

Abstract
Spinal muscular atrophy (SMA) is caused by the low levels of survival motor neuron (SMN) protein and is characterized by motor neuron degeneration and muscle atrophy. Respiratory failure causes death in SMA but the underlying molecular mechanism is unknown. The zinc finger protein ZPR1 interacts with SMN. ZPR1 is down regulated in SMA patients. We report that ZPR1 functions downstream of SMN to regulate HoxA5 levels in phrenic motor neurons that control respiration. Spatiotemporal inactivation of Zpr1 gene in motor neurons down-regulates HoxA5 and causes defects in the function of phrenic motor neurons that results in respiratory failure and perinatal lethality in mice. Modulation in ZPR1 levels directly correlates and influences levels of HoxA5 transcription. In SMA mice, SMN-deficiency causes down-regulation of ZPR1 and HoxA5 that result in degeneration of phrenic motor neurons. Identification of ZPR1 and HoxA5 as potential targets provides a paradigm for developing strategies to treat respiratory distress in SMA.
Introduction
Spinal muscular atrophy (SMA) is a neurodegenerative disease caused by mutation of the survival of motor neuron 1 (SMN1) gene. In humans, a duplicated copy of the SMN2 gene produces insufficient levels of full-length SMN protein due to alternative splicing that causes skipping of the last coding exon (exon7) and results in the truncated transcript1. The low levels of SMN result in degeneration of spinal motor neurons that causes muscle atrophy followed by symmetric limb paralysis and respiratory distress. Patients with severe SMA require ventilation for life support. Death of SMA patients is caused by respiratory failure2, 3. The molecular mechanism of respiratory failure caused by SMN deficiency in SMA is unknown. Currently, there is no treatment to prevent or alleviate respiratory distress in SMA.
ZPR1 is evolutionary conserved and ubiquitously expressed in mammalian cells4–6. ZPR1 binds to inactive receptor tyrosine kinases (RTKs), epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) receptors, translocates from the cytoplasm to the nucleus in response to mitogen or serum treatment and is a component of downstream signaling of RTKs4, 7. The function of ZPR1 is unclear. Biochemical studies have shown that ZPR1 interacts with SMN protein and is required for accumulation of SMN in the sub-nuclear bodies, gems and Cajal bodies (CB)8, 9. The severity of SMA disease negatively correlates with the number of SMN containing nuclear bodies10. ZPR1 may contribute to the nuclear function of SMN in RNA biogenesis such as snRNP assembly11 and splicing9 associated with sub-nuclear bodies12, 13. Interaction of ZPR1 with SMN is disrupted in SMA patients that have SMN1 mutations9. SMA patients express low levels of ZPR1. Reduced expression of Zpr1 increases severity of SMA disease and Zpr1 is a modifier of SMA14, 15. The low levels of ZPR1 in mice with SMA-like disease have been shown to increase the severity of disease and reduce the lifespan of SMA mice14.
Disruption of the Zpr1 gene in mice results in embryonic lethality8. However, the function of ZPR1 in mammalian embryogenesis and development is unknown. In this study, we show that the Zpr1 is critical for the development and function of the respiratory motor neuron system. Mutation of Zpr1 in motor neurons during early embryogenesis causes progressive loss of phrenic motor neurons and impairs the function of phrenic nerve that result in diaphragmatic paralysis, and respiratory failure, and stillbirth of mice. Temporal analysis of ZPR1 function unraveled, (a) selective vulnerability of phrenic motor neurons to the low levels of ZPR1 and (b) sustained Zpr1 gene activity is critical for the function of respiratory motor neurons. Molecular analysis shows that ZPR1 deficiency in motor neurons causes deregulation of HoxA5 expression. Modulation in ZPR1 levels directly correlates and influences levels of HoxA5 transcription. In SMA mice, SMN-deficiency causes deregulation of ZPR1 and HoxA5 and results in degeneration of phrenic motor neurons that regulate respiration16. Expression of HoxA5 is required for the normal development and function of the phrenic motor neuron system17. Results of genetic and molecular analysis suggest that the ZPR1 functions downstream of SMN to regulate HoxA5 levels in phrenic motor neurons. ZPR1 binds to HoxA5 gene promoter and regulates its transcription. These findings provide insight into the molecular mechanism of respiratory failure in SMA and identify ZPR1 and HoxA5 as potential therapeutic targets for developing therapies to reduce the burden of respiratory distress in SMA.
Results
Zpr1 mutation in motor neurons causes anatomical defects and respiratory failure
Reduced Zpr1 gene dosage in mice has been shown to cause progressive loss of motor neurons, gait abnormalities and defects in the peripheral nervous system18. However, the function of ZPR1 in growth, differentiation and survival of motor neurons is unknown. To gain insight into the function of ZPR1, we created mice with a conditional Zpr1 allele by flanking exon1 with loxP sites, Zpr1 F1/F1 (Supplementary Figure S1). To examine the function of ZPR1 in motor neurons, we generated mutant mice in which Zpr1 was selectively mutated in motor neurons by crossing conditional Zpr1 F1/F1 mice with transgenic Mnx1-Cre (Cre driven by Mnx1 or Hlxb9 or Hb9 promoter) mice. The Hb9 gene expresses during early embryogenesis (E8.5–9.5) in the spinal cord motor neuron and required for consolidation of motor neuron identity during embryonic development19. Mutant mice with homozygous deletion of Zpr1 exon 1 (Zpr1 Δ1/Δ1) in motor neurons, in the presence of transgene Hb9-Cre, are referred as (Zpr1 Hb9MNΔ).
The Zpr1 mutation in motor neurons causes defects in posterior axial growth and anatomical abnormalities. Mutant mice were born without tail, were cyanotic and found dead at birth (Fig. 1a). Anatomical defect suggested that there might also be defects in the skeleton development. Skeleton analysis of E18.5 embryos with X-ray (Supplementary Figure S2a,b) and staining with alcian blue and alizarin red shows that the skeleton is smaller but the skull of mutant (Zpr1 Hb9MNΔ) embryos is larger than control (Supplementary Figure S2c), however, the total numbers of vertebrae for the cervical, thoracic and lumbar regions of the spinal cord were same in mutant and control mice (Supplementary Figure S2d). Interestingly, skeleton defects noted in the sacral region, including underdeveloped ischium bones and fused sacral vertebrae (Supplementary Figure S2d–f) show similarity to congenital defects found in autosomal dominant Currarino syndrome (also known as hereditary sacral agenesis (HSA) or caudal regression syndrome) patients that have HLXB9 mutation20, 21. Examination of thoracic rib cage showed smaller sternum with reduced ossification of lower sternebrae (Supplementary Figure S2g,h). However, the presence of anatomical and skeleton defects Zpr1 Hb9MNΔ mice did not justify the severity that would lead to perinatal lethality. To determine whether death occurred in utero or at birth, we examined E18.5 embryos. Normal embryos were alive and started breathing, in contrast, rarely mutant embryos attempted to breathe but failed to initiate breathing and died within minutes (Supplementary Movie S1). This observation suggests that most of the mutant Zpr1 Hb9MNΔ mice died in utero and respiratory failure may be the cause of death.
Figure 1.
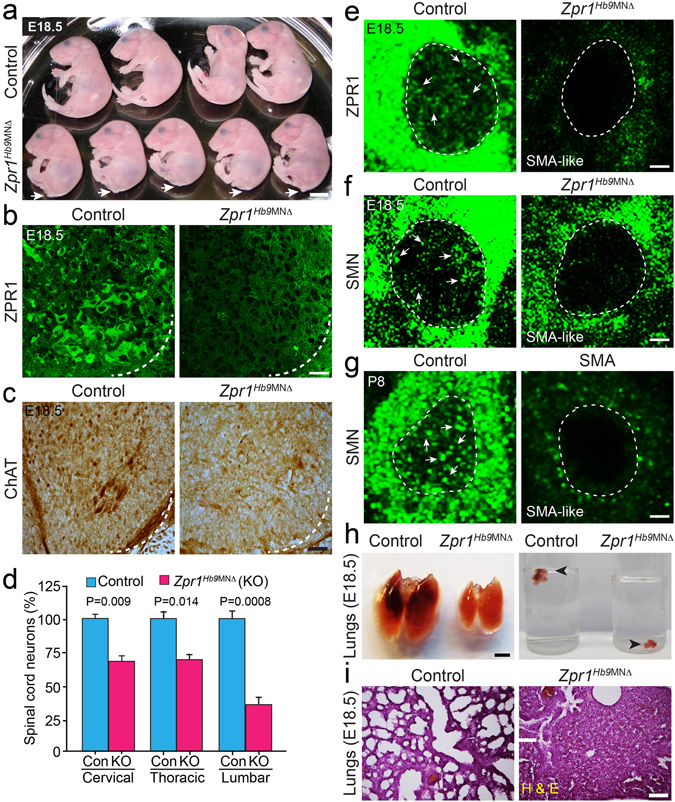
Mutation of the Zpr1 gene in motor neurons causes developmental defects and respiratory failure in Zpr1 Hb9MNΔ mice. (a) Photograph of E18.5 control and Zpr1 Hb9MNΔ embryos showing developmental defects in mutant embryos, including smaller size and loss of tail. Arrows indicate the absence of tail in mutant embryos. All mutant Zpr1 Hb9MNΔ mice were cyanotic and dead at birth (stillbirth). Scale bar is 5 mm. (b) Spinal cord sections from the thoracic region (T9-T12) of E18.5 control and Zpr1 Hb9MNΔ embryos were stained with monoclonal antibody against murine ZPR1 (Clone LG-D5). Zpr1 mutation in motor neurons results in reduced ZPR1 expression in spinal cord neurons of mutant embryos. Scale bar is 25 μm. (c) ZPR1 deficiency in motor neurons results in the loss spinal cord neurons. Spinal cord sections stained with antibody against choline acetyl transferase (ChAT). Scale bar is 100 μm. (d) Loss of neurons (mean ± s.e.m.; n = 3 mice/group) in different region of the spinal cords from Zpr1 Hb9MNΔ embryos, cervical (31.96 ± 6.74%, p = 0.009), thoracic (30.52 ± 7.38%, p = 0.014) and lumbar (64.46 ± 7.10%, p = 0.0008) regions. (e) Zpr1 mutation causes reduction in the cytoplasmic and nuclear staining of ZPR1 and (f) SMN in spinal cord neurons and show SMA-like phenotype. (g) Spinal cord motor neurons from SMA mice show phenotype similar to Zpr1 Hb9MNΔ mice. Arrows indicate accumulation of ZPR1 and SMN in sub-nuclear bodies in control neurons but sub-nuclear bodies missing in Zpr1 mutant and SMA mice. Dotted arcs represent the orientation of view from the anterior horn side of the spinal cord. Scale bar is 5 μm. (h) Lungs from E18.5 Zpr1 Hb9MNΔ embryos were smaller than control and lungs of Zpr1 Hb9MNΔ embryos immediately sank in water, indicating lungs were not inflated with air. Scale bar is 2.5 mm. (i) Histological analysis shows defects in morphogenesis of lungs from Zpr1 Hb9MNΔ embryos and lungs were collapsed. Scale bar is 200 μm.
ZPR1 deficiency causes degeneration of motor neurons and defects in the development of the respiratory system
To investigate the effect of ZPR1 deficiency on motor neurons, we examined the spinal cord motor neurons in control and Zpr1 Hb9MNΔ mice. ZPR1 staining of the spinal cord sections show reduced expression of ZPR1 in Zpr1 Hb9MNΔ mice compared to control mice (Fig. 1b, quantitative analysis presented in Fig. 8). Next, we examined the effect of ZPR1 deficiency on the number of motor neuron in different regions of the spinal cord. Spinal cord sections stained with choline acetyltransferase (ChAT) show reduced number of neurons in the ventral horns (Fig. 1c). Neuron count analysis shows the loss of neurons in the cervical (31.96 ± 6.74%, p = 0.009), thoracic (30.52 ± 7.38%, p = 0.014) and lumbar (64.46 ± 7.10%, p = 0.000) regions of the spinal cord from E18.5 Zpr1 Hb9MNΔ embryos compared to control embryos (Fig. 1d), suggesting that ZPR1 is required for neuron growth and survival. Increased loss of neurons in the lumbar region supports anatomical defects, loss of tail and sacral agenesis, found in Zpr1 Hb9MNΔ mice. Noticeably, the primary pathogenesis in SMA is the loss of motor neurons in the lumbar region. Further examination of motor neurons shows marked reduction in cytoplasmic and nuclear staining of ZPR1 and absence of ZPR1+ sub-nuclear bodies in neurons of mutant mice compared to control mice (Fig. 1e). ZPR1 deficiency causes reduced staining of SMN and the loss of SMN+ nuclear bodies in motor neurons and results in a SMA-like phenotype (Fig. 1f). Comparison of SMN stained motor neurons from Zpr1 Hb9MNΔ and SMA mice (Fig. 1f,g) shows ZPR1 deficiency results in a phenotype similar to SMA.
Figure 8.
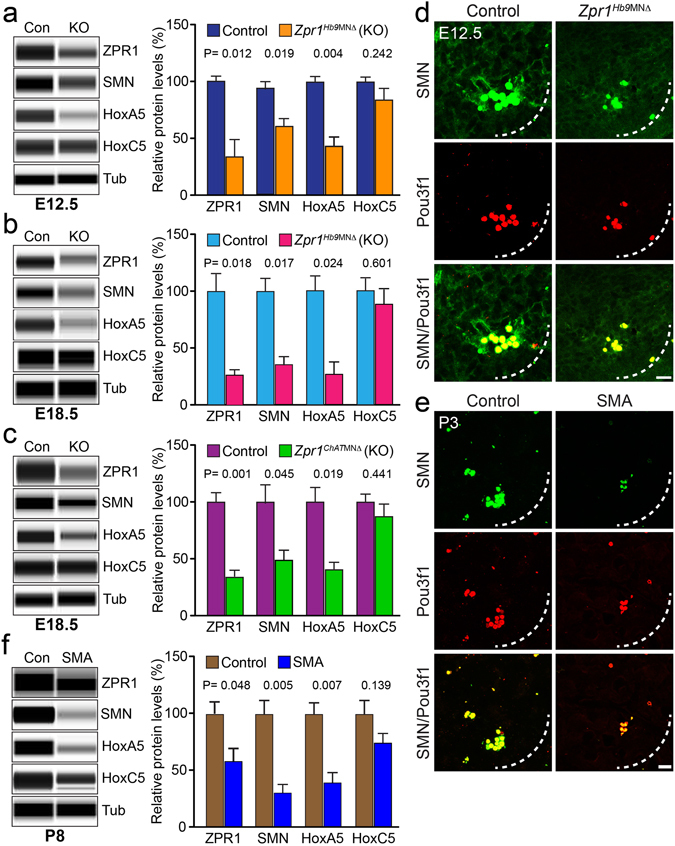
Deregulation of ZPR1 results in down regulation of HoxA5 that causes respiratory distress in Zpr1 mutant and SMA mice. Protein levels of ZPR1, SMN, HoxA5 and HoxC5 were examined in the spinal cords from control and mutant E12.5 and E18.5 embryos using automated Protein Wes System (ProteinSimple) and quantification was performed using Compass software. Quantitative analysis (mean ± s.e.m., n = 3 mice/group) of protein levels show that Zpr1 mutation resulted in reduced protein levels in (a) E12.5 embryos ZPR1 (47.44 ± 11.98%, p = 0.012), SMN (73.13 ± 5.14%, p = 0.019), HoxA5 (43.62 ± 8.84%, p = 0.004) and HoxC5 (87.23 ± 8.81%, p = 0.242) and (b) E18.5 embryos ZPR1 (27.67 ± 4.97%, p = 0.010), SMN (35.33 ± 7.86%, p = 0.011), HoxA5 (29.00 ± 10.44%, p = 0.024) and HoxC5 (89.00 ± 14.22%, p = 0.637) of Zpr1 Hb9MNΔ mice compared to control mice. (c) Protein levels in Zpr1 ChATMNΔ mice also shows reduced levels for ZPR1 (33.67 ± 3.75%, p = 0.001), SMN (50.33 ± 8.21%, p = 0.045), HoxA5 (42.33 ± 5.84%, p = 0.019) and HoxC5 (88.00 ± 12.17%, p = 0.441) compared to control at E18.5 stage. (d) Spinal cord sections of the cervical region (C3-C5) from E12.5 control and Zpr1 Hb9MNΔ embryos were stained with Pou3f1 and SMN antibodies. Immunofluorescence analysis shows ZPR1-deficiency causes loss of SMN+ and Pou3f1+ phrenic motor neurons in Zpr1 Hb9MNΔ mice. (e) Cervical region spinal cord sections from 3-days old (P3) severe SMA mice show that SMN-deficiency causes loss of SMN+ and Pou3f1+ phrenic motor neurons similar to ZPR1-deficiency in Zpr1 Hb9MNΔ mice. Scale bar is 10 μm. (f) Quantification of protein levels in the spinal cords from P8 SMAΔ7 mice show that SMN deficiency (69.10 ± 12.42%, p = 0.005) reduces protein levels to: ZPR1 (58.87 ± 10.43%, p = 0.048), HoxA5 (39.41 ± 9.03%, p = 0.007) and HoxC5 (74.90 ± 7.06%, p = 0.139) proteins in SMAΔ7 mice compared to control mice (Full-length blots are included in Supplementary Figures S8 and S9).
To investigate the cause of death as respiratory failure in Zpr1 Hb9MNΔ mice, first we examined lungs. The lungs from mutant Zpr1 Hb9MNΔ mice were smaller compared to control and failed to float on the surface upon submerging into water (Fig. 1h), suggesting that lungs from mutant mice were not inflated with air. Histochemical staining of lungs from Zpr1 Hb9MNΔ mice shows defects in lungs morphogenesis such as lack of alveolization (Fig. 1i). These findings suggest that the normal levels of ZPR1 in motor neurons are required for lungs development during mammalian embryogenesis. Similar defects in lungs development have been reported in mice with mutation of Hox5 group of genes that result in respiratory failure and death of mice17, 22, 23. These findings indicate that the Zpr1 and Hox5 genes may be components of common pathway involved in the development of mammalian respiratory system. Lack of air in lungs suggests that diaphragmatic paralysis might be the cause of respiratory failure in Zpr1 Hb9MNΔ mice.
Defects in the phrenic nerve development and innervation of diaphragm in Zpr1Hb9MNΔ mice
To further investigate the cause of respiratory failure, we examined the effect of Zpr1 mutation on the development of phrenic nerves and innervation of the diaphragm. Diaphragms were stained with neurofilament (NF) and α-bungarotoxin (BTX) and NF and synaptophysin (SYN) from control and Zpr1 Hb9MNΔ mice. Examination of whole mount diaphragm from E18.5 embryos shows severe defect in the development of phrenic nerves in Zpr1 Hb9MNΔ mice. Defects included absence of secondary and tertiary branches; only a single primary branch was detected on one side of the diaphragm in most severe cases (Fig. 2a,b). Longitudinal spinal cord sections containing C3, C4 vertebrae show marked reduction in the diameter of three phrenic nerve branches exiting from the spinal cord in Zpr1 Hb9MNΔ mice compared to control mice (Fig. 2c,d). Comparison of diameter of primary nerve in the diaphragm between control (21.44 ± 0.017 μm) and Zpr1 Hb9MNΔ (5.88 ± 0.028 μm) shows marked reduction (72.23 ± 3.27%, p = 0.000) that is consistent with the loss of neurons in the cervical region of the spinal cord (Fig. 1d). Further, examination of diaphragm stained with NF and BTX shows severe defects in branching of phrenic nerve and innervation of neuromuscular junctions (NMJs) suggesting that the phrenic nerve was able to innervate diaphragm muscle but neurons failed to generate sufficient secondary and tertiary branches to innervate NMJs in Zpr1 Hb9MNΔ mice (Fig. 2e–h). ZPR1 deficiency resulted in degeneration of secondary and tertiary nerve branches in mutant mice (Fig. 2h). Examination of neuromuscular synapses shows co-localization of NF and SYN in NMJs in control mice (Fig. 2i,k). In contrast, diaphragms from mutant Zpr1 Hb9MNΔ mice showed a severe lack of synapse formation, SYN failed to localize to NMJs and was accumulated in primary branch of phrenic nerve (Fig. 2i–l). These data suggest that defects in synapse formation by phrenic nerve may be the cause of diaphragmatic paralysis that leads to respiratory failure and perinatal mortality in Zpr1 Hb9MNΔ mice.
Figure 2.
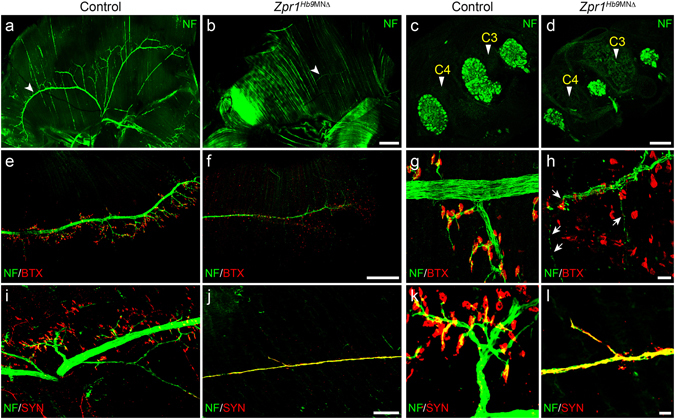
Defects in the development of phrenic nerve and diaphragm innervation in Zpr1 Hb9MNΔ mice. (a,b) Whole diaphragms from E18.5 embryos were stained with neurofilament (NF) antibody. Images shown are composite of individual panels acquired by TileScan using confocal microscope. Immunofluorescence image analysis revealed defects in the development of phrenic nerve in Zpr1 Hb9MNΔ embryos (arrowheads indicate phrenic nerve). (c,d) Analysis of longitudinal sections of spinal cord shows cross section of three bundles of respiratory motor neurons exiting from the C3-C5 region of the spinal cord that form phrenic nerve (arrowheads indicate cervical vertebrae). ZPR1 deficiency results in marked reduction in the size of all three bundles of motor axons, indicating ZPR1 is required for the development of phrenic motor neurons. (e,h) Analysis of diaphragms stained with NF and α-bungarotoxin (BTX) shows defects in the innervation of neuromuscular junctions (NMJs) by motor neurons in mutant embryos. The phrenic nerve reached diaphragm muscle but failed to generate secondary and tertiary branches and innervate NMJs in mutant embryos. Phrenic nerve degeneration was detected in mutant embryos (arrows). (i-l) Staining with NF and synaptophysin (SYN) show severe defects in the formation synapse in mutant embryos, a likely cause of stillbirth of Zpr1 Hb9MNΔ mice. Scale bars are 500 μm (a,b), 250 μm (c,d,e,f), 100 μm (i,j) and 12.5 μm (g,h,k,l).
ZPR1 deficiency causes progressive loss of phrenic motor neurons in Zpr1Hb9MNΔ mice
Phrenic motor column (PMC) neurons are located between cervical C3-C5 segments and exit spinal cord to form phrenic nerves that innervate diaphragm24. Decrease in diameter of phrenic nerve in mutant compared to control mice suggests that ZPR1 deficiency might have caused loss of phrenic neurons (Fig. 2c,d). We examined the effect of Zpr1 mutation on PMC neurons in Zpr1 Hb9MNΔ mice. PMC neurons can be identified and distinguished from other neurons in cervical region using Pou3f1 (also known as Scip or Oct6), a POU-class transcription factor25, as a marker17. Pou3f1+ PMC neurons express high levels of Hoxa5, Hoxc5, Isl1 and Hb917. Notably, Pou3f1+ PMC neurons do not express FoxP117, a transcription factor required for Hox genes activity, and presence of FoxP1 allows distinguishing between limb-innervating or lateral motor column (LMC) neurons from other subtypes, including PMC neurons26–28. Double stained immunofluorescence images of the spinal cords from E12.5 (Fig. 3a–d) and E18.5 (Fig. 3e–h) control and mutant embryos show location of Pou3f1+ PMC neurons in the context of neurons stained positive for Hoxa5, Hoxc5, Islet1/2 and FoxP1. Quantitative analysis of Pou3f1+ PMC neurons at E12.5 stage shows Zpr1 mutation causes loss (64.29 ± 4.78%, p = 0.000) of neurons in Zpr1 Hb9MNΔ compared to control mice. At E18.5 stage the loss of Pou3f1+ neurons increases to (90.00 ± 15.81%, p = 0.001) and neurons were barely detectable in Zpr1 Hb9MNΔ embryos, suggesting a progressive loss of PMC neurons between E12.5 to E18.5 stages of embryonic development (Supplementary Figure S3). Zpr1 mutation also causes the loss of Hoxa5+, Hoxc5+ and FoxP1+ neurons during development (Fig. 3). However, the loss of Hoxa5+ neurons was non-progressive, E12.5 (64.64 ± 15.54%, p = 0.003) and E18.5 (59.35 ± 11.60%, p = 0.002). The loss of Hoxc5+ neurons is ~2-folds less than Hoxa5+ neurons and was also non-progressive, E12.5 (31.93 ± 4.80%, p = 0.000) and E18.5 (24.51 ± 9.81%, p = 0.036). Comparison of neuron loss shows highest loss of Pou3f1+ (~90%) followed by Hoxa5+ (~60%) and Hoxc5+ (~25%) neurons at E18.5 stage in Zpr1 Hb9MNΔ mice. Comparison of average losses of Hoxa5+ neurons at E12.5 and E18.5 stages did not show significant change suggesting that ZPR1 is critical for the development and differentiation of Hoxa5+ neurons during early stages of embryogenesis. Notably, progressive loss of Pou3f1+ neurons between E12.5 and E18.5 stages suggests that sustained Zpr1 gene activity is required for differentiation and maintenance PMC respiratory motor neurons throughout embryonic development.
Figure 3.
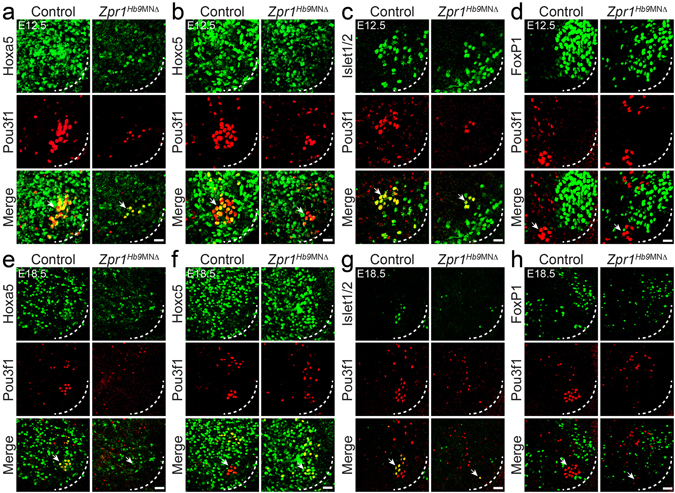
ZPR1 deficiency causes progressive loss of PMC neurons in Zpr1 Hb9MNΔ mice. Spinal cord sections of the cervical region (C3-C5) from (a–d) E12.5 and (e–h) E18.5 control and Zpr1 Hb9MNΔ embryos were stained with antibodies against Pou3f1, Hoxa5, Hoxc5, Islet1/2 and FoxP1 proteins. Immunofluorescence images reveal the effect of Zpr1 mutation on the loss and disorganization of different populations of neurons in the cervical region, including PMC and LMC groups of motor neurons. Quantification of Pou3f1+, Hoxa5+, Hoxc5+, and FoxP1+ neurons at E12.5 and E18.5 stages is presented in Supplementary Figure S3. Neurons were counted in serial sections of C3-C5 region. Neuron losses are presented as (mean ± s.e.m.; n = 3 mice/group). At E12.5 stage, images (a–d) and quantification shows loss (64.29 ± 4.78%, p < 0.0001) of Pou3f1+ motor neurons. By E18.5 stage (e–h), Pou3f1+ neurons were barely detectable and quantification shows greater loss (90.00 ± 15.81%, p = 0.0013) of neurons in Zpr1 Hb9MNΔ embryos. Image analysis and quantification of Hoxa5+ neurons shows non-progressive loss of neurons at E12.5 (64.64 ± 15.54%, p = 0.0032) and at E18.5 (59.35 ± 11.60%, p = 0.0022) stages in Zpr1 Hb9MNΔ embryos. A smaller loss of Hoxc5+ neurons at E12.5 (31.93 ± 4.80%, p = 0.0002) and E18.5 (24.51 ± 9.81%, p = 0.0369) stages was noted in Zpr1 Hb9MNΔ mice. A relatively smallest decrease in FoxP1+ LMC neurons at E12.5 (13.59 ± 10.94%, p = 0.2605) that increases to a statistically significant loss at E18.5 (28.17 ± 2.42%, p = 0.0001) was found in Zpr1 Hb9MNΔ mice. Images (d,h) show disorganization and the loss of compactness of FoxP1+ LMC neurons. Dotted arcs represent the orientation of view from the anterior horn side of the spinal cord. Arrows indicate phrenic nerve motor neurons. Scale bars are 25 μm (a–d) and 50 μm (e–h).
Zpr1 mutation results in disorganization of FoxP1+ neurons in Zpr1 Hb9MNΔ embryos compared to control at E12.5 (Fig. 3d,h). Neuron counting did not show significant decrease in FoxP1+ neurons at E12.5 (13.59 ± 10.94%, p = 0.260). Interestingly, at E18.5 FoxP1+ neurons loss increased to (28.17 ± 2.42%, p = 0.0001) (Supplementary Figure S3d,h). To find whether ZPR1 deficient FoxP1+ neurons have limbs innervation defects similar to PMC neurons, we examined forelimbs and hindlimbs from E18.5 embryos stained with NF and BTX. Both forelimb and hindlimb muscles were innervated by motor neurons but reduced secondary and tertiary branches were noted in Zpr1 Hb9MNΔ compared to control mice (Supplementary Figure S4). Defects were found in the innervation of NMJs in the hindlimb of Zpr1 Hb9MNΔ mice. ZPR1 deficiency resulted in degeneration of secondary and tertiary nerve branches in forelimbs of Zpr1 Hb9MNΔ mice. Comparison of innervation defect between diaphragm and limbs shows that limb innervation defects were less severe compared to diaphragm. These data suggest that the ZPR1 deficiency causes less severe defects in arborization of limb innervating motor neurons however ZPR1 is required for maintenance and function of terminal nerve branches.
Temporal delay in Zpr1 mutation preserves anatomical and skeleton development
To further investigate the role of ZPR1 in the functioning of the respiratory system, we examined the effect of temporal delay in Zpr1 mutation in motor neurons using choline acetlytransferase (ChAT)-Cre mice. The ChAT gene expression begins shortly after differentiation of cervical motor neurons at mid-gestation embryonic stage ~E10.5–11.5 days. The ChAT driven Cre recombinase has been shown to effectively remove floxed gene from motor neurons by E13.5 stage17. We generated mutant mice in which Zpr1 was selectively deleted from motor neurons by crossing conditional allele Zpr1 F1/F1 mice with transgenic ChAT-Cre mice. Mice with homozygous deletion of Zpr1 exon 1 (Zpr1 Δ1/Δ1) in motor neurons, in the presence of transgene ChAT-Cre, are referred as (Zpr1 ChATMNΔ).
Temporal delay in Zpr1 mutation in motor neurons resulted in reduced severity and preserved anatomical and skeleton development in Zpr1 ChATMNΔ mice (Fig. 4a). Interestingly, mutant Zpr1 ChATMNΔ mice were born live but died within minutes of birth. This finding suggested that Zpr1 ChATMNΔ mice might have also died because of respiratory failure similar to Zpr1 Hb9MNΔ mice. Therefore, first we examined the lungs from newborn control and Zpr1 ChATMNΔ mice. Lungs from mutant Zpr1 ChATMNΔ mice were smaller compared to control mice and failed to float to the surface upon submerging into water suggesting that the lungs were not inflated with air (Fig. 4b). Histological examination shows defects in the lungs morphogenesis in Zpr1 ChATMNΔ mice (Fig. 4c) however the severity of defect was less compared to Zpr1 Hb9MNΔ mice (Fig. 1i). Together, findings from Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice suggest that the sustained expression of ZPR1 in motor neurons during E8.5-E13.5 window of embryogenesis is critical for the development of normal respiratory system, including lungs morphogenesis.
Figure 4.
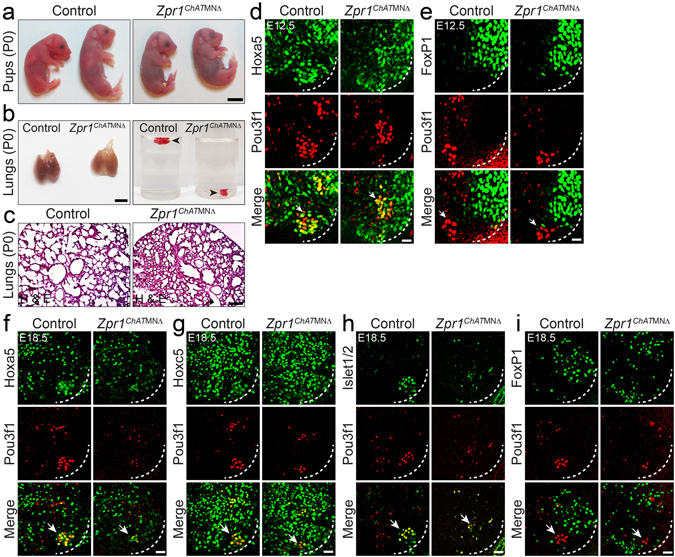
Temporal delay in mutation of Zpr1 preserves anatomical development but causes loss of PMC neurons and respiratory failure in Zpr1 ChATMNΔ mice. (a) Photograph of control and Zpr1 ChATMNΔ pups (P0) showing normal anatomical development. All mutant Zpr1 ChATMNΔ mice (n > 20) were died immediately after birth. Scale bar is 5 mm. (b) Lungs from Zpr1 ChATMNΔ pups (P0) were smaller than control and lungs of mutant embryos immediately sank in water, indicating lungs were not inflated with air suggesting mutant mice failed to breathe after birth. Scale bar is 3 mm. (c) Histological analysis shows defects in morphogenesis of lungs from Zpr1 ChATMNΔ pups (P0) and lungs were underdeveloped. Scale bar is 200 μm. (d,e) Spinal cord sections of the cervical region (C3-C5) from E12.5 control and Zpr1 ChATMNΔ embryos were stained with antibodies against Pou3f1, Hoxa5 and FoxP1 proteins. Scale bar is 25 μm (d,e). Quantification of Pou3f1+, Hoxa5+ and FoxP1+ neurons (mean ± s.e.m.; n = 3 mice/group) at E12.5 stage is presented in Supplementary Figure S5a–c. (f–i) Spinal cord sections of the cervical region (C3-C5) from E18.5 control and Zpr1 ChATMNΔ embryos were stained with antibodies against Pou3f1, Hoxa5, Hoxc5, Islet1/2 and FoxP1 proteins. Immunofluorescence images show that the delayed Zpr1 mutation causes loss of Pou3f1+ PMC motor neurons and disorganization of FoxP1+ LMC group of motor neurons. Quantification of Pou3f1+, Hoxa5+ and Hoxc5+ neurons at E18.5 stage is presented in Supplementary Figure S5d–f. Quantification of neurons shows the loss of Pou3f1+ neurons (61.90 ± 12.59%, p = 0.0026), Hoxa5+ neurons (48.34 ± 12.67%, p = 0.0189) and Hoxc5+ neurons (12.97 ± 1.93%, p = 0.0027) in E18.5 Zpr1 ChATMNΔ embryos. Scale bar is 50 μm (d–i).
Temporal delay in Zpr1 mutation causes reduced loss of PMC neurons in Zpr1ChATMNΔ mice
The death of newborn Zpr1 ChATMNΔ mice due to respiratory failure might be a consequence of defects in the functioning of the phrenic nerve and diaphragm system. The delayed Zpr1 mutation shows a smaller loss of Pou3f1+ (23.50 ± 9.77%, p = 0.037) and Hoxa5+ (35.56 ± 7.89%, p = 0.006) neurons (Fig. 4d,e and Supplementary Figure S5a–c) in Zpr1 ChATMNΔ compared to Zpr1 Hb9MNΔ, Pou3f1+ (64.29 ± 4.78%, p = 0.000) and Hoxa5+ (64.64 ± 15.54%, p = 0.003) in E12.5 embryos (Supplementary Figure S3). Interestingly, the loss of FoxP1+ (7.70 ± 7.89%, p = 0.1241) neurons was not significant at E12.5 stage suggesting that ZPR1 deficiency might have caused selective loss of Pou3f1+ and Hoxa5+ neurons at early stage due to delayed Zpr1 mutation (Fig. 4d,e). Comparison of neuron loss at E12.5 stage between two mutant mice shows reduction in the loss of Pou3f1+ (~2.7-folds) and Hoxa5+ (~1.8-folds) neurons in Zpr1 ChATMNΔ mice compared to Zpr1 Hb9MNΔ mice suggesting delayed Zpr1 mutation preserves development and differentiation of PMC motor neurons. Analysis at E18.5 stage shows increased loss of Pou3f1+ (61.90 ± 12.59%, p = 0.002) and Hoxa5+ (48.34 ± 12.67%, p = 0.018) neurons in Zpr1 ChATMNΔ mice (Fig. 4f–i and Supplementary Figure S5d–f). Comparison of Pou3f1+ neuron loss at E12.5 and E18.5 stages shows increased (~2.6-fold) loss of Pou3f1+ neurons at E18.5 that suggest progressive loss of PMC neurons during development in Zpr1 ChATMNΔ mice that is consistent with Zpr1 Hb9MNΔ mice (Supplementary Figures S3 and S5). The major loss of Pou3f1+ neurons among other neurons in both Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mutant mice suggest that the PMC neurons are particularly sensitive to low levels of ZPR1 during development of the respiratory system. Comparison of neuron loss at E18.5 between two mutant mice shows that the late removal of Zpr1 helps preserve ~28% more PMC neurons in Zpr1 ChATMNΔ than Zpr1 Hb9MNΔ mice, however, Zpr1 ChATMNΔ mice failed to breathe after birth. These data suggest that the PMC neurons in Zpr1 ChATMNΔ mice might not have potential to perform respiratory function.
Temporal delay in Zpr1 mutation improves diaphragm innervation in Zpr1ChATMNΔ mice
To determine whether PMC neurons in Zpr1 ChATMNΔ mice had potential to innervate and arborize in the diaphragm muscle and generate neuromuscular synapses, we examined whole diaphragms from control and Zpr1 ChATMNΔ mice, and compared with Zpr1 Hb9MNΔ mice. Comparison of diaphragms shows that the late removal of Zpr1 in motor neurons reduces severity of defects in the development of phrenic nerves and improves innervation of diaphragm (Fig. 5). Analysis of diaphragms from E18.5 control and Zpr1 ChATMNΔ embryos revealed difference in size and defects in innervation of the phrenic nerve (Fig. 5). Comparison of phrenic nerve diameter between control (21.66 ± 0.014 μm) and Zpr1 ChATMNΔ (10.08 ± 0.035 μm) of E18.5 embryos shows (53.28 ± 3.77%, p = 0.000) reduction in diameter and is consistent with the loss of PMC neurons in the spinal cord. However, comparison of phrenic nerve diameter between Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice show ~70% increase in the diameter of nerve in Zpr1 ChATMNΔ mice that is consistent with the reduced loss of PMC neurons. Diaphragms show improved primary innervation by phrenic nerve in Zpr1 ChATMNΔ compared to Zpr1 Hb9MNΔ mice but reduced secondary and tertiary branches compared to control (Fig. 6a,b). Reduced numbers of innervated NMJs in Zpr1 ChATMNΔ compared to control suggest that the motor neurons lacked potential to fully arborize in the diaphragm muscle. Degeneration of secondary and tertiary nerve branches in the diaphragms of Zpr1 ChATMNΔ embryos suggests that the neurons with ZPR1 deficiency lacked ability to sustain innervation of NMJs (Fig. 6c–f). Comparison of total number of NMJs between Zpr1 ChATMNΔ (12.92 ± 2.04%) and Zpr1 Hb9MNΔ (9.88 ± 1.34%) mice did not show significant improvement in Zpr1 ChATMNΔ mice. However, comparison of innervated NMJs between Zpr1 ChATMNΔ (19.44 ± 6.012%) and Zpr1 Hb9MNΔ (9.560 ± 0.7942%) mice shows small improvement in Zpr1 ChATMNΔ mice but not significant (p = 0.154) enough to support restoration of respiration (Supplementary Figure S6). Further, examination of neuromuscular synapse shows normal formation of synapse and co-localization of NF and SYN in neuromuscular junctions in control embryos (Fig. 6g,i). In contrast, diaphragm from mutant Zpr1 ChATMNΔ embryos shows severe defects in synapse formation (Fig. 6h,j). Comparison of synapse formation between Zpr1 ChATMNΔ and Zpr1 Hb9MNΔ mice shows improvement in Zpr1 ChATMNΔ mice but degeneration of secondary and tertiary nerve branches suggest the loss of functional ability of motor axons to generate synapse. These data suggest that the defects in the formation of synapses may be the cause of respiratory failure and neonatal mortality in Zpr1 ChATMNΔ mice. Despite overall reduced severity, including normal anatomical development, reduced loss of PMC neurons and improved innervation of diaphragm by phrenic nerves, all mutant Zpr1 ChATMNΔ mice died because of respiratory failure. These data suggest that sustained Zpr1 gene activity is required for the function of the mammalian respiratory motor neuron system.
Figure 5.
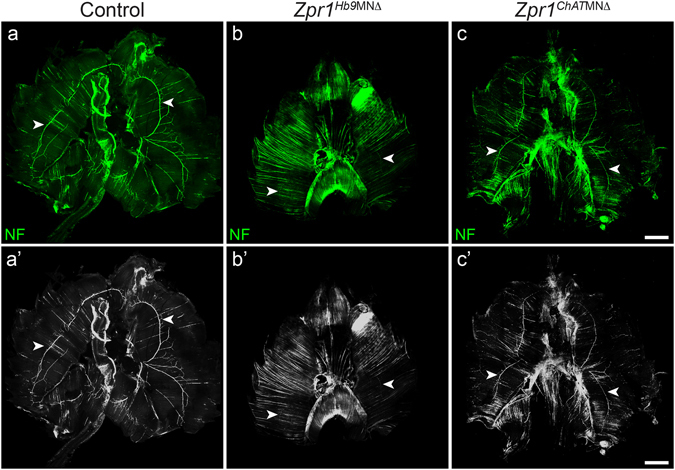
Temporal delay in Zpr1 mutation reduces severity of defects in the development of respiratory system. Whole diaphragms from E18.5 embryos were stained with neurofilament (NF) antibody. Images shown are composite of individual panels acquired by TileScan using confocal microscope. Diaphragms from (a,a’) control (b,b’) Zpr1 Hb9MNΔ and (c,c’) Zpr1 ChATMNΔ mice showing innervation of phrenic nerve in the muscle. Comparison of the phrenic nerve staining between Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice shows remarkable improvement in the development and innervation of phrenic nerve upon temporal delay in Zpr1 mutation in motor neurons. Arrowheads indicate phrenic nerve. Scale bar is 1000 μm.
Figure 6.
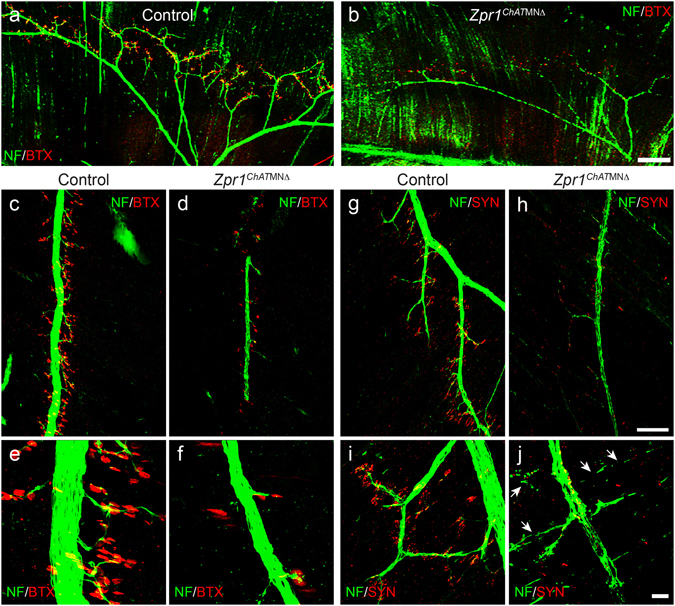
Temporal delay in Zpr1 mutation causes phrenic nerve degeneration and defects in synapse formation in Zpr1 ChATMNΔ mice. (a,b) Whole diaphragms from control and Zpr1 ChATMNΔ E18.5 embryos were stained with neurofilament (NF) antibody and α-bungarotoxin (BTX). Image analysis show defects in branching of the phrenic nerve in Zpr1 ChATMNΔ embryos. (c–f) Analysis of diaphragms stained with NF and BTX shows defects in the formation of NMJs in mutant embryos. Primary innervation of phrenic nerve was improved in Zpr1 ChATMNΔ compared to Zpr1 Hb9MNΔ mice but reduced numbers of secondary and tertiary branches were generated in Zpr1 ChATMNΔ mice. (g–j) Staining of diaphragms with NF and synaptophysin (SYN) shows marked reduction in the tertiary branches and formation of neuromuscular synapses in Zpr1 ChATMNΔ mice. Degeneration of secondary and tertiary branches was detected in mutant embryos (arrows). Lack of terminal branches and functional synapses indicate requirement of ZPR1 in motor neurons for the normal functioning of the respiratory system. Scale bars are 250 μm (a,b), 100 μm (c,d,g,h) and 12.5 μm (e,f,I,j).
ZPR1 interacts with promoter region of HoxA5 gene and regulates its expression
The loss of HoxA5+ neurons and reduced staining of HoxA5 in the spinal cords in Zpr1 mutant mice (Fig. 3a,e) suggest ZPR1-defciency might be a cause of deregulation of HoxA5 levels because sustained levels of HoxA5 are required for the survival and function of phrenic motor neurons17. It is possible that ZPR1 might be a regulator of HoxA5 expression because ZPR1 is a C4-type zinc finger protein and has been shown to be involved in the regulation of transcription29. To test whether ZPR1 acts as a transcription factor and regulates the expression of HoxA5 gene, we examined (a) interaction of ZPR1 with HoxA5 promoter region and (b) the effect of change in ZPR1 levels on the expression of HoxA5 gene. Immunoprecipitation of chromatin using antibodies against ZPR1 shows interaction of ZPR1 with genomic DNA (Fig. 7a). Electrophoretic mobility-shift assay shows that ZPR1 interacts with one of the four fragments (235 bp) tested within ~1 kb region of the human HoxA5 promoter (Fig. 7b). To test the effect of ZPR1 on the transcription, we utilized in vitro approach and used a plasmid containing Luciferase gene under the control of human HoxA5 promoter and transfected HeLa cells to measure the luciferase enzyme activity. The levels of ZPR1 were (i) down-regulated by knockdown with antisense oligonucleotide29 and (ii) up-regulated by transfection of plasmid containing recombinant human Flag-ZPR1 cDNA14. Knockdown of ZPR1 levels in cells expressing HoxA5-Luciferase results in reduced luciferase activity (32.77 ± 7.91%, p = 0.000) compared to cells treated with scrambled oligonucleotide (Fig. 7c,d). The decrease (~67%) in HoxA5 promoter activity is consistent with in vivo reduction in the levels of HoxA5 protein caused by Zpr1 gene mutation in Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice. Importantly, overexpression of ZPR1 results in increase of luciferase activity (42.80 ± 13.72%, p = 0.011) compared to cells without ZPR1 overexpression (Fig. 7e,f). These data show that the modulation of ZPR1 levels results in alteration of HoxA5 expression in direct correlation with ZPR1 levels. These findings suggest that modulation in ZPR1 levels directly correlates and influences levels of HoxA5 transcription and ZPR1 may be a transcriptional regulator of the HoxA5 gene.
Figure 7.
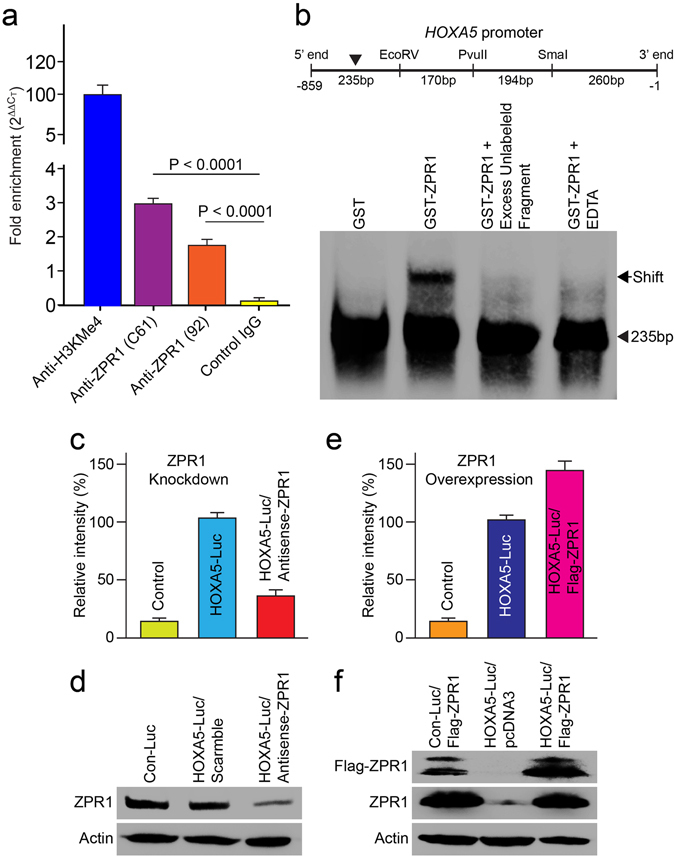
ZPR1 binds to HoxA5 promoter and regulates expression of the HoxA5 gene. (a) ZPR1 is a part of the chromatin complex. Chromatin samples were prepared using mouse brain tissue. ChIP assay was performed using antibodies against H3KMe4 (positive control) and monoclonal anti-mouseZPR1 (Clone: C61), rabbit polyclonal anti-mouseZPR1 (#92) and anti-Flag M2 (negative control). Real time PCR amplification was performed using universal Taqman PCR reagents and mouse genomic GAPDH primers. Levels of chromatin precipitated with each antibody were quantified using comparative CT (ΔΔCT) method for fold enrichment. (b) ZPR1 binds human HoxA5 promoter. EMSA shows that ZPR1 interacts with one of the four fragments (235 bp) tested within ~1 kb region of the human HoxA5 promoter. (c–f) Change in levels of ZPR1 influences expression of HoxA5-Luciferase reporter gene. (c,d) Effect of ZPR1 knockdown on ectopic Hox gene expression in cultured HeLa cells. Cells were transfected with either human HoxA5-Luc or Empty control reporter vector (Con-Luc). Transfected cells were re-transfected after 24 h with scrambled or Zpr1 antisense oligos to knockdown the levels of ZPR1. Cells were harvested after 24 h post-second transfection for determination of luciferase activity or immunoblot analysis. Quantification of luciferase activity (mean ± s.e.m.; n = 3) shows ZPR1 knockdown reduced levels of luciferase activity to 32.77 ± 7.91%, p = 0.000 in cells treated with antisense oligos. (e,f) To examine the effect of ZPR1 overexpression, HeLa cells were transfected with combination of two plasmids (i) Con-Luc + pcDNA3-FlagZPR1, (ii) HoxA5-Luc + pcDNA3 (empty) and (iii) HoxA5-Luc + pcDNA3FlagZPR1. After 30 h post-transfection, cells were harvested for determining luciferase activity or immunoblot analysis. Quantification of luciferase activity shows ZPR1 overexpression results in increase of luciferase activity (42.80 ± 13.72%, p = 0.011) compared to cells without ZPR1 overexpression (Full-length blots are included in Supplementary Figure S7).
ZPR1 deficiency causes down regulation of SMN and HoxA5 in motor neurons
To unravel the molecular mechanism of respiratory failure in mutant Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice caused by ZPR1 deficiency and its correlation with respiratory distress in SMA pathogenesis, we examined protein levels of SMN, HoxA5 and HoxC5 in the spinal cords from E12.5 and E18.5 control and mutant embryos. Quantitative analysis of protein levels in E12.5 spinal cords shows that the Zpr1 gene mutation reduced levels of proteins to, ZPR1 (47.44 ± 11.98%, p = 0.012), SMN (73.13 ± 5.14%, p = 0.019), HoxA5 (43.62 ± 8.84%, p = 0.004) and HoxC5 (87.23 ± 8.81%, p = 0.242) in Zpr1 Hb9MNΔ mice compared to control mice (Fig. 8a). Protein levels were further reduced to ZPR1 (27.67 ± 4.97%, p = 0.010), SMN (35.33 ± 7.86%, p = 0.011), HoxA5 (29.00 ± 10.44%, p = 0.024) and HoxC5 (89.00 ± 14.22%, p = 0.637) in E18.5 Zpr1 Hb9MNΔ mice compared to control mice (Fig. 8b). Similarly, estimation of protein levels in Zpr1 ChATMNΔ mice also shows reduced levels for ZPR1 (33.67 ± 3.75%, p = 0.001), SMN (50.33 ± 8.21%, p = 0.045), HoxA5 (42.33 ± 5.84%, p = 0.019) and HoxC5 (88.00 ± 12.17%, p = 0.441) compared to control at E18.5 stage (Fig. 8c). These data suggest that Zpr1 mutation causes significant reduction in the levels of SMN and HoxA5 but not in the levels of HoxC5 that shows the specificity of down-regulation HoxA5 in Zpr1 mutant mice. Reduction in levels of SMN ~65% and HoxA5 ~71% (Zpr1 Hb9MNΔ) and SMN ~50% and HoxA5 ~52% (Zpr1 ChATMNΔ) in mutant mice correlate with the greater severity of phenotype in Zpr1 Hb9MNΔ compared to Zpr1 ChATMNΔ mice. It is established that reduction in SMN protein levels by ~70%, ~50% and ~30% correlates with severity of SMA disease in type I, II and III patients, respectively30, 31. Therefore, reduction in SMN protein levels by ~65% (Zpr1 Hb9MNΔ) and ~50% (Zpr1 ChATMNΔ) suggest development of severe to moderate SMA-like disease in Zpr1 mutant mice.
SMN deficiency causes down regulation of ZPR1 and HoxA5 and degeneration of phrenic motor neurons in SMA mice
To test whether SMN deficiency causes degeneration of phrenic motor neurons and contributes to the respiratory distress associated with SMA pathogenesis, we examined cervical region of the spinal cord from mice with severe SMA that has mean survival of 4–5 days. We exploited the opportunity to examine phrenic motor neurons in postnatal (P3) mice because it was not possible to examine Zpr1 mutant mice due perinatal lethality. SMN deficiency causes the loss of SMN+ phrenic motor neurons in SMA mice similar to ZPR1 deficiency (Fig. 8d,e) suggesting that ZPR1 and SMN deficiency cause similar degeneration of phrenic motor neurons that may contribute to respiratory distress in SMA. To identify common molecular targets involved in phrenic motor neuron degeneration caused by ZPR1 and SMN deficiencies that may contribute to respiratory distress in SMA, we examined the levels of ZPR1, HoxA5 and HoxC5 in a less severe SMA mouse model SMAΔ7. Quantitative analysis of protein levels in spinal cords from postnatal (P8) mice show that SMN deficiency (69.10 ± 12.42%, p = 0.005) reduces levels of ZPR1 (58.87 ± 10.43%, p = 0.048), HoxA5 (39.41 ± 9.03%, p = 0.007) and HoxC5 (74.90 ± 7.06%, p = 0.139) proteins in SMAΔ7 mice compared to control mice (Fig. 8f). Small and non-significant change in HoxC5 is consistent with data from Zpr1 mutant mice and serves as a control to support the specificity of defect caused by either ZPR1 or SMN deficiency. Comparison of data from mutant Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice with SMAΔ7 mice show that ZPR1 deficiency results in deregulation SMN and HoxA5 levels and SMN deficiency results in down-regulation of ZPR1 and HoxA5. Together, these findings suggest that the low levels of SMN, ZPR1 and HoxA5 collectively contribute to the respiratory distress associated with SMA pathogenesis.
Discussion
Breath (Prana in Sanskrit or Spiritus in Latin) or the act of breathing, respiration, is essential for all vertebrates living on the planet Earth. Respiratory failure is the cause of death in SMA3. Molecular mechanisms underlying respiratory distress in SMA are unknown. Respiration rhythm in mammals is maintained by motor activity of diaphragmatic muscle controlled by motor neurons of the phrenic nerve, which originates in the cervical region (C3-C5) of the spinal cord and is the only source of motor neuron supply to the diaphragm32, 33. The Hox genes are known to control the differentiation, identity and columnar organization of specific groups of motor neurons in the spinal cord34. Genetic studies on mutation of the Hox5 group of genes (Hoxa5, Hoxb5 and Hoxc5) in mice have shown that Hoxa5 is critical for the development and function of the phrenic nerve motor neurons17, 22, 23. In this study, we found that ZPR1 is critical for the function of phrenic motor neurons that regulate respiration. ZPR1 functions downstream of SMN and transcriptionally regulates HoxA5 levels in motor neurons. In SMA, SMN-deficiency causes deregulation of ZPR1 and HoxA5 that lead to respiratory failure.
ZPR1 is an evolutionary conserved signaling protein
The Zpr1 gene (Chr 11q23.2 in humans) is evolutionary conserved in eukaryotes, yeast to mammals, and encodes protein with unique modular architecture consisting of two C4-type Zn2+ fingers (ZnF1 and ZnF2) with a common topology and two domains termed A and B, which are homologous to each other but not in other proteins. These domains occur in a tandem order (ZnF1-A domain-ZnF2-B domain) that is broadly conserved in eukaryotic organisms6. Genetic studies have shown that the core function of ZPR1 is preserved during evolution and the Zpr1 gene is essential for viability in yeast and mice5, 8, 35. However, the biological function of ZPR1 is unknown. Biochemical studies have provided insight that ZPR1 domains interact with distinct proteins: A-domain interacts with eEF1A and the B-domain interacts with SMN protein5, 9. In addition, both Zinc-Fingers interact with cytoplasmic domain of EGFR and PDGFR that is highly conserved among RTKs4. In mammalian cells, ZPR1 binds to inactive EGFR and PDGFR and upon receptor activation by EGF or serum, ZPR1 translocate to the nucleus4, 35. In the nucleus, ZPR1 accumulates in sub-nuclear bodies, SMN+ gems, histone-locus body and CB9, 29. Genetic mutation study in Drosophila has shown that the ZPR1 mediates effects of EGFR and fibroblast growth factor receptor (FGFR) signaling required for lumen formation in terminal cells7. Together these findings, including evolutionary conservation and ubiquitous expression suggest that ZPR1 may be a common downstream target of multiple RTKs, including EGFR, PDGFR and FGFR that mediates growth signaling in cells and tissues throughout the lifespan.
ZPR1 is critical for the development and function of respiratory motor neurons
Zpr1 mutation resulted in defects in the development and function of the respiratory system in Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice. These defects included under developed lungs, loss of PMC neurons, inability of motor neurons to innervate diaphragm muscle and populate NMJs and lack of neuromuscular synapse. Remarkable extent of severity was displayed by complete absence of phrenic nerve or partial presence of nerve only on one side of the diaphragm in Zpr1 Hb9MNΔ mice suggesting that the development and targeting of phrenic nerve neurons to the diaphragm was impaired by ZPR1 deficiency. Respiratory failure and perinatal lethality have been reported in mice with mutation of the Hb9 −/− gene19 and Hox5 MNΔ (Hoxa5 loxP/loxP; Hoxc5 −/−; Olig2::Cre mice)17. Defects in lungs morphogenesis observed in Zpr1 Hb9MNΔ mice have been reported in other mutant mice, Hb9 −/−, Hoxa5 −/−, Hox5 MNΔ, (Hoxa5 −/−; Hoxb5 −/−)17, 19, 23, 36, however, the severity of defects is most pronounced in Zpr1 Hb9MNΔ mice compared to other mutant mice. Similarity of defects in lungs morphogenesis in mice with conventional knockouts and conditional knockouts in motor neurons suggest that the upper spinal cord neurons may be involved in the regulation of lungs morphogenesis during embryogenesis16, 37. However, which specific region and group of neurons controls lungs development remains to be identified and warrants further studies? It is possible that the paralogs of Hox genes and Zpr1 may be components of a common mechanism that control development of mammalian respiratory system, including lungs morphogenesis.
Temporal delay in Zpr1 mutation preserved anatomical and skeleton development in Zpr1 ChATMNΔ mice. Although the severity of defects in the innervation of diaphragm was reduced but the perinatal lethality of mice could not be rescued and mice died immediately after birth because of respiratory failure. Temporal analysis of ZPR1 function and comparison of phenotypes of Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice revealed that phrenic motor neurons are most sensitive to the low levels of ZPR1 and sustained Zpr1 gene activity is required for the function of respiratory motor neurons.
Molecular mechanism of respiratory failure in SMA
Severe SMA Type I (Werdnig-Hoffman syndrome) patients require ventilation for life support after birth and live ~2 years. Primary SMA pathogenesis shows degeneration of motor neurons in the lumbar region that results in muscle atrophy3. How SMN-deficiency leads to respiratory failure in SMA is unclear? Findings of this study provide insight into the molecular events stemming downstream of SMN-deficiency that lead to degeneration of phrenic nerve motor neuron and respiratory failure in SMA.
Defects in intracellular signaling downstream of growth factor receptors because of low levels of SMN and ZPR1 might account for degeneration of phrenic motor neurons and respiratory distress in SMA. Previous studies have shown that ZPR1 binds to the highly conserved cytoplasmic domain of inactive growth receptors (EGFR, PDGFR) and the treatment of quiescent cells with serum containing growth factors results in the formation ZPR1-SMN protein complex and translocation of ZPR1-SMN complex from the cytoplasm to the nucleus in mammalian cells4, 9. Drosophila FGF receptor (breathless, btl) was identified as a modifier of Smn-dependent lethality by a genetic screen, suggesting a link between SMN and FGF signaling pathway38. Further, activation of FGF signaling in muscle rescues synaptic defects in neuromuscular junctions caused by SMN deficiency39. Intracellular FGF-2 isoform interacts with SMN and regulates stability of SMN containing sub-nuclear bodies40. ZPR1 is shown to be a downstream component of EGFR and FGFR signaling pathways and mediates development of trachea (respiratory system) in Drosophila 7. Interestingly, down regulation of SMN levels in Drosophila also resulted in a phenotype similar to ZPR1 deficiency, suggesting both ZPR1 and SMN are components of a common pathway that regulates development of the respiratory system7.
The results of this study demonstrate that the low levels of ZPR1 cause down-regulation of HoxA5 that leads to degeneration of phrenic motor neurons and respiratory failure in mice. SMA patients express reduced levels of ZPR114, 15, our data indicate that low levels of ZPR1 may contribute to respiratory distress in SMA. In SMA mice, low levels of SMN cause deregulation of ZPR1 and HoxA5 that result in degeneration of phrenic motor neurons leading to respiratory distress and death. Respiratory distress caused by the low levels of HoxA5 in Zpr1 mutant mice and SMA mice is consistent with finding that HoxA5 deficiency causes degeneration of phrenic motor neurons and respiratory failure in mice17. The FGF/FGFR pathway has been shown to regulate Hox gene expression during development41, 42. Deregulation of HoxA5 in motor neurons of two Zpr1 mutant (Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ) mice and SMA mice suggest that the normal levels of ZPR1-SMN complex are required to maintain optimal levels of HoxA5 in motor neurons. Because ZPR1 binds to inactive RTKs and translocate from the cytoplasm to the nucleus in response to receptor activation, it is possible that ZPR1 acts downstream of FGFR signaling and form complex with SMN to regulate expression of Hoxa5 gene. Recently, SMN-Pol II complex has been shown to be involved in regulation of transcription termination43. We show that increase in ZPR1 levels enhances transcription of HoxA5 and because ZPR1-SMN complex is localized in the nucleus it is possible that ZPR1 and SMN proteins are parts of transcription complex that regulates expression of HoxA5. Further, zinc fingers of ZPR1 have similarity to nucleic acid interacting proteins6. A recent study showed that plant (Solanum tuberosum) StZPR1 binds to a DNA motif ‘CAACAGCATC’, present in the promoter of a clock-controlled double B-box StBBX24 gene and regulate its expression, and suggested that ZPR1 may be a transcription regulator44. However, this class of plants specific light-regulated circadian clock genes is absent in mammals. Similarly, plants lack Hox genes. Our initial analysis showed that ZPR1 interacts with a 235 bp DNA fragment of human HoxA5 promoter. Further, analysis identified a 20 bp DNA sequence ‘CTGCGGGCAGGATTTATTTCTCCAATT’ that contains Oct3/4 binding motif ‘AGGATTTATTT’. Interestingly, in silico analysis shows that a 17 bp DNA sequence ‘GGGCAGGATTTATTTCT’ containing Oct3/4 binding motif is present in the both mouse and human HoxA5 promoters but not in the HoxC5 promoters suggesting that ZPR1 might regulate expression of HoxA5 but not of HoxC5. This is consistent with our data from mutant Zpr1 Hb9MNΔ and Zpr1 ChATMNΔ mice that show ZPR1 deficiency causes significant decrease in levels of HoxA5 but not of HoxC5. SMN-deficiency causes deregulation of ZPR1 that result in down-regulation of HoxA5 but did not reduce levels of HoxC5 significantly suggesting combined deficiency of SMN and ZPR1 selectively decreases levels of HoxA5 in phrenic motor neurons. In conclusion, ZPR1 is a critical regulatory factor that may functions in collaboration or downstream of SMN to regulate levels of HoxA5 in phrenic motor neurons that control respiration. Together, these findings provide insight into the molecular basis of respiratory failure associated with SMA pathogenesis and identify ZPR1 and HoxA5 as potential targets that lay a foundation for developing therapeutic strategies to treat respiratory distress in SMA.
Methods
All experiments and procedures were approved and performed according to the guidelines and policies set by the Institutional Biosafety Committee (IBC). All animals were housed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Texas Tech University Health Sciences Center El Paso (TTUHSC EP). Animals were treated humanly and euthanasia was performed using methods approved by the American Veterinary Medical Association (AVMA).
Creation of mice with a conditional allele of the Zpr1 gene
The murine Zpr1 gene was isolated from a 129/SvJ mouse λ Fix II genomic library (Stratagene) using the mouse Zpr1 cDNA as a probe. Sequence analysis confirmed that the clone contains the Zpr1 gene8. A targeting vector was designed to replace Zpr1 exon 1 with loxP-flanked exon 1 and a loxP-flanked Neo R (floxed) cassette (Figure S1a). A thymidine kinase (TK) cassette was included for negative selection. The targeting vector was linearized with NotI and electroporated into 129SvEvTac (Taconic) embryonic stem cells and subsequently selected with G418 and gancyclovir (Ingenious Targeting Laboratory, New York). Targeted clones were identified by Southern blot analysis using a random-primed 32P-labeled 425 bp probe prepared by PCR using primer pair CTCAGACATGACCAGAGACC and CCGCTGTGGGGCCAGGCCCG and mouse Zpr1 cDNA as the template (Figure S1b). Targeted ES cells were used to create chimeric mice by blastocyst injection. Germ-line transmission of floxed exon 1 Zpr1 allele with floxed Neo R cassette was achieved by crossing chimeric mice to C57BL/6J strain (The Jackson Laboratory). The Neo R cassette was removed in vivo by crossing mice with disrupted Zpr1 allele containing floxed Neo cassette with Protamine-Cre (PC3-Cre) mice that expresses Cre recombinase in male germ-line45 to generate mice with a conditional allele containing floxed exon 1 (Zpr1 +/F1) (Supplementary Figure S1a). Genomic DNA was isolated from the mouse-tail with DNA isolation kit (Lamda Biotech) and used for genotyping by PCR method using primers; Forward (EX1-F4: CCCTCAGCGCCGAGGATGAG) and Reverse (EX2-R4: GCAGGAAAAGGAGCTCACGA) that generate 249 bp fragment (wild-type allele) and 289 bp fragment (floxed allele) (Supplementary Fig. S1c). Heterozygous mice Zpr1 +/F1 were crossbred to generate mice with homozygous conditional allele Zpr1 F1/F1. Transgenic mice Hb9-Cre (B6;129S1-Mnx1 tm4(cre)Tmj/J, Stock No: 006600) and ChAT-Cre (B6;129S1-ChAT tm2(cre)Lowl/J, Stock No: 006410) were purchased from the Jackson Laboratory. The other mouse models used in this study were, severe SMA [Smn −/+; SMN2 +/+] and SMAΔ7 [Smn −/+; SMN2 +/+; SMNΔ7 +/+]46, purchased from the Jackson Laboratory.
Histology
Mouse tissues, lungs, diaphragm and spinal cords from control and mutant E18.5 embryos were isolated, fixed in 4% paraformaldehyde (PFA) at 4 °C for overnight, washed with PBS and then soaked in 30% sucrose solution for 48 h. Tissues were processed and embedded in OCT for frozen sections. Thin serial sections (10 µm) were cut from the cervical region (C2-C6), thoracic region (T9-T12) and lumbar region (L1-L5) of the spinal cords and stained with hematoxylin and eosin or processed for immunohistochemical staining. Motor neurons were counted in every fifth section (20 sections) of the regions of the spinal cord14, 18. For immunohistochemical staining, sections were processed for antigen retrieval, briefly incubated in SDS (1%) solution for 5 min, and another 5–10 min in 0.1 M sodium citrate buffer maintained at 95 °C. Sections were rinsed and incubated with primary antibodies against ZPR1 (Clone # LG-D5), ChAT (#ab6168, Abcam), diluted 1:100 with antibody diluent from the M.O.M kit (Vector Laboratories). Immune complexes were detected by using a biotinylated secondary antibody, streptavidin-conjugated horseradish peroxidase and the substrate 3,3′-diaminobenzene followed by brief counterstaining with hematoxylin.
Immunofluorescence analysis
Control or mutant E12.5 and E18.5 days-old embryos were collected and fixed overnight in 4% PFA. Next day fixed embryos were washed with PBS for 4 h and spinal cord, fore and hind limbs, whole diaphragm were carefully isolated and washed with PBS and soaked in (30%) sucrose solution for 48 h. Tissues were embedded in OCT for frozen sections. Thin serial spinal cord (10 μm) and hind limb (30 μm) sections were blocked in 3% BSA with 0.1% Triton-X100 for an hour and then incubated overnight with primary antibodies against HoxA5 and HoxC5 (Dr. T. Jessell, Columbia University)28, Pou3f1 (Dr. D. Meijer, University of Edinburgh)47, Islet1/2 (Developmental Study Hybridoma Bank, DSHB, #39.4D5), FoxP1 (#ab16645, Abcam), ZPR1 (Clone LG-D5) diluted (1:100) in 3% BSA. Sections were washed 5 times in PBST, incubated for an hour with Alexa 488-conjugated anti-mouse or rabbit IgG secondary antibody diluted to 1:400 in 3%BSA, washed 4X with PBST, and mounted with DAPI containing Vectashield (Vector Lab). Whole diaphragm was blocked in Mouse-blocking solution (MOM kit; Vector Labs) containing 0.3% Triton-X100 for 4 h. Diaphragms were incubated in neurofilament primary antibody (NF-M, 145 kDa) protein (#MAB1621, Millipore) for 24 h followed by Alexa488-conjugated anti-mouse secondary antibody after washing them in PBS (5 × 10 min). For double staining the diaphragms were again incubated in Mouse-blocking solution for 30 min, rinsed with PBS (3 × 5 min) and then incubated with α-Bungarotoxin (1:100, Abcam) conjugated with Alexa596 for 2 h and washed 5x with PBS before mounting on glass slides using fluorescence mounting medium (Vector Labs) and cover-slipped. For double staining of diaphragms with NF and synaptophysin, the protocol for blocking and NF staining was similar as mentioned above. Synaptophysin (1:100, ab14692, Abcam) staining was done overnight, tissues washed with PBS (5 × 10 min), incubated with anti-rabbit Alexa596 conjugated secondary antibody for 4 h and washed 5 times with PBS before mounting on microscope slides using florescence mounting medium (Vector Labs) and cover-slipped48. Whole diaphragm images were captured with 10x objective using TileScan mode and were auto-stitched and merged using Leica Software. For z-stack and other images, the z-volume and other acquisition parameters were constant for each experiment. Images were captured using confocal microscope (Leica-TCS-SP5, Leica Microsystems) equipped with AOBS and UV (405 nm), IR (633 nm) and Visible range lasers.
Immunoblot Analysis
Spinal cords were isolated from control and mutant E18.5 embryos. Tissue lysates were prepared and diluted to protein concentration of 0.4 μg/μl using sample preparation kit (ProteinSimple). Tissue lysates were examined using automated western blot system, WES System (ProteinSimple) as per manufacturer’s protocol. The following primary antibodies were used for analysis: ZPR1 (Clone LG-C61), Flag M2 (#F1804, Sigma), HoxA5, HoxC5, α-tubulin (#T8203, Sigma) and β-Actin (#A5316, Sigma). Data analysis and quantitation of protein levels were performed using Compass Software (Protein Simple). For standard immunoblot method, cell lysates were prepared from control and transfected HeLa cells using Triton lysis buffer5. Proteins were separated by SDS-PAGE and electrotransferred to PVDF membrane (Millipore). Proteins were detected using primary antibodies (1:100), ZPR1 (Clone LG-C61), Flag M2 and α-tubulin (#T6074, Sigma) followed by HRP-conjugated donkey anti-mouse or rabbit IgG (1:5000) secondary antibodies. Chemiluminescence and quantitation of immunoblots was performed using ImageQuant LAS4000. The relative levels of proteins (mean ± s.e.m.) normalized to either actin or tubulin, are presented as bar graphs.
ChIP-Real-time PCR Assay
Chromatin samples were prepared using mouse brain tissue. ChIP assay was performed using antibodies against H3KMe4 as a positive control (#NB21–1023, Novas Biologicals) and monoclonal anti-mouseZPR1 (Clone: LG-C61), rabbit polyclonal anti-mouseZPR1 (#92) and anti-Flag M2 (negative control) according to manufacturer's protocol of the Magna ChIP™ HiSens kit (Millipore, 17–10460). Real-time PCR amplification was performed using universal Taqman PCR reagents and inventoried mouse genomic GAPDH primers (AB-4167A, Life Technologies). Levels of chromatin precipitated with each antibody were quantified using comparative CT (ΔΔCT) method for fold enrichment.
Electrophoretic Mobility-Shift Assay
A 235 bp DNA fragment from human HoxA5 promoter was amplified by PCR using primers, Forward: 5′-ATATTCACACGAAAGAAAAATCG-3′ and Reverse: 5′-ATCACCTTCGGGG AAGG-3′. A small 20 bp oligonucleotide (CTGCGGGCAGGATTTATTTCTCCAATT) contains Oct3/4 binding motif from the 235 bp sequence was synthesized (IDT). DNA fragments were end-labeled using Biotin 3′-end DNA labeling kit (Thermofisher Scientific). Recombinant GST and GST-ZPR1 proteins were produced in bacteria (BL21) using PGEX-5x-2 (GST) and PGEX-5x-2/GST-humanZPR1 vectors and purified using GST-Agarose beads column5. Electrophoretic Mobility-Shift (EMSA) assay was performed using labeled DNA probe and GST and GST-ZPR1 proteins using light shift chemiluminescent EMSA kit according to the manufacturer’s instruction (Thermofisher Scientific). Briefly, biotin labeled HoxA5 DNA probe was added to 10X binding buffer, 100 mM MgCl2, 1% NP 40, 50% Glycerol, 1 ug/uL poly (dI.dC) and purified GST or GST-ZPR1 protein. Excess (100-folds) unlabeled probe was used for competition and 20 mM EDTA (final concentration) was used for chelating Zn2+ to collapse zinc fingers and disrupt protein-DNA interaction. The reaction mixture was incubated for 30 min and complexes were separated on a 5% native polyacrylamide gel by electrophoresis, electro-transferred to a nylon membrane at 100 V for 1 h. Transferred DNA-protein complexes were crosslinked by UV (120 mJ/cm2 for 60 sec) using a UV-light crosslinker (Spectrolinker XL-1500, Spectronics Corp.). Biotin-labeled DNA probes were detected using the streptavidin-HRP conjugate and chemilumeniscence.
Ectopic Hox Gene Expression Analysis
To examine the effect of change in the levels of ZPR1 on alteration of the Hox gene expression, we used HeLa cells co-transfected with human HoxA5 promoter driven luciferase reporter vector (HoxA5-Luc) (Affymetrix) and either pcDNA3-FlagZPR1 (ZPR1 overexpression)14 or with Zpr1-antisense oligonucleotides (ZPR1 knockdown)29. To examine the effect of ZPR1 knockdown, cultured HeLa cells were transfected with either human HoxA5-Luc or Empty control reporter vector (Con-Luc) using lipofectamine reagent (Invitrogen). Transfected cells were re-transfected after 24 h with scrambled or Zpr1 antisense oligos to knockdown the levels of ZPR1. Cells were harvested after 30 h post-second transfection for determination of luciferase activity or immunoblot analysis. To examine the effect of ZPR1 overexpression, HeLa cells were transfected with combination of two plasmids (i) Con-Luc + pcDNA3-FlagZPR1, (ii) HoxA5-Luc + pcDNA3 (empty) and (iii) HoxA5-Luc + pcDNA3FlagZPR1 using the lipofectamine reagent. After 30 h post-transfection, cells were harvested for determining luciferase activity or immunoblot analysis. Luciferase activity was measured using the Luciferase assay system (Promega) in the synergy H1 hybrid multi-mode microplate reader (BioTek Instruments). Relative levels of luciferase activity (mean ± s.e.m.) are presented as bar graphs.
Statistical analysis
The quantitative data is presented as mean ± s.e.m. Statistical analysis performed using Student’s t-test (two tailed) with GraphPad Prism (version 5.0d). The value p = 0.05 or less was considered significant. In all experiments with mice, ‘n’ represents the number of embryos of mice used per group and in experiments with tissues or cells ‘n’ represents the number of times experiment was performed. A minimum of n = 3 embryos of mice per genotype or number of times experiment performed was used in all the experiments, unless otherwise specified in an experiment.
Data availability
All data generated and analysed during this study are included in this published article (and its Supplementary Information files).
Electronic supplementary material
Acknowledgements
We thank Marisol Ramirez and Javier Ordonez for administrative assistance. We gratefully acknowledge and thank Dr. Tom Jessell, Columbia University for antibodies against HoxA5 and HoxC5, and Dr. Dies Meijer, University of Edinburgh, UK for Pou3f1 antibody. We thank Dr. Dana Branzei, IFOM, Institute of Molecular Oncology, Milan, Italy for reading manuscript and suggestions. This study was supported in part by research grants to L.G. from the Muscular Dystrophy Association (MDA4209) and the US National Institutes of Health (R01 NS064224).
Author Contributions
L.G. conceived project, created mice with conditional Zpr1 F1/F1 allele and designed study. N.K.G., S.A., K.B. and A.K. performed experiments, collected and organized data. K.B. also performed in silico analysis. X.J. provided technical assistance with maintaining mouse colony, genotyping and timed mating. N.K.G., S.A., A.K., K.B. and L.G. analyzed data and prepared figures. L.G. wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-07603-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy- determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Munsat TL, Davies KE. International SMA consortium meeting. (26–28 June 1992, Bonn, Germany) Neuromuscul Disord. 1992;2:423–428. doi: 10.1016/S0960-8966(06)80015-5. [DOI] [PubMed] [Google Scholar]
- 3.Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update. Pediatr Neurol. 2012;46:1–12. doi: 10.1016/j.pediatrneurol.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Galcheva-Gargova Z, et al. Binding of zinc finger protein ZPR1 to the epidermal growth factor receptor. Science. 1996;272:1797–1802. doi: 10.1126/science.272.5269.1797. [DOI] [PubMed] [Google Scholar]
- 5.Gangwani L, Mikrut M, Galcheva-Gargova Z, Davis RJ. Interaction of ZPR1 with translation elongation factor-1alpha in proliferating cells. J Cell Biol. 1998;143:1471–1484. doi: 10.1083/jcb.143.6.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mishra AK, Gangwani L, Davis RJ, Lambright DG. Structural insights into the interaction of the evolutionarily conserved ZPR1 domain tandem with eukaryotic EF1A, receptors, and SMN complexes. Proc Natl Acad Sci USA. 2007;104:13930–13935. doi: 10.1073/pnas.0704915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz OE, Nikolova LS, Metzstein MM. Drosophila Zpr1 (Zinc finger protein 1) is required downstream of both EGFR and FGFR signaling in tracheal subcellular lumen formation. PLoS One. 2012;7:e45649. doi: 10.1371/journal.pone.0045649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gangwani L, Flavell RA, Davis RJ. ZPR1 is essential for survival and is required for localization of the survival motor neurons (SMN) protein to Cajal bodies. Mol Cell Biol. 2005;25:2744–2756. doi: 10.1128/MCB.25.7.2744-2756.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gangwani L, Mikrut M, Theroux S, Sharma M, Davis RJ. Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat Cell Biol. 2001;3:376–383. doi: 10.1038/35070059. [DOI] [PubMed] [Google Scholar]
- 10.Lefebvre S, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 11.Narayanan U, Ospina JK, Frey MR, Hebert MD, Matera AG. SMN, the spinal muscular atrophy protein, forms a pre-import snRNP complex with snurportin1 and importin beta. Hum Mol Genet. 2002;11:1785–1795. doi: 10.1093/hmg/11.15.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gubitz AK, Feng W, Dreyfuss G. The SMN complex. Exp Cell Res. 2004;296:51–56. doi: 10.1016/j.yexcr.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 13.Singh RN, Howell MD, Ottesen EW, Singh NN. Diverse role of survival motor neuron protein. Biochim Biophys Acta. 2017;1860:299–315. doi: 10.1016/j.bbagrm.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmad S, Wang Y, Shaik GM, Burghes AH, Gangwani L. The zinc finger protein ZPR1 is a potential modifier of spinal muscular atrophy. Hum Mol Genet. 2012;21:2745–2758. doi: 10.1093/hmg/dds102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helmken C, et al. Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet. 2003;114:11–21. doi: 10.1007/s00439-003-1025-2. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell RA. Neural regulation of respiration. Clin Chest Med. 1980;1:3–12. [PubMed] [Google Scholar]
- 17.Philippidou P, Walsh CM, Aubin J, Jeannotte L, Dasen JS. Sustained Hox5 gene activity is required for respiratory motor neuron development. Nat Neurosci. 2012;15:1636–1644. doi: 10.1038/nn.3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doran B, et al. Deficiency of the zinc finger protein ZPR1 causes neurodegeneration. Proc Natl Acad Sci USA. 2006;103:7471–7475. doi: 10.1073/pnas.0602057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arber S, et al. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23:659–674. doi: 10.1016/S0896-6273(01)80026-X. [DOI] [PubMed] [Google Scholar]
- 20.Ross AJ, et al. A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet. 1998;20:358–361. doi: 10.1038/3828. [DOI] [PubMed] [Google Scholar]
- 21.Currarino G, Coln D, Votteler T. Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol. 1981;137:395–398. doi: 10.2214/ajr.137.2.395. [DOI] [PubMed] [Google Scholar]
- 22.Aubin J, Lemieux M, Tremblay M, Berard J, Jeannotte L. Early postnatal lethality in Hoxa-5 mutant mice is attributable to respiratory tract defects. Dev Biol. 1997;192:432–445. doi: 10.1006/dbio.1997.8746. [DOI] [PubMed] [Google Scholar]
- 23.Boucherat O, et al. Partial functional redundancy between Hoxa5 and Hoxb5 paralog genes during lung morphogenesis. Am J Physiol Lung Cell Mol Physiol. 2013;304:L817–830. doi: 10.1152/ajplung.00006.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goshgarian HG, Rafols JA. The phrenic nucleus of th albino rat: a correlative HRP and Golgi study. J Comp Neurol. 1981;201:441–456. doi: 10.1002/cne.902010309. [DOI] [PubMed] [Google Scholar]
- 25.Bermingham JR, Jr., et al. Tst-1/Oct-6/SCIP regulates a unique step in peripheral myelination and is required for normal respiration. Genes Dev. 1996;10:1751–1762. doi: 10.1101/gad.10.14.1751. [DOI] [PubMed] [Google Scholar]
- 26.Dasen JS, De Camilli A, Wang B, Tucker PW, Jessell TM. Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell. 2008;134:304–316. doi: 10.1016/j.cell.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 27.Rousso DL, Gaber ZB, Wellik D, Morrisey EE, Novitch BG. Coordinated actions of the forkhead protein Foxp1 and Hox proteins in the columnar organization of spinal motor neurons. Neuron. 2008;59:226–240. doi: 10.1016/j.neuron.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dasen JS, Tice BC, Brenner-Morton S, Jessell TM. A Hox regulatory network establishes motor neuron pool identity and target-muscle connectivity. Cell. 2005;123:477–491. doi: 10.1016/j.cell.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 29.Gangwani L. Deficiency of the zinc finger protein ZPR1 causes defects in transcription and cell cycle progression. J Biol Chem. 2006;281:40330–40340. doi: 10.1074/jbc.M608165200. [DOI] [PubMed] [Google Scholar]
- 30.Crawford TO, et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One. 2012;7:e33572. doi: 10.1371/journal.pone.0033572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kolb SJ, et al. A novel cell immunoassay to measure survival of motor neurons protein in blood cells. BMC Neurol. 2006;6:6. doi: 10.1186/1471-2377-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith JC, Abdala AP, Koizumi H, Rybak IA, Paton JF. Spatial and functional architecture of the mammalian brain stem respiratory network: a hierarchy of three oscillatory mechanisms. J Neurophysiol. 2007;98:3370–3387. doi: 10.1152/jn.00985.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Philippidou P, Dasen JS. Hox genes: choreographers in neural development, architects of circuit organization. Neuron. 2013;80:12–34. doi: 10.1016/j.neuron.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galcheva-Gargova Z, et al. The cytoplasmic zinc finger protein ZPR1 accumulates in the nucleolus of proliferating cells. Mol Biol Cell. 1998;9:2963–2971. doi: 10.1091/mbc.9.10.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandeville I, et al. Impact of the loss of Hoxa5 function on lung alveogenesis. Am J Pathol. 2006;169:1312–1327. doi: 10.2353/ajpath.2006.051333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wigglesworth JS, Winston RM, Bartlett K. Influence of the central nervous system on fetal lung development. Experimental study. Arch Dis Child. 1977;52:965–967. doi: 10.1136/adc.52.12.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang HC, et al. Modeling spinal muscular atrophy in Drosophila. PLoS One. 2008;3:e3209. doi: 10.1371/journal.pone.0003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sen A, et al. Modeling spinal muscular atrophy in Drosophila links Smn to FGF signaling. J Cell Biol. 2011;192:481–495. doi: 10.1083/jcb.201004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruns AF, et al. Fibroblast growth factor-2 regulates the stability of nuclear bodies. Proc Natl Acad Sci USA. 2009;106:12747–12752. doi: 10.1073/pnas.0900122106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dubrulle J, McGrew MJ, Pourquie O. FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell. 2001;106:219–232. doi: 10.1016/S0092-8674(01)00437-8. [DOI] [PubMed] [Google Scholar]
- 42.Parrish, M., Nolte, C. & Krumlauf, R. In Encyclopedia of Neurosciences (ed L. R. Squire) 1221–1231 (Oxford Academic Press, 2009).
- 43.Zhao DY, et al. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature. 2016;529:48–53. doi: 10.1038/nature16469. [DOI] [PubMed] [Google Scholar]
- 44.Kielbowicz-Matuk A, Czarnecka J, Banachowicz E, Rey P, Rorat T. “Solanum tuberosum ZPR1 encodes a light-regulated nuclear DNA-binding protein adjusting the circadian expression of StBBX24 to light cycle”. Plant Cell Environ. 2016 doi: 10.1111/pce.12875. [DOI] [PubMed] [Google Scholar]
- 45.O’Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci USA. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le TT, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 47.Jaegle M, et al. The POU factor Oct-6 and Schwann cell differentiation. Science. 1996;273:507–510. doi: 10.1126/science.273.5274.507. [DOI] [PubMed] [Google Scholar]
- 48.Genabai NK, et al. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum Mol Genet. 2015;24:6986–7004. doi: 10.1093/hmg/ddv401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and analysed during this study are included in this published article (and its Supplementary Information files).