important therapeutic approach for diabetes since islet beta cell damage plays a key role in the pathogenesis of diabetes (Jain et al., 2015). On the one hand, STZ exerts toxicity on islet beta cells of mice triggering destruction, which is manifest as significantly atrophic islets and decreased insulin secretion (Wang et al., 1993; Kumar and Kar, 2015), and DKS26 effectively ameliorated beta cell damage by restoring islet areas and plasma insulin levels in STZ‐induced diabetic mice. Metformin and OA did not show insulinotropic effects despite promoting beta cell proliferation, which is in accord with the reports that they ameliorate hyperglycaemia by increasing insulin sensitivity without augmenting insulin secretion in diabetic mice (Junien et al., 1981; Hundal and Inzucchi, 2003; Wang et al., 2011). On the other hand, the impaired islets beta cells along with hyperinsulinaemia resulting from insulin resistance, which are compensatory enlarged by the stimulation of long‐term hyperglycaemia, can be reversed at the early stage of diabetes in db/db mice (Do et al., 2016). Intriguingly, DKS26 evidently elevated plasma insulin levels in db/db mice, which might be attributed to the amelioration of hypertrophic islet beta cells promoting insulin production. In contrast, metformin and OA had no significant effect on hypertrophic islets in spite of slightly increasing plasma insulin levels. Hence, the amelioration of islet beta cell damage and improved insulin secretion induced by DKS26 in both diabetic models indicates that it has more potential benefits compared to metformin and OA for diabetes care.
Dyslipidaemia is the main cause for the development of cardiovascular disease, which is a major and fatal complication of diabetes, and therefore, lipid control is an effective option to alleviate diabetic symptoms (Beckman et al., 2013). Studies have shown that plasma TG and TC levels are significantly elevated in STZ‐induced diabetic mice, accompanied by an increase in LDL‐C and decrease in HDL‐C (Qiu et al., 2012); these findings were confirmed in our study. Also, db/db mice are widely utilized to study the dyslipidaemia associated with diabetes (Jung et al., 2012). We showed that plasma TG, TC, LDL‐C and the ratio of LDL‐C/HDL‐C were decreased by DKS26 in both diabetic dyslipidaemia models, although no escalation in HDL‐C was observed, indicating that DKS26 may be an excellent agent for the therapy of diabetic complications associated with changes in blood lipid levels.
Liver damage with a marked rise in plasma ALT and AST occurs when excessive fat accumulates in liver due to hyperglycaemia and hyperlipidaemia in diabetes (Kim et al., 2009; Mohamed et al., 2016). The hepatic lipid overload is associated with an abnormal metabolism of lipid synthesis and oxidation in the liver, which can induce and exacerbate liver inflammation that is accompanied by increased hepatic expression of TNF‐α, IL‐6 and IL‐1β, the major pro‐inflammatory cytokines in non‐alcoholic fatty liver disease (Paz et al., 2013; Kwon et al., 2016). Based on the hepatoprotective action of OA (Kim et al., 2005), the effects of DKS26 on hepatic function in diabetic mice were assessed. DKS26, as well as metformin and OA, ameliorated the hepatic abnormalities of STZ‐induced diabetic mice; they significantly reduced the increased plasma levels of ALT and AST, necrotic hepatocytes, expression levels of TNF‐α, IL‐6 and IL‐1β and lipid accumulation in the liver. Previous studies have shown that db/db mice display severe hepatic dysfunction, numerous fatty degeneration hepatocytes, hepatic lipid accumulation and liver inflammation (Sleeman et al., 2003; Li et al., 2015), and have high levels of plasma ALT and AST and liver lipids, numerous fatty vacuolated hepatocytes and hepatic pro‐inflammatory cytokines; all these latter effects were alleviated by DKS26, metformin or OA in our db/db mice. SREBP‐1c and FAS are two lipogenic key genes involved in triglyceride synthesis and accumulation (Eberle et al., 2004), and the nuclear receptor PPARα, which is highly expressed in liver, modulates lipid metabolism‐related gene expression, mainly CPT1, the key enzyme involved in fatty acid oxidation (Gulick et al., 1994). In the present study it was shown that DKS26, metformin and OA enhance lipid oxidation and diminish lipid synthesis by down‐regulating the expression of SREBP‐1c and FAS and up‐regulating PPARα and CPT1 to reduce liver lipid accumulation in both diabetic models. Further exploration has shown that DKS26 stimulates hepatic AMPK phosphorylation in vitro (data not shown). As an enhanced sensitivity of hepatic insulin contributes to decreased hepatic lipid accumulation (Hwang et al., 2015), and a GLP‐1 receptor agonist ameliorates steatohepatitis (Wang et al., 2014; Yamamoto et al., 2016), we speculate that the hepatoprotection of DKS26 mediated by its effects on AMPK might be as a consequence of it directly or indirectly improving insulin sensitivity or activating the GLP‐1 receptor to alleviate hepatic fat accumulation and inflammation, but this remains to be further confirmed.
It is important to note that in STZ‐induced diabetic mice, the total plasma bile acids (TBA) levels were elevated by OA in comparison with STZ control regardless of the lack of statistical significance (Supporting Information Figure S2), suggesting that OA has the potential to induce hepatic cholestasis. OA, when used as a repeated medication, has been reported to induce liver injury in normal mice by causing hepatic cholestasis through an adverse effect on the bile acid metabolism balance (Liu et al., 2013), so we investigated the effect of DKS26 on hepatic function in normal ICR mice. Surprisingly, DKS26, at a dose of 100 mg·kg−1·day−1 i.g. for 8 days, showed no hepatotoxicity, whereas OA at the same dose produced hepatic damage accompanied by significantly increased levels of plasma ALT, AST and TBA (Supporting Information Figure S3). The mechanism of the differences between DKS26 and OA in the modulation of bile acid metabolism and hepatic function needs to be further clarified.
As OA has been shown to affect the secretion of GLP‐1, we investigated the effects of DKS26 on GLP‐1. DKS26 increased plasma GLP‐1 levels and the expressions of intestinal GLP‐1 protein and related genes (gcg and PC3) in both diabetic mouse models, and these effects were more marked than those of OA. These findings together with the decreased plasma levels of glucose and lipid and improved islets beta cells function in diabetic mice further indicate that the antidiabetic mechanism of DKS26 is related to GLP‐1, as GLP‐1 is involved in glucolipid homeostasis and can improve islet functions (Sandoval and D'Alessio, 2015). Regarding GLP‐1 production, PKA is highly involved as a crucial target protein of cAMP produced by the activation of GPCRs highly expressed in the ileum and colon (Ong et al., 2009). The activation of PKA (a key target of Wnt signalling pathway) is followed by gcg gene expression and then active GLP‐1 is produced after being activated by prohormone converting enzyme 3 (PC3) (Damholt et al., 1999; Yu and Jin, 2010). DKS26 elevated GLP‐1 release and increased levels of GLP‐1 protein expression in situ and the mRNA of related genes (gcg and PC3), along with an elevation in cAMP and activation of PKA, in NCI‐H716 cells, as well as in diabetic mice, illustrating that DKS26 might enhance GLP‐1 secretion and biosynthesis by activating the cAMP/PKA signalling pathway.
Interestingly, the GLP‐1 secretory effect of DKS26 was equivalent to that of OA in vitro but significantly stronger than that of OA in vivo. Since the weak hypoglycaemic efficacy of OA may be related to its low oral bioavailability, as described previously (Jeong et al., 2007; Cao et al., 2013), pharmacokinetic tests of DKS26 and OA were done and showed that the oral bioavailability of DKS26 was much better than that of OA (40.46 vs. 0.77%, detailed data not shown). As OA is a selective TGR5 ligand, the agonistic effect of DKS26 on TGR5 was investigated. Unexpectedly, DKS26 exerted no agonistic effect on human TGR5, while OA activated it (Supporting Information Figure S4), indicating that the GLP‐1 regulating effects of DKS26 might be associated with the activation of other GPCRs or a different mechanism is involved, which remains to be further verified. TGR5 when activated in the gallbladder can cause gallbladder filling, a common adverse reaction for many synthetic TGR5 agonists (Bunnett, 2014; Briere et al., 2015). DKS26 at a dose of 100 mg·kg−1·day−1 did not display this side effect in either diabetic model, further indicating that it maybe devoid of TGR5‐mediated adverse effects (Supporting Information Figure S5). In summary, there are differences between DKS26 and OA not only with their pharmacokinetics but also pharmacodynamics and the mechanisms through which they affect GLP‐1.
In conclusion, DKS26 exerts hypoglycaemic, hypolipidaemic and islet protective effects in vivo by promoting the secretion and expression of GLP‐1 and this is mediated by an effect on the cAMP/PKA signalling pathway. In addition, DKS26 probably also ameliorates the hepatic damage associated with diabetes by reducing liver lipids and inflammation. All these findings suggest that DKS26 could be developed into a novel potential therapeutic for the treatment of diabetes and related chronic metabolic diseases.
Author contributions
L.T. and H.‐S.W. conceived and designed this study and revised the manuscript. F.‐F.C. did the majority of the in vitro experiments, carried out the in vivo experiments and together with J.‐T.W., L.‐X.Z., S.‐F.X., Y.‐X.W., K.W. and H.‐S.W., analysed the research data and wrote the manuscript with the help of S.‐L.D. and J.‐Q.Z.
Conflict of interest
The authors delcare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effects of DKS26 on the glucose consumption in human hepatic HepG2 Cells. OA represented oleanolic acid, MET represented metformin. Values are expressed as mean ± SEM (n = 3). Compared with control group, * P < 0.05, ** P < 0.01. Glucose consumption = glucose concentrations of blank wells − glucose concentrations of cell plated wells. The glucose consumption values were adjusted by SRB test. This glucose lowering activity in vitro showed that DKS26 might have hypoglycemic effects in vivo.
Figure S2 Effects of DKS26 on plasma total bile acids (TBA) levels in STZ‐induced diabetic mice for 33 days of administration. OA represented oleanolic acid, MET represented metformin. Values are expressed as mean ± SEM. Normal Control (n = 5), STZ control (n = 7) and other groups (n = 8). Compared with normal control group, ## P < 0.01.
Figure S3 Effects of DKS26 on hepatic function and plasma total bile acids (TBA) levels in normal ICR mice for 8 days. OA represented oleanolic acid, MET represented metformin. Values are expressed as mean ± SEM (OA group n = 5 and other groups n = 6). Compared with control group, * P < 0.05, ** P < 0.01, *** P < 0.001. These findings indicated that the effect of DKS26 on hepatic injury in normal mice was significantly lower than OA.
Figure S4 Direct effects of DKS26 on TGR5 in HEK293/pGL4.29/hTGR5 cells. Effect (%) was calculated as relative effect of 20 μM INT‐777 regarded as 100%. EC50 was 1.77 ± 0.45 μM and 13.66 ± 1.81 μM for INT‐777 and OA respectively. OA represented oleanolic acid. Values are expressed as mean ± SD (n = 3).
Figure S5 Effects of DKS26 on gallbladder size in STZ‐induced and db/db diabetic mice. The gallbladders were removed 2 h after the last administration and measured by a vernier calliper or analytical balance. The size of gallbladder was expressed as the cross‐sectional area (length x width, mm2) or weight (mg) and corrected by body weight (g). (A) STZ‐induced diabetic mice (normal control n = 5, STZ control n = 7 and other groups n = 8). (B) Db/db diabetic mice (db/db and metformin control n = 7, and other groups n = 8). OA represented oleanolic acid, MET represented metformin. Values are expressed as mean ± SEM. Compared with normal control group, # P < 0.05, ## P < 0.01.
Table S1 Effects of DKS26 on blood glucose with a single administration in STZ‐induced diabetic mice.
Table S2 The primer sequences used in this study.
Acknowledgements
This work was supported by the Natural Science Foundation of Guizhou Province (no. JZ2015‐2001) and the Natural Science Foundation of Zhejiang Province (no. LY17H140004) of China. We sincerely thank Dr Ying Leng (State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica (SIMM), Chinese Academy of Sciences) for assisting with measuring the direct effects of DKS26 and OA on hTGR5 in vitro and thank Ke Wang (College of Pharmaceutical Sciences, Zhejiang University) for correcting the grammar of the revised manuscript.
Chen, F.‐F. , Wang, J.‐T. , Zhang, L.‐X. , Xing, S.‐F. , Wang, Y.‐X. , Wang, K. , Deng, S.‐L. , Zhang, J.‐Q. , Tang, L. , and Wu, H.‐S. (2017) Oleanolic acid derivative DKS26 exerts antidiabetic and hepatoprotective effects in diabetic mice and promotes glucagon‐like peptide‐1 secretion and expression in intestinal cells. British Journal of Pharmacology, 174: 2912–2928. doi: 10.1111/bph.13921.
Contributor Information
Lei Tang, Email: tlei1974@hotmail.com.
Hao‐Shu Wu, Email: fwhs@zju.edu.cn.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay L (2015). Incretin‐based drugs and adverse pancreatic events: almost a decade later and uncertainty remains. Diabetes Care 38: 951–953. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ (2007). Biology of incretins: GLP‐1 and GIP. Gastroenterology 132: 2131–2157. [DOI] [PubMed] [Google Scholar]
- Bala V, Rajagopal S, Kumar DP, Nalli AD, Mahavadi S, Sanyal AJ et al. (2014). Release of GLP‐1 and PYY in response to the activation of G protein‐coupled bile acid receptor TGR5 is mediated by Epac/PLC‐epsilon pathway and modulated by endogenous H2S. Front Physiol 5: 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JA, Paneni F, Cosentino F, Creager MA (2013). Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Eur Heart J 34: 2444–2452. [DOI] [PubMed] [Google Scholar]
- Briere DA, Ruan X, Cheng CC, Siesky AM, Fitch TE, Dominguez C et al. (2015). Novel small molecule agonist of TGR5 possesses anti‐diabetic effects but causes gallbladder filling in mice. PLoS One 10: e0136873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone S, Magnone M, Mannino E, Sociali G, Sturla L, Fresia C et al. (2015). Abscisic acid stimulates glucagon‐like peptide‐1 secretion from l‐cells and its oral administration increases plasma glucagon‐like peptide‐1 levels in rats. PLoS One 10: e0140588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnett NW (2014). Neuro‐humoral signalling by bile acids and the TGR5 receptor in the gastrointestinal tract. J Physiol 592: 2943–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, Drucker DJ (2013). Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 17: 819–837. [DOI] [PubMed] [Google Scholar]
- Cao F, Gao YH, Wang M, Fang L, Ping QN (2013). Propylene glycol‐linked amino acid/dipeptide diester prodrugs of oleanolic acid for PepT1‐mediated transport: synthesis, intestinal permeability, and pharmacokinetics. Mol Pharm 10: 1378–1387. [DOI] [PubMed] [Google Scholar]
- Castellano JM, Guinda A, Delgado T, Rada M, Cayuela JA (2013). Biochemical basis of the antidiabetic activity of oleanolic acid and related pentacyclic triterpenes. Diabetes 62: 1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crickx E, Marroun I, Veyrie C, Le Beller C, Schoindre Y, Bouilloud F et al. (2014). DPP4 inhibitor‐induced polyarthritis: a report of three cases. Rheumatol Int 34: 291–292. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damholt AB, Buchan AMJ, Holst JJ, Kofod H (1999). Proglucagon processing profile in canine L cells expressing endogenous prohormone convertase 1/3 and prohormone convertase 2. Endocrinology 140: 4800–4808. [DOI] [PubMed] [Google Scholar]
- Do OH, Gunton JE, Gaisano HY, Thorn P (2016). Changes in beta cell function occur in prediabetes and early disease in the Lepr(db) mouse model of diabetes. Diabetologia 59: 1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker DJ, Nauck MA (2006). The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet 368: 1696–1705. [DOI] [PubMed] [Google Scholar]
- Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F (2004). SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86: 839–848. [DOI] [PubMed] [Google Scholar]
- Ghosh P, Sahoo R, Vaidya A, Chorev M, Halperin JA (2015). Role of complement and complement regulatory proteins in the complications of diabetes. Endocr Rev 36: 272–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guariguata L, Whiting DR, Hambleton I, Beagley J, Linnenkamp U, Shaw JE (2014). Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pr 103: 137–149. [DOI] [PubMed] [Google Scholar]
- Gulick T, Cresci S, Caira T, Moore DD, Kelly DP (1994). The peroxisome proliferator‐activated receptor regulates mitochondrial fatty‐acid oxidative enzyme gene‐expression. P Natl Acad Sci USA 91: 11012–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundal RS, Inzucchi SE (2003). Metformin: new understandings, new uses. Drugs 63: 1879–1894. [DOI] [PubMed] [Google Scholar]
- Hwang KA, Hwang YJ, Kim GR, Choe JS (2015). Extracts from Aralia elata (Miq) seem alleviate hepatosteatosis via improving hepatic insulin sensitivity. Bmc Complem Altern M 15: 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D, Weber G, Eberhard D, Mehana AE, Eglinger J, Welters A et al. (2015). DJ‐1 protects pancreatic beta cells from cytokine‐ and streptozotocin‐mediated cell death. PLoS One 10: e0138535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong DW, Kim YH, Kim HH, Ji HY, Yoo SD, Choi WR et al. (2007). Dose‐linear pharmacokinetics of oleanolic acid after intravenous and oral administration in rats. Biopharm Drug Dispos 28: 51–57. [DOI] [PubMed] [Google Scholar]
- Jiang QK, Yang XX, Du P, Zhang HF, Zhang TH (2016). Dual strategies to improve oral bioavailability of oleanolic acid: enhancing water‐solubility, permeability and inhibiting cytochrome P450 isozymes. Eur J Pharm Biopharm 99: 65–72. [DOI] [PubMed] [Google Scholar]
- Jung UJ, Park YB, Kim SR, Choi MS (2012). Supplementation of persimmon leaf ameliorates hyperglycemia, dyslipidemia and hepatic fat accumulation in type 2 diabetic mice. PLoS One 7: e49030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junien JL, Chomette G, Guillaume M, Wajcman H, Sterne J (1981). Effect of long‐term metformin treatment on the development of diabetes in genetically diabetic mice (DBM) (author's transl). Diabete Metab 7: 251–258. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Park JY, Lee KU, Kim JH, Kim HK (2009). Association of serum gamma‐glutamyltransferase and alanine aminotransferase activities with risk of type 2 diabetes mellitus independent of fatty liver. Diabetes‐Metab Res 25: 64–69. [DOI] [PubMed] [Google Scholar]
- Kim NY, Lee MK, Park MJ, Kim SJ, Park HJ, Choi JW et al. (2005). Momordin Ic and oleanolic acid from Kochiae Fructus reduce carbon tetrachloride‐induced hepatotoxicity in rats. J Med Food 8: 177–183. [DOI] [PubMed] [Google Scholar]
- Kumar N, Kar A (2015). Pyrroloquinoline quinone (PQQ) has potential to ameliorate streptozotocin‐induced diabetes mellitus and oxidative stress in mice: a histopathological and biochemical study. Chem Biol Interact 240: 278–290. [DOI] [PubMed] [Google Scholar]
- Kwon H, Song K, Han C, Chen W, Wang Y, Dash S et al. (2016). Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with Nonalcoholic fatty liver disease. Hepatology 63: 1155–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Xu XZ, Chen C, Wang Y, Gruzdev A, Zeldin DC et al. (2015). CYP2J2 attenuates metabolic dysfunction in diabetic mice by reducing hepatic inflammation via the PPAR gamma. Am J Physiol‐Endoc M 308: E270–E282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lu YF, Zhang Y, Wu KC, Fan F, Klaassen CD (2013). Oleanolic acid alters bile acid metabolism and produces cholestatic liver injury in mice. Toxicol Appl Pharmacol 272: 816–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YX, Si MM, Lu W, Zhang LX, Zhou CX, Deng SL et al. (2015). Effects and molecular mechanisms of the antidiabetic fraction of Acorus calamus L. on GLP‐1 expression and secretion in vivo and in vitro. J Ethnopharmacol 166: 168–175. [DOI] [PubMed] [Google Scholar]
- McCall AL (2012). Insulin therapy and hypoglycemia. Endocrinol Metab Clin North Am 41: 57–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed J, Nazratun Nafizah AH, Zariyantey AH, Budin SB (2016). Mechanisms of diabetes‐induced liver damage: the role of oxidative stress and inflammation. Sultan Qaboos Univ Med J 16: e132–e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DM, Barrows HJ, Clapp DH, Ortman‐Nabi J, Whitehurst RM (1985). Glycosylated serum protein levels in diabetic and nondiabetic pregnant patients: an indicator of short‐term glycemic control in the diabetic patient. Am J Obstet Gynecol 151: 1042–1047. [DOI] [PubMed] [Google Scholar]
- Ong WK, Gribble FM, Reimann F, Lynch MJ, Houslay MD, Baillie GS et al. (2009). The role of the PDE4D cAMP phosphodiesterase in the regulation of glucagon‐like peptide‐1 release. Br J Pharmacol 157: 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz G, Mastronardi CA, Parker BJ, Khan A, Inserra A, Matthaei KI et al. (2013). Molecular pathways involved in the improvement of non‐alcoholic fatty liver disease. J Mol Endocrinol 51: 167–179. [DOI] [PubMed] [Google Scholar]
- Qiu L, Ye H, Chen L, Hong Y, Zhong F, Zhang T (2012). Red clover extract ameliorates dyslipidemia in streptozotocin‐induced diabetic C57BL/6 mice by activating hepatic PPARalpha. Phytother Res 26: 860–864. [DOI] [PubMed] [Google Scholar]
- Ramirez‐Espinosa JJ, Rios MY, Paoli P, Flores‐Morales V, Camici G, de la Rosa‐Lugo V et al. (2014). Synthesis of oleanolic acid derivatives: in vitro, in vivo and in silico studies for PTP‐1B inhibition. Eur J Med Chem 87: 316–327. [DOI] [PubMed] [Google Scholar]
- Sandoval DA, D'Alessio DA (2015). Physiology of proglucagon peptides: role of glucagon and GLP‐1 in health and disease. Physiol Rev 95: 513–548. [DOI] [PubMed] [Google Scholar]
- Sato H, Genet C, Strehle A, Thomas C, Lobstein A, Wagner A et al. (2007). Anti‐hyperglycemic activity of a TGR5 agonist isolated from Olea europaea . Biochem Bioph Res Co 362: 793–798. [DOI] [PubMed] [Google Scholar]
- Schwartz SS, Epstein S, Corkey BE, Grant SFA, Gavin JR, Aguilar RB (2016). The time is right for a new classification system for diabetes: rationale and implications of the beta‐cell‐centric classification schema. Diabetes Care 39: 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleeman MW, Garcia K, Liu R, Murray JD, Malinova L, Moncrieffe M et al. (2003). Ciliary neurotrophic factor improves diabetic parameters and hepatic steatosis and increases basal metabolic rate in db/db mice. P Natl Acad Sci USA 100: 14297–14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein SA, Lamos EM, Davis SN (2013). A review of the efficacy and safety of oral antidiabetic drugs. Expert Opin Drug Saf 12: 153–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li YL, Wu H, Liu JZ, Hu JX, Liao N et al. (2011). Antidiabetic effect of oleanolic acid: a promising use of a traditional pharmacological agent. Phytother Res 25: 1031–1040. [DOI] [PubMed] [Google Scholar]
- Wang Y, Parlevliet ET, Geerling JJ, van der Tuin SJL, Zhang H, Bieghs V et al. (2014). Exendin‐4 decreases liver inflammation and atherosclerosis development simultaneously by reducing macrophage infiltration. Br J Pharmacol 171: 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZY, Dohle C, Friemann J, Green BS, Gleichmann H (1993). Prevention of high‐dose and low‐dose STZ‐induced diabetes with D‐glucose and 5‐thio‐D‐glucose. Diabetes 42: 420–428. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nakade Y, Yamauchi T, Kobayashi Y, Ishii N, Ohashi T et al. (2016). Glucagon‐like peptide‐1 analogue prevents nonalcoholic steatohepatitis in non‐obese mice. World J Gastroentero 22: 2512–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YL, Hao G, Zhang QY, Hua WY, Wang M, Zhou WJ et al. (2015). Berberine induces GLP‐1 secretion through activation of bitter taste receptor pathways. Biochem Pharmacol 97: 173–177. [DOI] [PubMed] [Google Scholar]
- Yu ZW, Jin TR (2010). New insights into the role of cAMP in the production and function of the incretin hormone glucagon‐like peptide‐1 (GLP‐1). Cell Signal 22: 1–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of DKS26 on the glucose consumption in human hepatic HepG2 Cells. OA represented oleanolic acid, MET represented metformin. Values are expressed as mean ± SEM (n = 3). Compared with control group, * P < 0.05, ** P < 0.01. Glucose consumption = glucose concentrations of blank wells − glucose concentrations of cell plated wells. The glucose consumption values were adjusted by SRB test. This glucose lowering activity in vitro showed that DKS26 might have hypoglycemic effects in vivo.
Figure S2 Effects of DKS26 on plasma total bile acids (TBA) levels in STZ‐induced diabetic mice for 33 days of administration. OA represented oleanolic acid, MET represented metformin. Values are expressed as mean ± SEM. Normal Control (n = 5), STZ control (n = 7) and other groups (n = 8). Compared with normal control group, ## P < 0.01.
Figure S3 Effects of DKS26 on hepatic function and plasma total bile acids (TBA) levels in normal ICR mice for 8 days. OA represented oleanolic acid, MET represented metformin. Values are expressed as mean ± SEM (OA group n = 5 and other groups n = 6). Compared with control group, * P < 0.05, ** P < 0.01, *** P < 0.001. These findings indicated that the effect of DKS26 on hepatic injury in normal mice was significantly lower than OA.
Figure S4 Direct effects of DKS26 on TGR5 in HEK293/pGL4.29/hTGR5 cells. Effect (%) was calculated as relative effect of 20 μM INT‐777 regarded as 100%. EC50 was 1.77 ± 0.45 μM and 13.66 ± 1.81 μM for INT‐777 and OA respectively. OA represented oleanolic acid. Values are expressed as mean ± SD (n = 3).
Figure S5 Effects of DKS26 on gallbladder size in STZ‐induced and db/db diabetic mice. The gallbladders were removed 2 h after the last administration and measured by a vernier calliper or analytical balance. The size of gallbladder was expressed as the cross‐sectional area (length x width, mm2) or weight (mg) and corrected by body weight (g). (A) STZ‐induced diabetic mice (normal control n = 5, STZ control n = 7 and other groups n = 8). (B) Db/db diabetic mice (db/db and metformin control n = 7, and other groups n = 8). OA represented oleanolic acid, MET represented metformin. Values are expressed as mean ± SEM. Compared with normal control group, # P < 0.05, ## P < 0.01.
Table S1 Effects of DKS26 on blood glucose with a single administration in STZ‐induced diabetic mice.
Table S2 The primer sequences used in this study.