

Abstract
Microglial cells are activated in response to different types of injuries or stress in the CNS. Such activation is necessary to get rid of the injurious agents and restore tissue homeostasis. However, excessive activation of microglial cells is harmful and contributes to secondary injury. Pertinently, microglial cell activity was targeted in many preclinical and clinical studies but such strategy failed in clinical trials. The main reason behind the failed attempts is the complexity of the injury mechanisms which needs either a combination therapy or targeting a process that is involved in multiple pathways. Cofilin is a cytoskeleton associated protein involved in actin dynamics. In our previous study, we demonstrated the role of cofilin in mediating neuronal apoptosis during OGD conditions. Previous studies on microglia have shown the involvement of cofilin in ROS formation and phagocytosis. However, additional studies are needed to delineate the role of cofilin in microglial cell activation. Therefore, in the current study, we investigated the role of cofilin in LPS-induced microglial cell activation using cofilin siRNA knockdown paradigms. The viability of differentiated PC12 cells was used as a measure of the neurotoxic potential of conditioned medium derived from cofilin siRNA-transfected and LPS-activated microglial cells. Cofilin knockdown significantly inhibited LPS-induced microglial cell activation through NF-κB and JAK-STAT pathways. The release of proinflammatory mediators (NO, TNF-α, iNOS and COX2) as well as microglial proliferation and migration rates were significantly reduced by cofilin knockdown. Furthermore, differentiated PC12 cells were protected from the neurotoxicity induced by conditioned medium derived from cofilin-transfected and LPS-activated microglial cells. In conclusion, we demonstrated that cofilin is involved in the cascade of microglial cell activation and further validates our previous study on cofilin’s role in mediating neuronal apoptosis. Together, our results suggest that cofilin could present a common target in neurons and microglial cells and might prove to be a promising therapy for different brain injury mechanisms including stroke.
Keywords: Cofilin, Microglia, LPS, Inflammation, Neurotoxicity, ROS
Introduction
Microglia, the resident CNS macrophage, are the first line of defense against various infectious and injurious agents that breach the CNS. To perform their defense mechanism, microglia are equipped with different weapons like phagocytic activity, production of proinflammatory cytokines and chemokines as well as production of cytotoxic reactive oxygen (ROS) and nitrogen species (RNS). Paradoxically, excessive microglial activation is deleterious and is associated with brain injury which aggravates the initial trigger [1].
Different pathologies and different injurious agents elicit microglial cell activation by different mechanisms. For example, ischemia leads to oxygen and glucose deprivation (OGD) which initiates neuronal cell death, ROS and RNS generation, and complement activation. As a consequence, dying cells release many danger-associated molecular patterns including ATP, nucleic acids, neurotransmitters and high mobility group box 1 (HMGB1) into the extracellular environment. These molecules have an important role in mediating microglial cell activation during ischemia [2,3]. In addition to danger signals, many other substances including lipopolysaccharide (LPS), a bacterial endotoxin [4], hemin and thrombin released during hemorrhage [5,6], β-Amyloid (Aβ) [7], interferon γ (IFN)-γ and other proinflammatory cytokines [8] can stimulate microglial activation and contribute to brain injury. However, different stimuli interact differently with the microglial cells and control proinflammatory cytokine expression by regulating the activity of one or more of the major proinflammatory transcription factors namely nuclear fctor kappa B (NF-κB), activator protin 1 (AP-1) and Signal transducer and activator of transcription 1 (STAT1) [9,10]. Such signaling differences contribute to change in the intensity of activation as well as in the identity of the proinflammatory mediators released [11].
Microglial activation is associated with several morphological and functional changes like transforming from a ramified to an amoeboid morphology and upregulation of proinflammatory proteins, with subsequent increase in cell proliferation, migration and phagocytic activity [12]. Furthermore, cytokines and chemokines released from activated microglia induce leukocytes and macrophage infiltration from systemic circulation [13]. Collectively, these cells have an important role in mediating neuroinflammation and subsequent neurotoxicity through releasing different proinflammatory and cytotoxic substances [ROS, RNS, tumot necrosis factor-α (TNF-α), Interleukin-1β (IL-1β), inducible nitric oxide synthease (iNOS), cyclooxygenase-2 (COX2) and various matrix metalloproteinases (MMPs), which induce neuronal and glial cell death as well as blood brain barrier (BBB) disruption [14].
To reduce neuroinflammation and associated neuronal injury, various treatment modalities have been tested targeting different pathways involved in microglial activation [15]. In preclinical studies, these agents were effective in reducing inflammation by inhibiting microglial cell activation. However, none of these agents were effective in clinical trials so far. This is mainly because other brain components like neurons, astrocyte and BBB are also affected directly by injury in addition to the indirect effect mediated by microglial activation. Therefore, targeting microglial cells only will produce limited effect due to inability in preventing the direct effect. Accordingly, identifying and targeting a common pathway involved in neuronal cell death and microglial cell activation during brain injury will be promising.
Cofilin, an actin associated protein, has an important role in maintaining rapid recycling of globular actin (G-actin) monomers by depolymerizing and severing filamentous actin (F-actin) [16]. Multiple regulatory kinases and phosphatases modulate cofilin activity through phosphorylation/dephosphorylation processes. Cofilin is inactivated by phosphorylation at Ser 3 by LIM kinase and TES kinase and reactivated through dephosphorylation by the phosphatases like slingshot (SSH), chronophin and phosphoprotein phosphatases (PP1/PP2A) [17]. Studies have shown that cofilin hyperactivation during stressful conditions leads to both cofilin-actin rods formation and initiation of apoptosis which are involved in many neurodegenerative diseases [18,19]. Similarly, we have shown in our previous study that cofilin inhibition restores the viability of primary cortical neurons subjected to OGD [20]. However, few studies investigated the role of cofilin in microglial cells focusing mainly on a single aspect. Therefore, further information is required on the role of cofilin in the processes associated with microglial activation like morphological changes, migration, proliferation, and proinflammatory mediator expression.
Our objective in the present study is to investigate the role of cofilin in microglial cell activation and associated neurotoxicity. Initially we employed OGD and LPS as a microglial cell activator but we dropped OGD later due to inconsistency in data and used LPS only in subsequent experiment. Cofilin1 siRNA was used to knockdown cofilin in microglial cells and test the impact on LPS induced microglial cell activation, proliferation, migration, phagocytosis and viability. Furthermore, the viability of differentiated PC12 cells was used as a measure for the neurotoxic potential of conditioned medium derived from siRNA transfected and LPS activated microglia.
Materials and Methods
Cell Culture
Spontaneously immortalized microglial cells (SIM-A9 Cells), a generous gift from Dr. Kumi Nakamoto-Combs at the University of North Dakota, were grown in DMEM/F12 medium (HyClone, Thermo scientific, West Palm Beach, FL, USA) supplemented with 5% horse serum (HS), 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. To be used for our experiments, they were plated at the density of 1×106 cells/6-cm plate and 5×104 cells/well in a 24-well plate.
PC12 cells (ATCC, Manassas, VA, USA) were grown in DMEM (HyClone, Thermo scientific, West Palm Beach, FL, USA) supplemented with 10% HS, 5% FBS and 1% penicillin-streptomycin. To induce differentiation of PC12 cells into neurons, cells were plated at the density of 2×104 cells/well in 24 well plate and then incubated overnight. After that, the growth medium was replaced with the differentiation medium (DMEM supplemented with 50 ng/ml nerve growth factor (NGF) and 1% FBS) for a period of 7 days to allow for complete differentiation. To study the impact of cofilin knockdown in LPS activated microglia on the survival of differentiated PC12 cells, a conditioned medium from microglial cells was added to differentiated PC12 cells. After 24 h of incubation, MTT viability assay was conducted.
Oxygen–Glucose Deprivation/Reperfusion Model of Ischemia
In this in vitro model of ischemia, SIM-A9 cells were deprived from oxygen and glucose as the growth medium was replaced with glucose free medium (HBSS phenol red medium) and then placed in oxygen free chamber that was rendered anaerobic by a sachet containing ascorbic acid (AnaeroGenTM, OXOID, Germany). Resazurin, an anaerobic indicator (OXOID, Germany) was used to sense for the oxygen level in the chamber. After that the chamber containing cell culture plate, ascorbic acid sachet and anaerobic indicator was tightly closed and placed in the incubator at 37 °C. The complete lack of oxygen in the chamber is indicated by the change in the color of the indicator from pink to white, and the onset time for OGD was started. In the OGD model, SIM-A9 cells were subjected to 1 h OGD only, whereas for OGD/reperfusion (OGD/R), cells were subjected to 1 h OGD followed by 24 h reperfusion period.
LPS Induced Microglial Activation
LPS (100 ng/ml) was used to activate microglia in all experiments. To study protein expression levels by western blotting (WB), SIM-A9 cells were plated in 6-cm plate and then stimulated with LPS for 24 h before cell lysis. In case of siRNA transfection experiments, cells were transfected with scrambled/cofilin siRNA for 72 h prior to LPS stimulation. To study phosphorylation/activation status of the transcription factors (NF-κB, SAPK/JNK and STAT1), scrambled/cofilin siRNA transfected SIM-A9 cells were stimulated with LPS for 1 h only before cell lysis. In case of MTT assay, NO assay and ELISA assay, SIM-A9 cells were plated in 24-well plate, transfected with siRNA and then stimulated with LPS for 24 h.
MTT-Cell Proliferation and Viability Assay
Cell viability and proliferation were determined using the 3-[4,5-dimethyl- thiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. In this assay, the viability of siRNA transfected SIM-A9 cells treated with OGD and LPS as well as differentiated PC12 cells, treated with microglia conditioned medium, were measured. Furthermore, the proliferation rate of transfected SIM-A9 cells at different posttransfection time intervals (12 h, 60 h and 84 h) was calculated. MTT assay protocol involves incubation of the cells (Microglia or differentiated PC12 cells) with MTT reagent (Promega Corporation, Madison, WI, USA) for 3 h in 5 % CO2 at 37 °C. After that, the whole medium was discarded and DMSO was added to dissolve formazan crystals. Viable cells were quantified by measuring the absorbance at 570 nm.
Nitrite Assay
SIM-A9 cells were plated in 24-well plate and then transfected with scram/cofilin siRNA for 72 h prior to LPS stimulation. Cell culture medium from the respective wells was mixed with equal volume of Griess reagent (Sigma-Aldrich) in 96-well plate at room temperature. The amount of nitric oxide released into cell culture medium was quantified calorimetrically at 540 nm according to the manufacturer instructions.
Morphological Changes Assay
SIM-A9 cells were plated in 24-well plate and then transfected with scram/cofilin siRNA for 72 h. Morphological changes associated with cofilin knockdown was quantified by measuring radius ratio which is equal to maximum radius over minimum radius [21].
ELISA Assay
SIM-A9 cells were plated in 24-well plate, transfected with scrambled/cofilin siRNA and then stimulated with LPS. After that, the levels of TNF-α and IL-1β released into cell culture medium were quantified using commercially available ELISA kits (eBioscience, San Diego, CA, USA) according to the manufacturer protocol.
Scratch migration Assay
SIM-A9 cells were cultured in 24-well plate and then transfected with scrambled/cofilin siRNA for 72 h. By using a sterile 200 μl pipette tip, a scratch was made in the middle of each well followed by washing with PBS to remove damaged cell remnants. At different time intervals (6 h, 12 h and 24 h), cells migrated to the scratched area were imaged and then the percent of area they occupied were measured using Image J software.
Phagocytosis Assay
To test the impact of cofilin knockdown on the phagocytic activity of microglia, SIM-A9 cells were plated in 96-well plate at the density of 1*104 cells/well and then transfected with scram/cofilin siRNA for 72 h. LPS activation was continued for 24 h after transfection followed by incubation with 0.125 mg/ml FITC-conjugated E.coli bioparticles (Life Technologies, Carlsbad, CA, USA) for additional 6 h. The unphagocytosed bioparticles were quenched by washing for 1 minute with 0.25 mg/ml trypan blue and the amount of phagocytosed FITC labeled particles were quantified by measuring fluorescence excitation/emission at 494/518. Furthermore, cells with phagocytosed fluorescent bioparticles were imaged using fluorescent microscope.
Immunocytochemistry
SIM-A9 cells subjected to OGD and LPS treatment were fixed using 4 % paraformaldehyde and then treated with 0.3 % of Triton X-100. After that, cells are blocked with 3% bovine serum albumin and then incubated with rabbit anti-cofilin (1:400; Cell Signaling Technology, Danvers, MA, USA) primary antibody overnight at 4 °C. Texas red-labeled donkey anti-rabbit secondary IgG antibody (1:500; Jackson ImmunoResearch, West Grove, PA, USA) was used at room temperature for 1 h. F-actin staining was performed using one unit of phalloidin (Invitrogen, Carlsbad, CA, USA). After rinsing with PBS, the coverslip was mounted with DAPI (Santa Cruz Biotechnology, Santa Cruz, CA, USA) over the glass slide, and the gap was sealed. Cells were imaged after that using confocal fluorescent microscopy and the corrected total cell fluorescence was measured using image J software.
Subcellular Fractionation and Western Blotting
To obtain subcellular fractions, SIM-A9 cells were harvested using ice- cold lysis buffer [250 mM sucrose, 1.5 MgCl2, 10 mM KCl, 20 mM HEPES (pH 7.5), 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 50 mM NaF, 10 mM Na vanadate, 20 mM Na pyrophosphate and protease inhibitor cocktail (Thermo Scientific)]. Cell homogenates were kept on ice for 15 min and then NP-40 was added at 0.5% for 2 min more. Nuclear pellet was sedimented after that by centrifugation at 14000×g for 10 min. The resultant supernatant represents the cytosolic fraction. Nuclear pellets were resuspended in the nuclear lysis buffer [20 mM HEPES pH 7.9, 20 % glycerol, 0.5 M NaCl, 1.5 mM MgCl2, 1 % Triton-X-100, 1 mM DTT, 1 mM PMSF, 50 mM NaF, 10 mM Na vanadate, 20 mM Na pyrophosphate, and protease inhibitor cocktail (Thermo Scientific)]. Nuclear suspension was kept on ice for 30 min and then centrifuged at 9000×g for 30 min to obtain nuclear fractions. Protein concentrations were determined using Bradford reagent (Bio-Rad Laboratories, CA, USA), and samples were analyzed by loading equivalent amounts of protein (5–50 μg) onto 10–15 % SDS-polyacrylamide gels. Proteins separated on the gels were transferred onto a pretreated PVDF membrane and were blocked with 3% BSA for 1 h to prevent nonspecific binding. Afterward, the membrane was incubated overnight at 4 °C with the following primary antibodies: rabbit anti-phosphocofilin (1:1000; Abcam, Cambridge, MA, USA), rabbit anti-cofilin, rabbit anti-α-tubulin, rabbit anti-histone H3, rabbit anti-phospho-SAPK/JNK, rabbit anti-NF-κB p-65, and rabbit anti-phospho-STAT1 (1:2000; Cell Signaling Technology, Danvers, MA, USA). Following the incubation, the blots were washed and incubated with horseradish peroxidase (HRP) conjugated goat anti-rabbit secondary antibody (1:6000; Jackson ImmunoResearch) for 1 h at room temperature. α-Tubulin was used as a loading control for cytosolic proteins, and histone H3 for nuclear proteins. The images were analyzed using Bio-Rad ChemiDoc™ XRS Image Lab Software.
Cofilin siRNA Microglial Transfection
SiGENOME SMARTpool mouse cofilin1 siRNA with the following sequences AGACAAGGACUGCCGCUAU, GGUGGCAGCGCCGUCAUUU, GUUCGCAAGUCUUCAACAC, and GAGAAUGCACCCCUCAAGA was ordered from GE Healthcare Dharmacon, Inc. Furthermore, scrambled siRNA was used as a control. SIM-A9 cells were plated at the density of 2×105 cells/ml and then transfected with scrambled/cofilin1 siRNA at 25 and 50 nM concentrations. Xfect transfection reagent (Clontech Laboratories, Inc) was used to perform the transfection experiment according to the manufacturer’s instructions. Briefly, in two separate sets of microtubes, siRNA and xfect transfection reagent were diluted using Xfect buffer. Meanwhile, the transfection reagent was added to the siRNA and kept at room temperature for 20 min to ensure complex formation. After that, transfection mix was added to the cell culture and then incubated back for 72 h before starting OGD and LPS treatment.
Statistical Analysis
The experimental results were expressed as the mean ± SEM and are accompanied by the number of observations (independent preparations of cultured cells). Data were analyzed by ANOVA followed by Newman-Keuls using GraphPad Prism (GraphPad Software, San Diego, CA). Furthermore, student’s unpaired t-test was used to determine significant differences between two groups. A value of p<0.05 is considered to be statistically significant.
Results
Cofilin is highly activated during OGD, OGD/R and LPS treatment
To investigate cofilin activation status in SIM-A9 cells during OGD, OGD/R and LPS treatment, cofilin and phosphocofilin expression were tested by WB. Although cofilin expression did not show any change (Fig.1A), phosphocofilin expression was significantly reduced (P < 0.05) during OGD, OGD/R and LPS treatment relative to total cofilin and α-tubulin (Fig. 1 B and C, respectively). Furthermore, LPS treatment induced highly significant cofilin activation (P < 0.05) compared with OGD/R group (Fig. 1 B and C). Compared to OGD/R group, the trend of cofilin activation was higher with OGD (Fig. 1 B and C) and therefore OGD condition only, together with LPS will be used to activate microglia in the subsequent experiments. To test for any possibility of translocation between nuclear and cytosolic fraction, nuclear cofilin level was tested. Among the treatment groups tested, no significant changes in nuclear cofilin levels were detected with regard to control group (Fig. 1 D). To confirm our western blotting findings, an immunocytochemical staining of cofilin in SIM-A9 cells stimulated with OGD and LPS was conducted. In unstimulated SIM-A9 cells, cofilin was localized mainly in the cytosol at cell periphery (Fig 1 E, control). However, microglial cells activation with OGD and LPS induced cofilin redistribution in the whole cellular area (Fig 1 E, OGD and LPS). Analysis of the total cofilin immunofluorescence intensity with image J software showed statistically non-significant differences (figure not shown).
Figure 1. Phosphocofilin and cofilin expression levels in response to OGD, OGD/R and LPS.
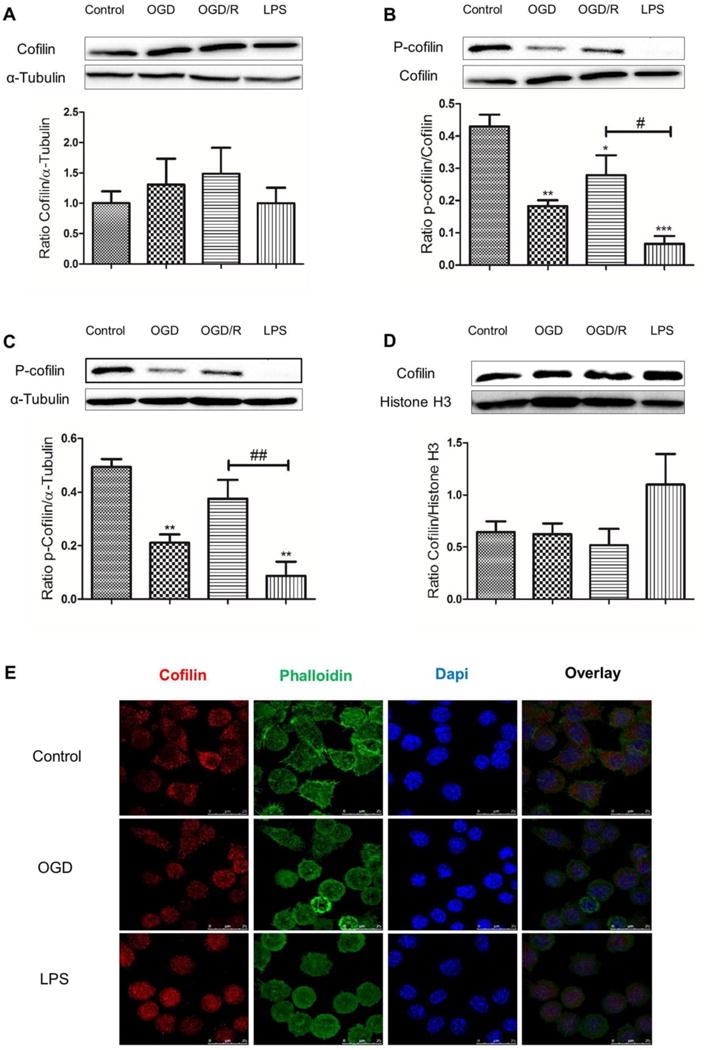
SIM-A9 microglial cells were treated with OGD for 1 h, OGD/R for 24 h and LPS 100 ng/ml for 24 h and then protein expression level was evaluated by WB. Although cytosolic (A) and nuclear (D) total cofilin level did not show any significant change in all treatment groups, p-cofilin level was significantly downregulated relative to total cofilin (B) and α-tubulin (C). Furthermore, p-cofilin level was significantly downregulated in LPS treated group compared with OGD/R (B and C). α- tubulin and histone H3 were used as a loading control for cytosolic and nuclear proteins, respectively. E. Representative immunoflorescence pictures show cofilin (stained red) is localized predominantly in the cell periphery. After OGD and LPS treatment, cofilin is redistributed in the whole cell area. Scale bar is 25 μm. Data are expressed as mean±SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 relative to control group. #P < 0.05, ##P < 0.01, ###P < 0.001 relative to OGD/R treated groups.
Cofilin knockdown changed Microglial cells morphology
Since we detected significant activation of cofilin during OGD, OGD/R and LPS treatment, our next step was to inhibit cofilin activity. Because no cofilin inhibitor is available so far, we used cofilin siRNA technique to knockdown cofilin and then test the impact of LPS and OGD challenge on microglial cell activation. In the beginning, two control groups were used with and without scrambled siRNA, but later we continued with scrambled siRNA only since no difference was detected (data not shown). Anyway, cofilin siRNA at 25 and 50 nM concentrations showed highly significant reduction in cofilin expression level (P < 0.01). However, no significant difference was detected between the two concentrations used (P = 0.26) although the trend with 50 nM concentration was more (Fig. 2 A). Therefore, cofilin siRNA at 50 nM concentration was used in the subsequent experiments. Additionally, cofilin siRNA-transfected cells were almost round in shape compared to scrambled siRNA-transfected cells (Fig 2 C). Such cofilin associated morphological changes was highlighted by measuring the radius ratio which was significantly less (P < 0.05) in cofilin siRNA transfected cells (Fig. 2 B).
Figure 2. Cofilin knockdown by siRNA and its associated effect on microglial cell morphology.
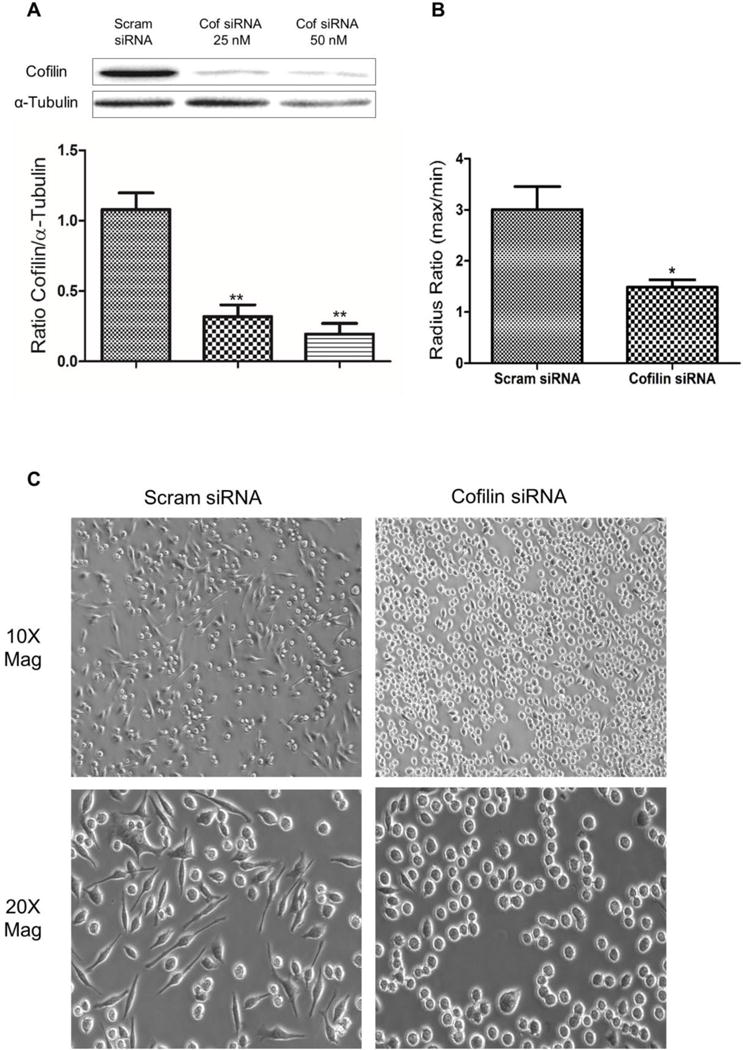
A. Microglial cells were transfected with scrambled/cofilin siRNA at 25 and 50 nM concentration for 72 h and then harvested for WB analysis. Cofilin siRNA at 25 and 50 nM concentrations reduced cofilin expression level significantly 72 h posttransfection. Although no significant difference was detected between the two concentrations used, the trend with cofilin siRNA 50 nM was higher. α- tubulin was used as a loading control for cytosolic proteins. B and C. Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM for 72 h and then the changes in cell morphology associated with cofilin knockdown were imaged (C) and quantified (B). Cofilin knockdown reduced the radius ratio (maximum radius over minimum radius) significantly relative to scrambled littermate (B). Furthermore, cofilin transfected cells imaged at 10× and 20× magnification were almost round in shape while their control showed different morphology (C). The 20× magnification images were used to measure the radius ratio in B. Data are expressed as mean±SEM of three independent experiments. *P < 0.05, **P < 0.01, relative to scrambled siRNA control group.
Cofilin knockdown reduces both microglial cell proliferation as well as LPS induced microglial cell death
Our experimental design is shown in Fig. 3 A. However, during inflammatory reaction activated microglia undergo proliferation and activation induced cell death. Therefore, our objective in this experiment was to test the effect of cofilin knockdown on microglial cell proliferation as well as LPS and OGD-induced cell death using MTT assay. Microglial cell proliferation at 60 h and 84 h posttransfection was significantly reduced (P < 0.001) by cofilin knockdown as shown in Fig. 3 B. However, no significant change in cell proliferation was detected 12 h postransfection. Such highly significant reduction in cell proliferation with cofilin knockdown was considered in the subsequent experiments. Regarding LPS and OGD-induced cell death, cofilin-transfected cells were resistant to cytotoxic LPS effect as shown in Fig. 3 C. In OGD experiment, both scrambled and cofilin transfected cells showed the same susceptibility to OGD and no significant difference was detected between them (Fig. 3 D).
Figure 3. The effect of cofilin knockdown on microglial cells proliferation and viability.
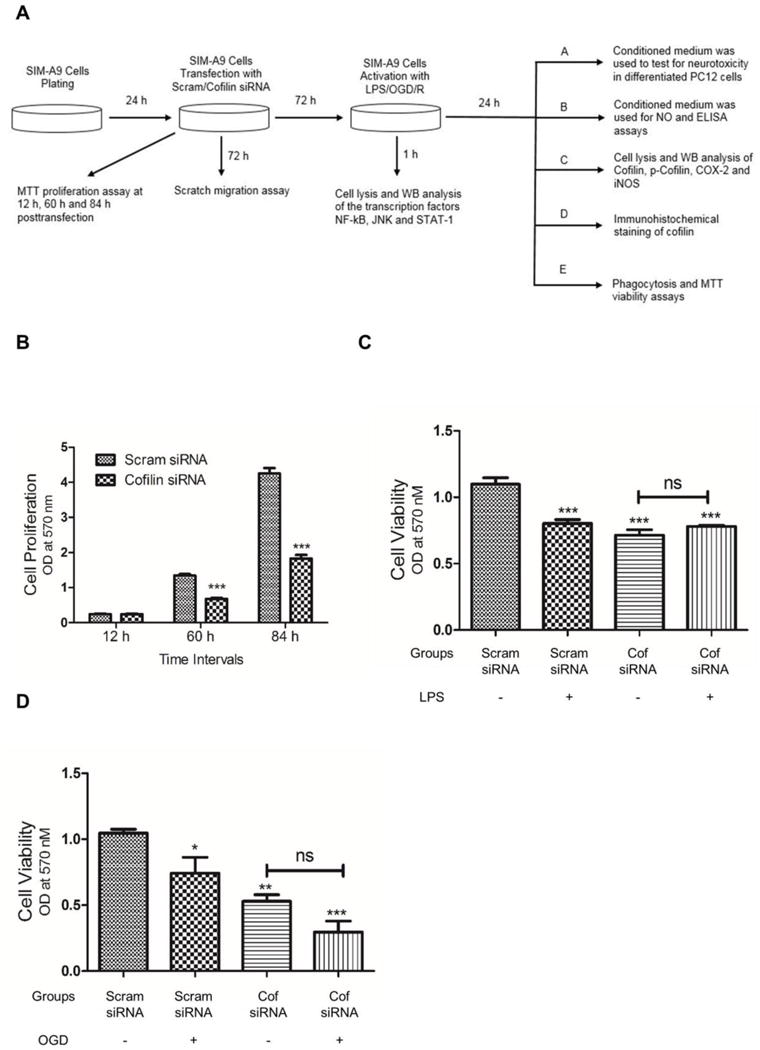
A. Summary of the experimental design. B. Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM concentration and then MTT proliferation assay was performed after 12 h, 60 h and 84 h. While the cell number was almost same for cofilin and scrambled groups after 12 h, cofilin knockdown reduced the cell number significantly after 60 and 84 h. C and D. Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM concentration for 72 h and then treated with LPS (100 ng/ml) for 24 h (C) or OGD for 1 h (D) followed by MTT viability assay. LPS induced significant microglial cells death in scrambled siRNA transfected group but not in cofilin group (C). Similarly, OGD induced significant cell death in scrambled but not in cofilin group (D). As shown in B, cofilin knockdown reduced cell numbers in all treatment groups (C and D). The data was expressed as Mean±SEM of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 relative to scrambled siRNA group without treatment (LPS and OGD). ns: non-significant.
Cofilin knockdown inhibits LPS induced microglial cell activation
Activated microglial cells release and express different proinflammatory substances which have an important role in mediating neuroinflammation. Accordingly, our next objective was to test the impact of cofilin knockdown on LPS-induced microglial cells activation. Using NO release assay, cofilin knockdown significantly inhibited LPS-induced NO release (P < 0.001), shown in Fig 4 A. Furthermore, considering cell density by dividing over MTT values did not change the result significantly (data not shown). However, OGD did not show any increase in NO release (Fig 4 B). Therefore, only LPS was used as a microglial cells activator in the subsequent experiments. Next, the effect of cofilin knockdown on LPS-induced iNOS and COX2 expression was tested by WB. The expression level of iNOS and COX2 was significantly reduced by cofilin knockdown as shown in Fig. 4 C and D, respectively. Using ELISA assay, the level of TNF-α but not IL-1β was significantly reduced (P < 0.001) by cofilin knockdown in LPS-stimulated groups (Fig E and F).
Figure 4. The effect of cofilin knockdown on LPS induced microglial cells activation.
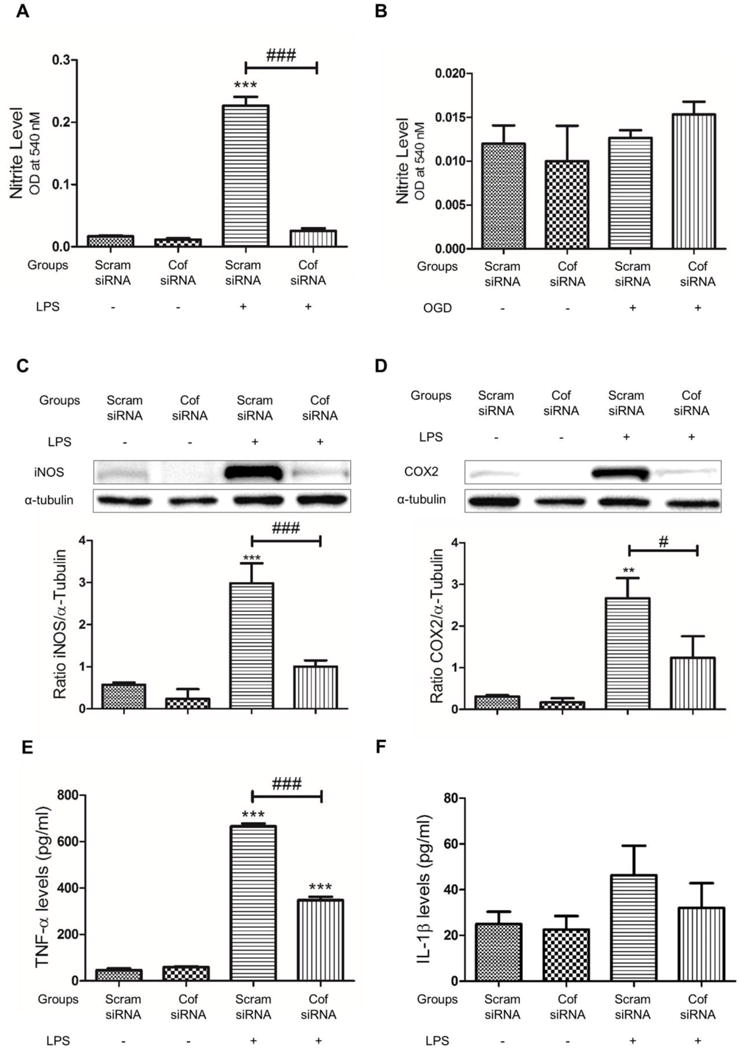
A and B. Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM concentration for 72 h and then treated with LPS (100 ng/ml) or OGD for 1 h. Nitrite release assay was done after that by mixing 100 μl of cell culture medium with 100 μl of griess reagent. Relative to LPS treated control, NO release was significantly reduced by cofilin knockdown. Furthermore, no significant difference in NO release was detected between LPS untreated groups (scrambled and cofilin) and LPS treated cofilin transfected group (A). In OGD treated groups, no significant increase in NO release was detected (B). C–F. Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM concentration for 72 h and then treated with LPS (100 ng/ml) for further 24 h. Cell culture medium was used for ELISA assay of TNF-α and IL-1β (E and F) and cell lysate was used for WB analysis of iNOS and COX2 expression (C and D). Relative to LPS stimulated control, cofilin knockdown reduced LPS induced iNOS (C) and COX2 (D) expression significantly. Furthermore, no significant differences in iNOS and COX2 levels was detected between LPS untreated groups (scrambled and cofilin) and LPS treated cofilin transfected group. α-tubulin was used as a loading control. (E and F). In ELISA assay, LPS treatment increased TNF-α level significantly in both groups (scrambled and cofilin transfected), compared to unstimulated scrambled control group. In LPS treated groups, TNF-α level was significantly less in cofilin transfected group (E). Similarly, IL-1β release was also reduced by cofilin knockdown but the reduction did not reach the level of significance (P = 0.2) (F). Data are represented as Mean±SEM of three independent experiments. **P < 0.01, ***P < 0.001 relative to scrambled siRNA group without LPS treatment. #P < 0.05, ###P < 0.001 relative to scrambled transfected and LPS treated group.
Cofilin knockdown inhibits microglial cell migration and modulate phagocytosis
Other characteristics of activated microglial cells include migration and phagocytosis, to get rid of the injurious agent. Scratch assay was conducted to test the effect of cofilin knockdown on microglial cells migration. At different time intervals, the number of microglial cells migrated to the scratched area (represented by the percent of area occupied) was significantly less (P < 0.01) in cofilin siRNA-transfected cells (Fig 5 A and B). Different time periods (6 h, 12 h and 24 h) were considered to reduce the impact of cell proliferation in the scratched area. Next, the effect of cofilin knockdown on microglial cells phagocytic activity was tested using FITC-labeled Ecoli bioparticles. Cofilin knockdown reduced microglial cells phagocytic activity significantly (P < 0.01) in both LPS-stimulated and unstimulated groups. However, LPS-stimulated group exhibited highly significant (P < 0.05) phagocytic activity compared with unstimulated one (Fig 5 C). Furthermore, the fluorescent intensity in LPS-activated groups (scrambled and cofilin siRNA) was higher than that in unstimulated groups (Fig 5 E). If we consider the damaging effect of LPS and the proliferation inhibitory effect of cofilin knockdown by dividing FITC fluorescence over MTT absorbance, phagocytic activity will be significantly higher (P < 0.01) in LPS-stimulated and cofilin-transfected group (Fig 5 D). LPS activation also increased phagocytic activity in scrambled siRNA-transfected cells although such increment was statistically non-significant (Fig 5 D).
Figure 5. The effect of cofilin knockdown on microglial cells migration and phagocytic activity.
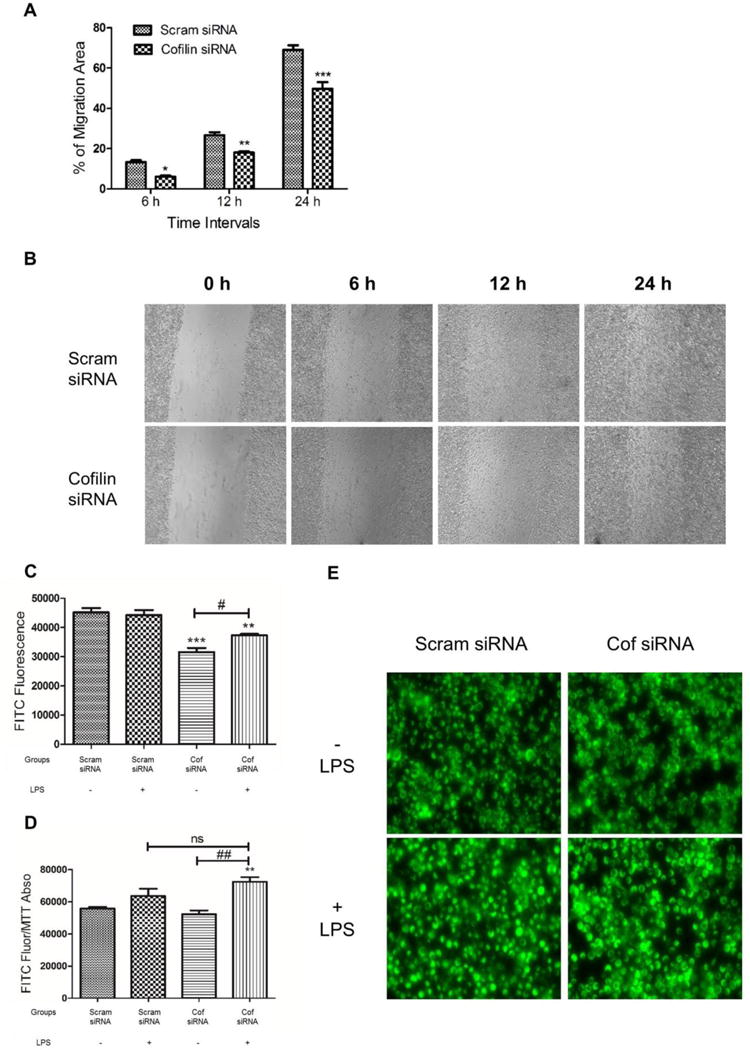
Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM concentration for 72 h and then a scratch was made in the middle of each well of 24-well plate. Microglial cells migrated to the scratched area were imaged at 20× magnification (B) and then the percentage of area occupied by migrated cells was calculated at 6 h, 12 h and 24 h (A) using image J software. Cofilin knockdown reduced microglial cell migration significantly at all time intervals tested (A). C–E. Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM concentration for 72 h and then treated with LPS (100 ng/ml) for 24 h more. After that FITC-labeled Ecoli bioparticles was added at 0.125 mg/ml and incubated for 6 h. Trypan blue was used to quench unphagocytosed particles and the fluorescence intensity of phagocytosed particles was imaged using fluorescent microscope (E) and quantified using Synergy H1 Hybrid Reader at fluorescence excitation/emission of 494/518 (C and D). Cofilin knockdown reduced microglial cell phagocytic activity significantly (C). However, by dividing FITC- fluorescence over MTT absorbance (to consider cell density), phagocytic activity was higher in cofilin transfected and LPS treated group (D). In general, LPS treatment increased phagocytic activity in all groups as shown in D and E. Furthermore, in cofilin transfected groups, phagocytic activity was significantly higher in LPS treated one (C and D). Data are expressed as mean±SEM of three independent experiments. *P <0.05, **P < 0.01, ***P < 0.001 relative to scrambled siRNA group without LPS treatment. #P < 0.05, ##P < 0.01 relative to cofilin transfected and LPS untreated group. ns: non-significant.
Cofilin knockdown in microglial cells protects differentiated PC12 cells from the neurotoxic conditioned medium of LPS-activated microglia
To test the neurotoxic potential of the activated microglia, a conditioned medium of siRNA-transfected and LPS-activated cells was added to differentiated PC12 cells and incubated for 24 h. The viability of treated PC12 cells was measured after that using MTT assay. The conditioned medium derived from microglia transfected with scrambled siRNA induced highly significant neurotoxicity (P < 0.01) in differentiated PC12 cells. However, no signs of toxicity were detected in cells treated with conditioned medium derived from cofilin-transfected microglia as shown in Fig 6.
Figure 6. The effect of conditioned medium derived from cofilin transfected and LPS treated microglia on differentiated PC12 cells viability.
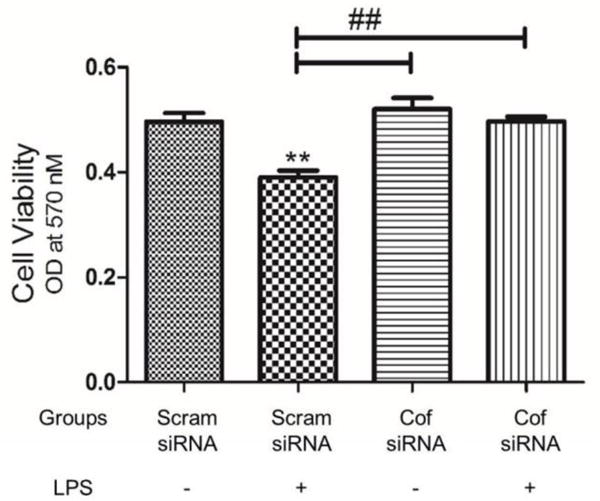
Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM concentration for 72 h and then treated with LPS (100 ng/ml) for 24 h more. Afterwards microglial cells conditioned medium was added to differentiated PC12 cells and incubated for 24 h. MTT viability assay was done to assay the neurotoxic potential of the conditioned medium. Differentiated PC12 cells viability was significantly reduced by treatment with the conditioned medium derived from scrambled transfected and LPS activated microglia. However, cofilin knockdown in microglial cells protect differentiated PC12 cells from the neurotoxic effect of the conditioned medium. Data are expressed as mean±SEM of three independent experiments. **P < 0.01 relative to scrambled siRNA group without LPS treatment. ##P < 0.01 relative to scrambled transfected and LPS treated group.
Cofilin knockdown inhibits LPS-induced NF-κB and STAT1 activation
As we showed in Fig 4, cofilin knockdown inhibited LPS-induced microglial cells activation by reducing NO, TNF-α and IL-1β secretion as well as iNOS and COX2 expression. However, cofilin regulatory mechanism on microglial cells activation need to be investigated. Accordingly, we tested the effect of cofilin knockdown on LPS-induced activation of the major transcription factors involved in microglial cells activation namely NF-κB, JNK and STAT1. Although cofilin knockdown did not show any significant effect on the expression level of the cytosolic NF-κB (Fig 7 A), nuclear translocation was significantly inhibited (P < 0.001) as shown in Fig 7 B. Similarly, stat1 activation was significantly inhibited (P < 0.01) by cofilin knockdown (Fig 7 D). However, active p-JNK did not change significantly by cofilin knockdown (Fig 7 C).
Figure 7. The effect of cofilin knockdown in microglia on LPS induced NF-κB, JNK and STAT1 activation.
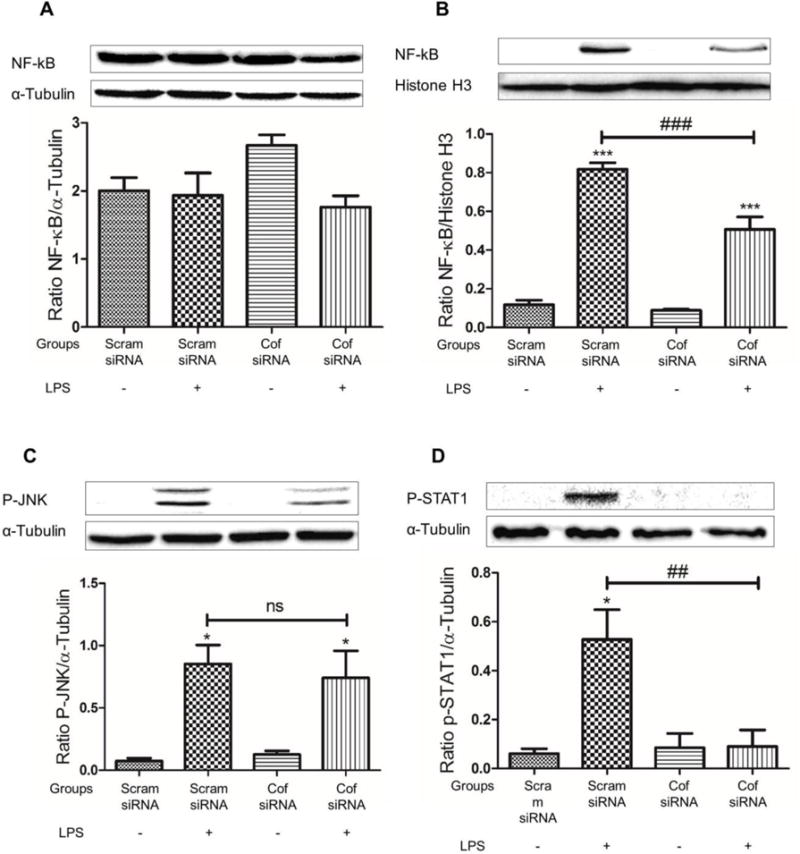
Microglial cells were transfected with scrambled/cofilin siRNA at 50 nM concentration for 72 h and then treated with LPS (100 ng/ml) for another hour. Protein expression levels for cytosolic NF-κB (A), nuclear NF-κB (B), p-JNK (C) and p-STAT1 (D) was evaluated using WB. Relative to LPS treated and scrambled transfected group, cofilin knockdown reduced nuclear translocation of NF-κB significantly (B). STAT1 activation was also significantly reduced by cofilin knockdown (D). However, no significant effect for cofilin knockdown on JNK activation (C) and cytosolic NF-κB expression (A) was detected. Data are expressed as mean±SEM of three independent experiments. *P < 0.05, ***P < 0.001 relative to scrambled siRNA group without LPS treatment. ##P < 0.01, ###P < 0.001 relative to scrambled transfected and LPS treated group. ns: non-significant.
Discussion
In our previous study, we highlighted the major role of active cofilin in mediating neuronal cell apoptosis triggered by OGD and chemical stressors [20]. Similarly, our present study emphasized the key role of active cofilin in mediating microglial cell activation triggered by LPS. All-together, our previous and present study identified cofilin as a common target in neurons and microglial cells and its inhibition might be a promising therapeutic option for different acute and chronic brain injuries [22].
Microglia – the guardians of the brain – maintains CNS homeostasis and integrity through their diverse and highly sensitive surface receptors. Normally, microglia survey and sense the external environment and clear any encountered foreign body or damaged cellular debris through their phagocytic activity [23]. Additionally, microglia regulate adult neurogenesis and synaptic plasticity [24,25]. However, excessive microglial activation or microgliosis is detrimental and involved in the pathogenesis of many acute brain injuries like stroke and spinal cord injury as well as chronic neurodegenerative diseases like Alzheimer’s disease, Huntington disease (HD), Parkinson’s disease and amyotrophic lateral sclerosis [26,27]. Furthermore, recent studies have shown that reactive microglia are also involved in cognitive impairment and psychiatric disorders [28]. Such consequences that follow microglial activation are mediated mainly by overexpression of cytotoxic proinflammatory cytokines, proteases and generation of ROS/RNS [14]. These cytotoxic agents target not only neurons but also other brain components including astrocytes, oligodendrocytes and BBB and aggravate the initial injury [29–31]. Accordingly, microglia are the major player in neuroinflammation and its targeting represent one of the important neuroprotective therapeutic strategies [32].
Phosphatases like SSH and chronophin are activated in response to different intracellular and extracellular signals like ROS [33], elevation in intracellular Ca2+ [34] and drop in intracellular ATP [35], resulting in cofilin activation and actin dynamic disruption. Therefore, cofilin activation in microglial cells subjected to OGD (Figure 1 B and C) is mainly attributed to OGD-induced ATP drop and ROS formation [36]. On the other hand, it has been shown that treatment of alveolar epithelial cells, lymphocytes, dendritic cells and macrophages with LPS induces actin reorganization and cofilin activation [37–39]. In lymphocytes, induction of SSH mediates LPS induced cofilin activation [40,38]. In a similar way, SSH may be involved in LPS induced cofilin dephosphorylation and activation in microglial cells (Fig 1 B and C).
Through controlling actin dynamics, cofilin has an important role in maintaining cell shape and morphology, proliferation, migration and phagocytic activity [41,42]. In resting cells, cofilin is localized to the regions with high actin turnover like leading edge of the plasma membrane [42,43]. Upon cell stimulation, cofilin is activated and redistributed in the cytoplasm and other compartments leading to change in cell morphology [44,45]. Similarly, cofilin knockdown is associated with the increased F-actin amounts and change in cell shape [42]. Such evidences are in concordance with our immunohistochemical findings that show cofilin redistribution and round cell morphology upon stimulation with OGD and LPS. The same pattern was found in our cofilin knockdown experiment which resulted in almost round cell morphology of microglia compared to scrambled littermate.
Actin has a key role in cell division and proliferation through constructing the contractile ring at the cleavage furrow [46]. In the later stage of cell division, cofilin is activated and mediates F-actin disassembly which is an essential step in cytokinesis [47,48]. Cofilin also has an important role in cell migration process through controlling the formation of lamellipodial protrusions [49]. In agreement with our findings in figure 3, n-cofilin null macrophage exhibited impairment in proliferation and migration [42]. Furthermore, primary microglia isolated from early postnatal HD mice exhibited reduction in cofilin expression and impairment in migration [50]. The correlation between phagocytosis and actin dynamics is well established [51] but the nature of this correlation still controversial. Some reports stated that cofilin inhibition enhances phagocytic activity [42,52] where as others reported reverse trend [53,54]. However, our findings showed that cofilin knockdown enhances phagocytic activity at the cellular level and reduces it at the whole culture level. This is mainly because of reduction in cell proliferation which is illustrated by decreased number of phagocytosing cells.
Beside its function in maintaining cell morphology, motility, migration and proliferation, actin is also involved in different signaling pathways [55,56]. The role of actin cytoskeleton in signal transduction is well studied using actin targeting natural compounds [57]. Latrunculin B and cytochalasin D, F-actin depolymerizing agents, enhance cytokine production in T lymphocyte through activation of nuclear factor of activated T cells (NFAT) [56]. Conversely, Jasplakinolide, F-actin stabilizing agent, inhibits T cells signaling [58]. Additionally, it has been shown that cofilin regulates nuclear translocation of NF-κB in thrombin induced endothelial cell inflammation [59]. In another recent study, SSH1 depletion impairs pathogen induced NOD1 signaling through inhibiting NF-κB activation [60]. Such evidences confirm the direct correlation between actin and cofilin and NF-κB activation. It has also been shown that LPS induces microglial cell activation via NF-κB, JAK-STAT and JNK stress kinase pathways [30]. However, the role of cofilin in LPS induced activation of JNK and STAT1 is still unclear. Interestingly, our findings showed that cofilin knockdown in microglia inhibited two (NF-κB and STAT1) out of three induced transcription factors which consequently lead to highly significant reduction in the levels of NO, iNOS, COX2 and TNF-α. Such reduction in these mediators may justify the protective effect of cofilin knockdown on LPS induced microglia cell death [61] as well as microglial conditioned medium induced differentiated PC12 cells death [62]. Accordingly, inhibiting two basic transcription factors involved in microglial cell activation may be a novel therapeutic strategy for different microglia associated CNS injuries [63]. However, further future in vivo studies are warranted to test the impact of cofilin inhibition on different models of microglia induced inflammatory diseases. Furthermore, the role of cofilin in astrocytes and BBB endothelial cells is still unknown and therefore future studies are recommended.
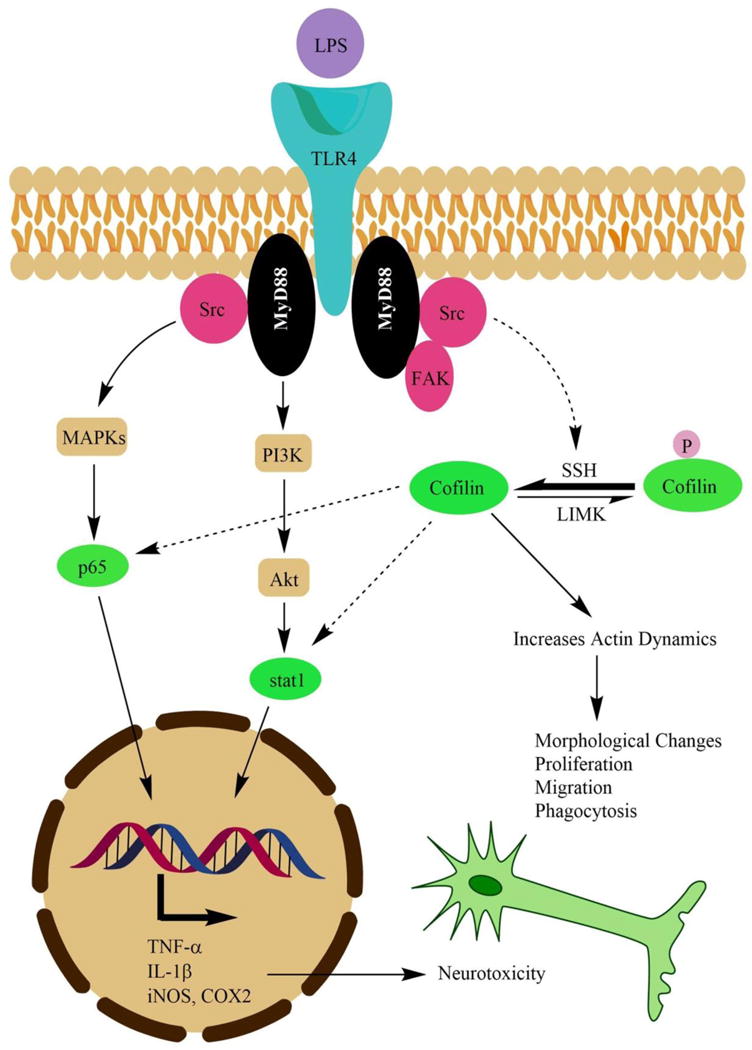
The proposed mechanism that clarifies the active role of cofilin in mediating LPS effect to proinflammatory transcription factors and actin cytoskeleton is illustrated in Fig 8. The figure demonstrates that Src kinase mediates the effect of LPS from toll like receptor 4 (TLR4) to NF-κB, STAT1 and cofilin activation [64,53]. Regarding Src effect on cofilin activation, active Src works together with the recruited focal adhesion kinase (FAK) to activate paxillin which can activate cofilin indirectly [53,65]. However, activation of phosphatases like SSH or inhibition of kinases like LIMK is the proposed mechanism by which Src kinase induces cofilin activity. Nevertheless, active cofilin enhances actin dynamics and lead to changes in cell morphology, proliferation, migration and phagocytic activity. Furthermore, active cofilin regulate nuclear translocation of NF-κB [66] but STAT1 activation mechanism by cofilin is unknown and need further investigation. Altogether, activated proinflammatory transcription factors induce the expression of wide range of neurotoxic proinflammatory mediators which results in neurotoxicity and neurodegeneration.
Figure 8. The proposed role of cofilin in mediating LPS stimulatory effect on microglia.
| Active Pathway |
|
| Inactive Pathway |
|
In conclusion, cofilin mediates LPS induced microglial cell activation and associated neurotoxicity which is significantly inhibited by cofilin knockdown. Furthermore, our current findings supported by our previous study recognized cofilin as a common target in neurons and microglial cells and its targeting will be a promising therapeutic option for different CNS injuries.
Acknowledgments
The study was partly funded by a grant from NIH-National Complimentary and integrative health (R00AT004197) and start-up funds from The University of Toledo to ZAS. Qasim Alhadidi was supported by the Higher Committee for Education Development in Iraq (www.hcediraq.org). We would like to thank Dr. Kumi Nagamoto-Combs from the University of North Dakota for the generous gift of SIM-A9 cells. The content is solely the responsibility of the authors and does not necessarily represent the official views of funding agencies.
References
- 1.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173(6):3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 2.Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. doi: 10.1038/nm.2399. doi:nm.2399 [pii] 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hur J, Lee P, Kim MJ, Kim Y, Cho YW. Ischemia-activated microglia induces neuronal injury via activation of gp91phox NADPH oxidase. Biochem Biophys Res Commun. 2010;391(3):1526–1530. doi: 10.1016/j.bbrc.2009.12.114. [DOI] [PubMed] [Google Scholar]
- 4.Dolga AM, Letsche T, Gold M, Doti N, Bacher M, Chiamvimonvat N, Dodel R, Culmsee C. Activation of KCNN3/SK3/K(Ca)2.3 channels attenuates enhanced calcium influx and inflammatory cytokine production in activated microglia. Glia. 2012;60(12):2050–2064. doi: 10.1002/glia.22419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee DY, Oh YJ, Jin BK. Thrombin-activated microglia contribute to death of dopaminergic neurons in rat mesencephalic cultures: dual roles of mitogen-activated protein kinase signaling pathways. Glia. 2005;51(2):98–110. doi: 10.1002/glia.20190. [DOI] [PubMed] [Google Scholar]
- 6.Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY, Yang QW. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation. 2012;9:46. doi: 10.1186/1742-2094-9-46. doi:1742-2094-9-46 [pii]10.1186/1742-2094-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jana M, Palencia CA, Pahan K. Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol. 2008;181(10):7254–7262. doi: 10.4049/jimmunol.181.10.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozenfeld C, Martinez R, Seabra S, Sant’anna C, Goncalves JG, Bozza M, Moura-Neto V, De Souza W. Toxoplasma gondii prevents neuron degeneration by interferon-gamma-activated microglia in a mechanism involving inhibition of inducible nitric oxide synthase and transforming growth factor-beta1 production by infected microglia. Am J Pathol. 2005;167(4):1021–1031. doi: 10.1016/s0002-9440(10)61191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng KW, Yu Q, Liao LX, Song FJ, Lv HN, Jiang Y, Tu PF. Anti-Neuroinflammatory Effect of MC13, a Novel Coumarin Compound From Condiment Murraya, Through Inhibiting Lipopolysaccharide-Induced TRAF6-TAK1-NF-kappaB, P38/ERK MAPKS and Jak2-Stat1/Stat3 Pathways. J Cell Biochem. 2015;116(7):1286–1299. doi: 10.1002/jcb.25084. [DOI] [PubMed] [Google Scholar]
- 10.Babu R, Bagley JH, Di C, Friedman AH, Adamson C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurgical focus. 2012;32(4):E8. doi: 10.3171/2012.1.FOCUS11366. [DOI] [PubMed] [Google Scholar]
- 11.Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol. 2013;2013:746068. doi: 10.1155/2013/746068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colton C, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9(2):174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44. doi: 10.1016/j.pneurobio.2013.11.003. doi:S0301-0082(13)00127-5 [pii] 10.1016/j.pneurobio.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 15.Rangarajan P, Eng-Ang L, Dheen ST. Potential drugs targeting microglia: current knowledge and future prospects. CNS Neurol Disord Drug Targets. 2013;12(6):799–806. doi: 10.2174/18715273113126660175. [DOI] [PubMed] [Google Scholar]
- 16.Bernstein BW, Bamburg JR. ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 2010;20(4):187–195. doi: 10.1016/j.tcb.2010.01.001. doi:S0962-8924(10)00002-4 [pii] 10.1016/j.tcb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 2008;87(8–9):649–667. doi: 10.1016/j.ejcb.2008.04.001. doi:S0171-9335(08)00069-1 [pii] 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Cichon J, Sun C, Chen B, Jiang M, Chen XA, Sun Y, Wang Y, Chen G. Cofilin aggregation blocks intracellular trafficking and induces synaptic loss in hippocampal neurons. J Biol Chem. 2012;287(6):3919–3929. doi: 10.1074/jbc.M111.301911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Posadas I, Perez-Martinez FC, Guerra J, Sanchez-Verdu P, Cena V. Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J Neurochem. 2012;120(4):515–527. doi: 10.1111/j.1471-4159.2011.07599.x. [DOI] [PubMed] [Google Scholar]
- 20.Madineni A, Alhadidi Q, Shah ZA. Cofilin Inhibition Restores Neuronal Cell Death in Oxygen-Glucose Deprivation Model of Ischemia. Mol Neurobiol. 2014 doi: 10.1007/s12035-014-9056-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong ZY, Shi XR, Zhu K, Wu TT, Zhu YZ. SCM-198 inhibits microglial overactivation and attenuates Abeta(1-40)-induced cognitive impairments in rats via JNK and NF-small ka, CyrillicB pathways. J Neuroinflammation. 2014;11:147. doi: 10.1186/s12974-014-0147-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alhadidi Q, Bin Sayeed MS, Shah ZA. Cofilin as a Promising Therapeutic Target for Ischemic and Hemorrhagic Stroke. Translational stroke research. 2016;7(1):33–41. doi: 10.1007/s12975-015-0438-2. [DOI] [PubMed] [Google Scholar]
- 23.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 24.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7(4):378–391. doi: 10.1016/j.nurt.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012;1(1):9. doi: 10.1186/2047-9158-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frick LR, Williams K, Pittenger C. Microglial dysregulation in psychiatric disease. Clinical & developmental immunology. 2013;2013:608654. doi: 10.1155/2013/608654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol. 1993;151(4):2132–2141. [PubMed] [Google Scholar]
- 30.Kacimi R, Giffard RG, Yenari MA. Endotoxin-activated microglia injure brain derived endothelial cells via NF-kappaB, JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond) 2011;8:7. doi: 10.1186/1476-9255-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. 2006;37(4):1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 32.Kim JY, Kim N, Yenari MA. Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci Ther. 2015;21(4):309–319. doi: 10.1111/cns.12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JS, Huang TY, Bokoch GM. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Molecular biology of the cell. 2009;20(11):2650–2660. doi: 10.1091/mbc.E09-02-0131. doi:E09-02-0131 [pii] 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Shibasaki F, Mizuno K. Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J Biol Chem. 2005;280(13):12683–12689. doi: 10.1074/jbc.M411494200. [DOI] [PubMed] [Google Scholar]
- 35.Huang TY, Minamide LS, Bamburg JR, Bokoch GM. Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Dev Cell. 2008;15(5):691–703. doi: 10.1016/j.devcel.2008.09.017. doi:S1534-5807(08)00399-7 [pii] 10.1016/j.devcel.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gouix E, Buisson A, Nieoullon A, Kerkerian-Le Goff L, Tauskela JS, Blondeau N, Had-Aissouni L. Oxygen glucose deprivation-induced astrocyte dysfunction provokes neuronal death through oxidative stress. Pharmacol Res. 2014;87:8–17. doi: 10.1016/j.phrs.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Verdijk P, van Veelen PA, de Ru AH, Hensbergen PJ, Mizuno K, Koerten HK, Koning F, Tensen CP, Mommaas AM. Morphological changes during dendritic cell maturation correlate with cofilin activation and translocation to the cell membrane. Eur J Immunol. 2004;34(1):156–164. doi: 10.1002/eji.200324241. [DOI] [PubMed] [Google Scholar]
- 38.Freeman SA, Jaumouille V, Choi K, Hsu BE, Wong HS, Abraham L, Graves ML, Coombs D, Roskelley CD, Das R, Grinstein S, Gold MR. Toll-like receptor ligands sensitize B-cell receptor signalling by reducing actin-dependent spatial confinement of the receptor. Nat Commun. 2015;6:6168. doi: 10.1038/ncomms7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Isowa N, Xavier AM, Dziak E, Opas M, McRitchie DI, Slutsky AS, Keshavjee SH, Liu M. LPS-induced depolymerization of cytoskeleton and its role in TNF-alpha production by rat pneumocytes. Am J Physiol. 1999;277(3 Pt 1):L606–615. doi: 10.1152/ajplung.1999.277.3.L606. [DOI] [PubMed] [Google Scholar]
- 40.Freeman SA, Lei V, Dang-Lawson M, Mizuno K, Roskelley CD, Gold MR. Cofilin-mediated F-actin severing is regulated by the Rap GTPase and controls the cytoskeletal dynamics that drive lymphocyte spreading and BCR microcluster formation. J Immunol. 2011;187(11):5887–5900. doi: 10.4049/jimmunol.1102233. [DOI] [PubMed] [Google Scholar]
- 41.Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science. 2009;326(5957):1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jonsson F, Gurniak CB, Fleischer B, Kirfel G, Witke W. Immunological responses and actin dynamics in macrophages are controlled by N-cofilin but are independent from ADF. PloS one. 2012;7(4):e36034. doi: 10.1371/journal.pone.0036034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maizels Y, Oberman F, Miloslavski R, Ginzach N, Berman M, Yisraeli JK. Localization of cofilin mRNA to the leading edge of migrating cells promotes directed cell migration. J Cell Sci. 2015;128(10):1922–1933. doi: 10.1242/jcs.163972. [DOI] [PubMed] [Google Scholar]
- 44.Samstag Y, Eckerskorn C, Wesselborg S, Henning S, Wallich R, Meuer SC. Costimulatory signals for human T-cell activation induce nuclear translocation of pp19/cofilin. Proc Natl Acad Sci U S A. 1994;91(10):4494–4498. doi: 10.1073/pnas.91.10.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Rheenen J, Condeelis J, Glogauer M. A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci. 2009;122(Pt 3):305–311. doi: 10.1242/jcs.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heng YW, Koh CG. Actin cytoskeleton dynamics and the cell division cycle. Int J Biochem Cell Biol. 2010;42(10):1622–1633. doi: 10.1016/j.biocel.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 47.Nagaoka R, Abe H, Kusano K, Obinata T. Concentration of cofilin, a small actin-binding protein, at the cleavage furrow during cytokinesis. Cell Motil Cytoskeleton. 1995;30(1):1–7. doi: 10.1002/cm.970300102. [DOI] [PubMed] [Google Scholar]
- 48.Abe H, Obinata T, Minamide LS, Bamburg JR. Xenopus laevis actin-depolymerizing factor/cofilin: a phosphorylation-regulated protein essential for development. J Cell Biol. 1996;132(5):871–885. doi: 10.1083/jcb.132.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiuchi T, Nagai T, Ohashi K, Mizuno K. Measurements of spatiotemporal changes in G-actin concentration reveal its effect on stimulus-induced actin assembly and lamellipodium extension. J Cell Biol. 2011;193(2):365–380. doi: 10.1083/jcb.201101035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R, Miller A, Weiss A, Giorgini F, Cheah C, Moller T, Stella N, Akassoglou K, Tabrizi SJ, Muchowski PJ. Mutant huntingtin impairs immune cell migration in Huntington disease. J Clin Invest. 2012;122(12):4737–4747. doi: 10.1172/JCI64484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.May RC, Machesky LM. Phagocytosis and the actin cytoskeleton. J Cell Sci. 2001;114(Pt 6):1061–1077. doi: 10.1242/jcs.114.6.1061. [DOI] [PubMed] [Google Scholar]
- 52.Adachi R, Takeuchi K, Suzuki K. Antisense oligonucleotide to cofilin enhances respiratory burst and phagocytosis in opsonized zymosan-stimulated mouse macrophage J774.1 cells. The Journal of biological chemistry. 2002;277(47):45566–45571. doi: 10.1074/jbc.M207419200. doi:10.1074/jbc.M207419200 M207419200 [pii] [DOI] [PubMed] [Google Scholar]
- 53.Gitik M, Kleinhaus R, Hadas S, Reichert F, Rotshenker S. Phagocytic receptors activate and immune inhibitory receptor SIRPalpha inhibits phagocytosis through paxillin and cofilin. Front Cell Neurosci. 2014;8:104. doi: 10.3389/fncel.2014.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hadas S, Spira M, Hanisch UK, Reichert F, Rotshenker S. Complement receptor-3 negatively regulates the phagocytosis of degenerated myelin through tyrosine kinase Syk and cofilin. J Neuroinflammation. 2012;9:166. doi: 10.1186/1742-2094-9-166. doi:1742-2094-9-166 [pii] 10.1186/1742-2094-9-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kustermans G, El Benna J, Piette J, Legrand-Poels S. Perturbation of actin dynamics induces NF-kappaB activation in myelomonocytic cells through an NADPH oxidase-dependent pathway. Biochem J. 2005;387(Pt 2):531–540. doi: 10.1042/BJ20041318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rivas FV, O’Keefe JP, Alegre ML, Gajewski TF. Actin cytoskeleton regulates calcium dynamics and NFAT nuclear duration. Mol Cell Biol. 2004;24(4):1628–1639. doi: 10.1128/MCB.24.4.1628-1639.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kustermans G, Piette J, Legrand-Poels S. Actin-targeting natural compounds as tools to study the role of actin cytoskeleton in signal transduction. Biochem Pharmacol. 2008;76(11):1310–1322. doi: 10.1016/j.bcp.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 58.Shen A, Puente LG, Ostergaard HL. Tyrosine kinase activity and remodelling of the actin cytoskeleton are co-temporally required for degranulation by cytotoxic T lymphocytes. Immunology. 2005;116(2):276–286. doi: 10.1111/j.1365-2567.2005.02222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fazal F, Bijli KM, Minhajuddin M, Rein T, Finkelstein JN, Rahman A. Essential role of cofilin-1 in regulating thrombin-induced RelA/p65 nuclear translocation and intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells. The Journal of biological chemistry. 2009;284(31):21047–21056. doi: 10.1074/jbc.M109.016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bielig H, Lautz K, Braun PR, Menning M, Machuy N, Brugmann C, Barisic S, Eisler SA, Andree M, Zurek B, Kashkar H, Sansonetti PJ, Hausser A, Meyer TF, Kufer TA. The cofilin phosphatase slingshot homolog 1 (SSH1) links NOD1 signaling to actin remodeling. PLoS Pathog. 2014;10(9):e1004351. doi: 10.1371/journal.ppat.1004351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mayo L, Stein R. Characterization of LPS and interferon-gamma triggered activation-induced cell death in N9 and primary microglial cells: induction of the mitochondrial gateway by nitric oxide. Cell Death Differ. 2007;14(1):183–186. doi: 10.1038/sj.cdd.4401989. [DOI] [PubMed] [Google Scholar]
- 62.Dai XJ, Li N, Yu L, Chen ZY, Hua R, Qin X, Zhang YM. Activation of BV2 microglia by lipopolysaccharide triggers an inflammatory reaction in PC12 cell apoptosis through a toll-like receptor 4-dependent pathway. Cell Stress Chaperones. 2015;20(2):321–331. doi: 10.1007/s12192-014-0552-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ivanenkov YA, Balakin KV, Lavrovsky Y. Small molecule inhibitors of NF-kB and JAK/STAT signal transduction pathways as promising anti-inflammatory therapeutics. Mini reviews in medicinal chemistry. 2011;11(1):55–78. doi: 10.2174/138955711793564079. [DOI] [PubMed] [Google Scholar]
- 64.Socodato R, Portugal CC, Domith I, Oliveira NA, Coreixas VS, Loiola EC, Martins T, Santiago AR, Paes-de-Carvalho R, Ambrosio AF, Relvas JB. c-Src function is necessary and sufficient for triggering microglial cell activation. Glia. 2015;63(3):497–511. doi: 10.1002/glia.22767. [DOI] [PubMed] [Google Scholar]
- 65.Chen YJ, Hsieh MY, Chang MY, Chen HC, Jan MS, Maa MC, Leu TH. Eps8 protein facilitates phagocytosis by increasing TLR4-MyD88 protein interaction in lipopolysaccharide-stimulated macrophages. The Journal of biological chemistry. 2012;287(22):18806–18819. doi: 10.1074/jbc.M112.340935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leonard A, Marando C, Rahman A, Fazal F. Thrombin selectively engages LIM kinase 1 and slingshot-1L phosphatase to regulate NF-kappaB activation and endothelial cell inflammation. Am J Physiol Lung Cell Mol Physiol. 2013;305(9):L651–664. doi: 10.1152/ajplung.00071.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]