

Abstract
Background
Bacteria of the genus Xanthomonas are economically important plant pathogens. Pathogenicity of Xanthomonas spp. depends on the type III-secretion system and additional virulence determinants. The number of sequenced Xanthomonas genomes increases rapidly, however, accurate annotation of these genomes is difficult, because it relies on gene prediction programs. In this study, we used a mass-spectrometry (MS)-based approach to identify the proteome of Xanthomonas euvesicatoria (Xe) strain 85–10 also known as X. campestris pv. vesicatoria, a well-studied member of plant-pathogenic Xanthomonadaceae.
Results
Using different culture conditions, MS-datasets were searched against a six-frame-translated genome database of Xe. In total, we identified 2588 proteins covering 55% of the Xe genome, including 764 hitherto hypothetical proteins. Our proteogenomic approach identified 30 new protein-coding genes and allowed correction of the N-termini of 50 protein-coding genes. For five novel and two N-terminally corrected genes the corresponding proteins were confirmed by immunoblot. Furthermore, our data indicate that two putative type VI-secretion systems encoded in Xe play no role in bacterial virulence which was experimentally confirmed.
Conclusions
The discovery and re-annotation of numerous genes in the genome of Xe shows that also a well-annotated genome can be improved. Additionally, our proteogenomic analyses validates “hypothetical” proteins and will improve annotation of Xanthomonadaceae genomes, providing a solid basis for further studies.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4041-7) contains supplementary material, which is available to authorized users.
Keywords: Xanthomonas, Proteogenome, Ortho proteogenomic, Genome re-annotation, Translational start sites, T3SS, T4SS, T6SS
Background
Since the first sequenced genome of phage ϕX174 in 1978 the number of sequenced genomes has steadily increased due to the development of new and efficient sequencing methods [1, 2]. Today, a major challenge is not the sequencing of new genomes, but the correct annotation of as many genes as possible, the basis for detailed functional analyses. Coding sequences (CDS) are usually annotated using gene prediction software such as Glimmer, Prodigal, Genemark and Easygene [3–6]. The high coding capacity (up to 90%) of bacterial, viral and archaeal genomes [3] require a high accuracy of gene prediction programs. An important quality parameter of prediction software is the sensitivity, i.e., how many of the known genes in a well-characterized genome are predicted [3]. One example is the 99% sensitivity of the first version of Glimmer (2.0) [7]. With respect to protein-coding genes, a major challenge is the correct prediction of the translation start sites (TSS) because homology often decreases in the vicinity of the TSS [8]. Gene annotation quality can be improved by the integration of transcriptome and, more importantly, proteome data using a mass-spectrometry based approach. Proteogenomics integrates shot-gun proteome information into the genome annotation process [9], thereby directly mapping MS-spectra to the six possible open reading frames. This helps to validate predicted protein-coding genes and improves genome annotation. Refinement of a given genome annotation can then be extended to related species using comparative genomics.
Our lab studies the Gram-negative γ-proteobacterium Xanthomonas euvesicatoria strain 85–10 (Xe), also termed X. campestris pv. vesicatoria [10, 11], which causes bacterial spot disease on pepper and tomato plants [12]. The genus Xanthomonas comprises economically important pathogens that together infect a wide range of crop plant species [13]. Xe enters the plant tissue via natural openings, e.g., stomata, or wounds and multiplies locally in the intercellular space [14]. Pathogenicity of Xe relies on the type III-secretion system (T3SS), which is encoded by the chromosomal hrp (hypersensitive response and pathogenicity)-gene cluster [15, 16] and translocates bacterial effector proteins (T3E) directly into the plant cell [17]. Expression of the T3SS components is induced during infection and in special minimal media (e.g., XVM2 [18]). The key regulator HrpG, an OmpR-type response regulator is activated by unknown plant signals and controls the expression of a large hrp-regulon, including many T3E [19]. The isolation of a point mutation in hrpG (termed hrpG*), which renders the HrpG protein constitutively active, was key for the analysis of the T3SS and the identification of new virulence factors [20].
The genome sequence of our model Xe strain 85–10 was published in 2005 [12] and has a G + C-content of 64.5%. Besides the 5.18 Mb chromosome, there are four plasmids, pXCV2, pXCV19, pXCV38 and pXCV183 (1.8 kb, 19 kb, 38 kb and 182.5 kb, respectively) [12]. In the original annotation, 4726 genes for proteins and functional RNAs were predicted. This number did not include yet the 24 genes for small non-protein coding RNAs (sRNAs) which were recently identified by an RNA-seq approach [21]. The latter approach also revealed unusually long 5′-UTRs for a number of T3E genes suggesting incorrectly annotated TSS. One confirmed example is the T3E XopD whose N-terminus had to be extended by 215 amino acids (aa) [22].
Here, we propose a re-annotation of the Xe 85–10 genome using proteogenomic data obtained in a large-scale experiment. This is the first study to propose a Xe genome refinement, which can be extended to other economically important bacterial genera.
Results
Proteogenomic analysis of Xe 85–10
The overall goal of this study was to identify as many proteins as possible that are expressed in the Xe strain 85–10 and its derivative 85*. 85* carries a point mutation in hrpG which renders the expression of the T3SS and effector genes constitutive in minimal media and complex medium NYG [20]. For MS analyses bacteria were grown to exponential and stationary phase, respectively, in three different media: NYG, minimal medium A (MA) pH 7 and XVM2. MA and XVM2 media induce the T3SS and T3E genes [18]. Bacterial cells were ruptured by French press, and the lysates analysed as shown in the flow-sheet (Fig. 1, for details see Methods). MS/MS analyses revealed 845,925 spectra which were assigned to peptide sequences using Sequest and an in silico translated six-frame database of Xe 85–10. The rationale behind this was the aim to cover all annotated coding sequences but also possible CDS missed in the original genome annotation [12]. Peptides were mapped to 2588 CDS thus covering 54.7% of the Xe 85–10 genome. Please note that 2500 CDS map to the chromosome (Additional file 1) and the remaining 88 CDS to the four Xe 85–10 plasmids. Given 1684 hypothetical CDS (termed hypothetical, or putative secreted or membrane proteins) in the originally annotated Xe 85–10 genome, we validated the expression of 764 CDS on the protein level (45%) (Additional file 2).
Fig. 1.
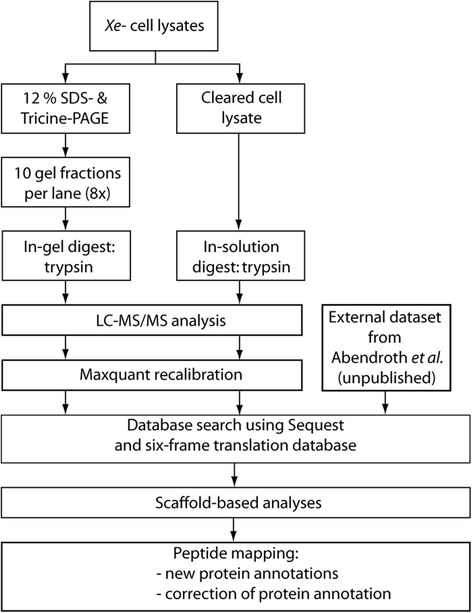
Experimental workflow of the proteogenomic analysis of Xe. The Xe strains 85–10, 85* and 85-10ΔsX13 were grown in NYG, Minimal medium A pH 7 and XVM2, respectively, at 30 °C until OD600 of either 0.5 (exponential), 0.8 (early stationary) or 1.2 (stationary). Proteins extracted from Xe 85–10, 85-10ΔsX13 and 85* cell lysates were separated by 12% SDS PAGE and Tricine PAGE. Gel fractions and cell lysate were digested by trypsin. Samples were analyzed by LC–MS/MS. A database search against a six-frame translation database of Xe 85–10 was performed. Peptides were mapped to the genome of Xe using a TBlastN-based approach. The dataset from Abendroth et al. is originally a comparative study between Xe strains 85–10 and 85-10ΔsX13 and is based on the original genome annotation. The MS spectra of this dataset were also searched against the six-frame database
Mapping of the peptides to the six-frame genome database revealed (i) 50 protein-coding regions with a longer N-terminal region than annotated and (ii) 30 new genes (Fig. 2 and Tables 1 and 2, for additional information see Additional file 3). If the annotation would be corrected based on the new data, 32 genes would overlap now with previously annotated CDS, e.g., the newly identified protein-coding genes XCV_PG10 and XCV_PG15 (Fig. 3a), and raxB and XCV0251 (Fig. 3b), for which the original annotation likely has to be revisited since MS-data for the raxB protein point toward a new TSS and raxA spectra are missing.
Fig. 2.
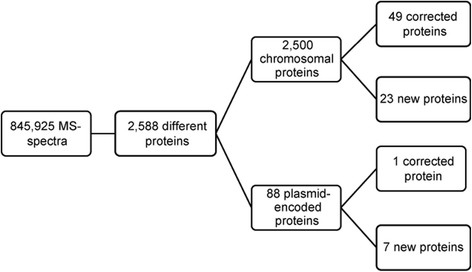
Overview of proteins identified in the proteogenomic analysis of Xe
Table 1.
Summary of incorrectly annotated genes
| IDa | Gene | Chromosomal/plasmid position | Detected length (aa) | Annotated length (aa) |
|---|---|---|---|---|
| Erroneously annotated CDS on Xe chromosome | ||||
| 0056329 | XCV0252 | 292545..293186 | 214 | 103 |
| 0056337 | dcp2 | 295098..297215 | 706 | 697 |
| 0162384 | XCV0352 | 403872..404171 c | 100 | 98 |
| 0107340 | hrpD6 | 469724..470224 c | 167 | 85 |
| 0002631 | xopD | 486784..488823 | 757 | 545 |
| 0057941 | hsdS1 | 576612..577958 | 449 | 419 |
| 0003072 | trpC | 584455..585336 | 294 | 265 |
| 0106644 | XCV0529 | 595352..597223 c | 624 | 532 |
| 0030344 | XCV0537 | 602135..603208 | 358 | 308 |
| 0133900 | XCV0557 | 624664..625527 c | 281 | 247 |
| 0161114 | XCV0564 | 632247..633056 c | 270 | 144 |
| 0003611 | XCV0612 | 693670..694671 | 334 | 326 |
| 0105852 | pheC | 736187..737158 c | 324 | 266 |
| 0032290 | XCV0855 | 974414..975196 | 261 | 260 |
| 0059889 | XCV0861 | 983577..986486 | 968 | 660 |
| 0061379 | XCV1116 | 1247631..1248212 | 194 | 193 |
| 0034580 | raxB | 1401839..1404616 | 926 | 718 |
| 0156885 | XCV1265 | 1423680..1424456 c | 259 | 208 |
| 0007939 | XCV1378 | 1558348..1558917 | 190 | 162 |
| 0156077 | XCV1397 | 1577580..1578143 c | 188 | 185 |
| 0036069 | dapD | 1672904..1674070 | 389 | 285 |
| 0036594 | grpE | 1761374..1762582 | 403 | 172 |
| 0154666 | hutU | 1889784..1891697 c | 638 | 555 |
| 0099525 | XCV1716 | 1935113..1936231 c | 373 | 272 |
| 0010368 | XCV1807 | 2036752..2038401 | 550 | 497 |
| 0098447 | XCV1885 | 2132024..2133079 c | 352 | 188 |
| 0067505 | XCV2100 | 2394132..2395619 | 469 | 306 |
| 0125297 | flgG | 2310361..2311182 c | 274 | 261 |
| 0040728 | exbB2 | 2584091..2584648 | 186 | 183 |
| 0150414 | XCV2312 | 2645805..2646305 c | 167 | 150 |
| 0122465 | XCV2513 | 2837767..2838294 c | 176 | 89 |
| 0094634 | cydD | 2857544..2859259 c | 572 | 570 |
| 0094553 | argB | 2874455..2876008 c | 518 | 426 |
| 0122245 | dksA | 2884537..2885304 c | 256 | 147 |
| 0093090 | gumE | 3162956..3164299 c | 448 | 433 |
| 0120648 | infC | 3173992..3174486 c | 165 | 156 |
| 0091857 | XCV2971 | 3378614..3379942 c | 443 | 375 |
| 0073386 | cheB2 | 3442416..3444254 | 613 | 369 |
| 0019248 | XCV3212 | 3657508..3659298 | 597 | 518 |
| 0020234 | XCV3377 | 3862711..3863067 | 119 | 103 |
| 0075628 | XCV3419 | 3905421..3907445 | 675 | 557 |
| 0114862 | xpsM | 4216381..4217121 c | 247 | 217 |
| 0114852 | xpsK | 4218136..4219170 c | 345 | 301 |
| 0114582 | rmlD | 4282300..4283133 c | 278 | 273 |
| 0086369 | XCV3785 | 4369448..4371499 c | 684 | 616 |
| 0085725 | rpoD | 4490900..4492780 c | 627 | 625 |
| 0078964 | rho | 4539894..4541690 | 599 | 420 |
| 0053728 | guaA | 4966625..4967413 | 266 | 256 |
| 0053999 | XCV4380 | 5042765..5043472 | 236 | 222 |
| Erroneously annotated CDS on Xe plasmid | ||||
| 0166278 | XCVd0050 | 56630..57289 | 220 | 217 |
a Number of the corresponding six-frame-database entry
c chromosomal position on the minus strand
For detailed information see Additional file 3
Table 2.
Novel genes identified in this study
| IDa | Name | Neighboring genes | Detected protein length (aa) | Plausible protein length (aa)b |
|---|---|---|---|---|
| New CDS found on Xe chromosome | ||||
| 0136836 | XCV_PG01 | XCV0062-XCV0063 | 242 | 256 |
| 0055942 | XCV_PG02 | XCV0209-XCV0210 | 114 | 116 |
| 0028571 | XCV_PG03 | XCV0214-XCV0215 | 241 | 306 |
| 0056540 | XCV_PG04 | XCV0282-XCV0283 | 77 | 98 |
| 0065083 | XCV_PG05 | parE-pyrG | 25 | 59 |
| 0094126 | XCV_PG06 | XCV2618-XCV2619 | 76 | 107 |
| 0043902 | XCV_PG07 | XCV2723-XCV2724 | 42 | 70 |
| 0089084 | XCV_PG08 | XCV3389-virB6 | 111 | 161 |
| 0020369 | XCV_PG09 | XCV3391-XCV3392 | 73 | 141 |
| 0143360 | XCV_PG10 | XCV3494 | 47 | 117 |
| 0087222 | XCV_PG11 | XCV3657-xpsD | 59 | 99 |
| 0022971 | XCV_PG12 | XCV3783-XCV3784 | 150 | 191 |
| 0050568 | XCV_PG13 | rsmC-XCV3801 | 131 | 157 |
| 0112004 | XCV_PG14 | kefC-XCV4167 | 125 | 148 |
| 0111304 | XCV_PG15 | xylB2-XCV4282 | 65 | ‡ |
| 0081693 | XCV_PG16 | XCV4416-XCV4417 | 112 | 141 |
| New CDS found on Xe plasmids | ||||
| 0175626 | XCV_PG17 | after XCVa0002 | 53 | 60 |
| 0173148 | XCV_PG18 | before XCVc0001 | 34 | 109 |
| 0172926 | XCV_PG19 | tnpR-XCVc0009 | 74 | 76 |
| 0174118 | XCV_PG20 | XCVc0025-XCVc0026 | 123 | 138 |
| 0169438 | XCV_PG21 | XCVd0054-XCVd0055 | 92 | 132 |
| 0166803 | XCV_PG22 | XCVd0124-XCVd0125 | 107 | 129 |
| New CDS found antisense to annotated CDS | ||||
| 0152041 | XCV_PG23 | anti-XCV2096† | 30 | 39 |
| 0122029 | XCV_PG24 | anti-XCV2593† | 258 | 258 |
| 0049655 | XCV_PG25 | anti-xadA1† | 1552 | 1597 |
| 0080326 | XCV_PG26 | anti-XCV4209† | 200 | 203 |
| 0166979 | XCV_PG27 | anti-XCVd0155† | 41 | 51 |
| 0013218 | XCV_PG28 | anti-glk1* | 143 | 162 |
| 0008300 | XCV_PG29 | anti-XCV1454* | 508 | ‡ |
| 0007106 | XCV_PG30 | anti-gcvP* | 820 | 837 |
a Number of the corresponding six-frame-database entry
† no MS-data for the annotated protein detected
* MS-data for the annotated protein detected
b Protein length till the next plausible translation start site (ATG, GTG, TTG)
‡ No plausible translation start site (ATG, GTG, TTG) between detected peptide and the next upstream stop codon
For detailed information see Additional file 3
Fig. 3.
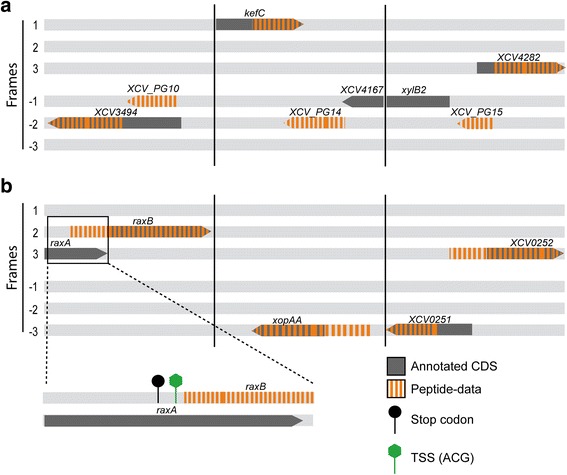
Schematic overview of chromosomal regions with detected new and corrected protein-coding genes. a Examples of three new protein-coding genes detected by proteogenomics, XCV_PG10, XCV_PG14 and XCV_PG15. b Examples of three corrected protein-coding genes detected by proteogenomics with a close-up of the raxA/B region. All six reading frames are shown. Grey: annotated CDS; orange dashes represent peptide-data detected by MS/MS. Black circle represents a stop codon; the green hexagon represents the possible translation start codon of raxB
Protein coding genes with longer N-terminal regions in Xe
The prediction of the most likely TSS is a critical point in genome annotations. In GC-rich genomes, ~60% of genes might have a incorrectly annotated TSS [23]. To identify erroneously annotated TSS we searched for peptides located upstream of and in the same frame as a previously annotated TSS. Out of the 50 longer genes 49 are encoded on the chromosome and one on pXCV183, the largest plasmid (Table 1). Among the longer genes is dksA, which now largely overlaps with XCV2557, encoded on the opposite strand (Fig. 4a) and not represented by any peptides in this study. Thus, we propose to delete XCV2557. That this appears to be justified is based on a previous transcriptome study which revealed a transcription start site for dksA overlapping with XCV2557 [21]. Given the dksA transcription start site and peptides covering this genomic region our new data suggest two possible TSS (Fig. 4a). Site-directed mutagenesis of the annotated and the possible TSS revealed that protein translation most likely starts at the first GTG (Fig. 4b). Using expression constructs whose expression is driven by the native promoter, we observed not only that the first GTG is used, but also a processed variant of DksA. We also experimentally analyzed TSS of XCV1265, encoding a D-alanyl-D-alanine carboxypeptidase. For XCV1265, peptide data suggest a TSS further upstream than in the annotation, which was confirmed by site-directed mutagenesis (Fig. 4c and d).
Fig. 4.
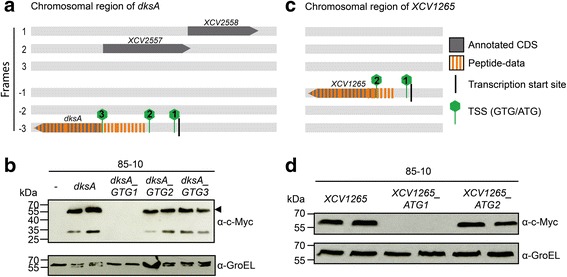
Gene organization of the dksA and XCV1265 regions. a and c dksA and XCV1265 loci of Xe. All six reading frames are shown. Grey: annotated CDS; orange dashes: peptide-data detected by MS/MS; green hexagons: possible translation start codons of dksA and XCV1265. b and d Analysis of potential translation start codons of DksA and XCV1265. Total protein extracts of Xe 85–10 containing pBRM-P(dksA), pBRM-P(dksA_GTG1), pBRM-P (dksA_GTG2), pBRM-P (dksA_GTG3), pBRM-P (XCV1265), pBRM-P (XCV1265_ATG1), pBRM-P (XCV1265_ATG2) or an empty vector (−) were separated by 12% SDS PAGE and analyzed by immunoblotting using a c-Myc-specific antibody. As loading control, membranes were reacted with a GroEL-specific antibody. Experiments were repeated at least twice with similar results
Another example of a longer than previously thought gene is dcp2 for which a peptide overlapping the annotated GTG TSS suggests an ATG start codon further upstream. This is supported by ortho-proteogenomic analysis of other members of the Xanthomonadaceae (Additional file 4). Similarly, our peptide data and an ortho-proteogenomic analysis indicate that exbB2 is incorrectly annotated (Additional file 4). Surprisingly, given the 64% G + C content in Xe, we found that for infC obviously the codon ATT is used as TSS. Here, we detected peptides further upstream of the previously annotated TSS. Additional sequence analyses showed that the only possible TSS is an ATT, because there is no alternative start codon (common TSS: ATG, GTG, TTG) between the last peptide-covered sequence and the stop codon (Additional file 4). InfC is well-analyzed in other bacteria, e.g., E. coli, where the same TSS codon is used [24, 25].
Identification and verification of novel protein-coding genes
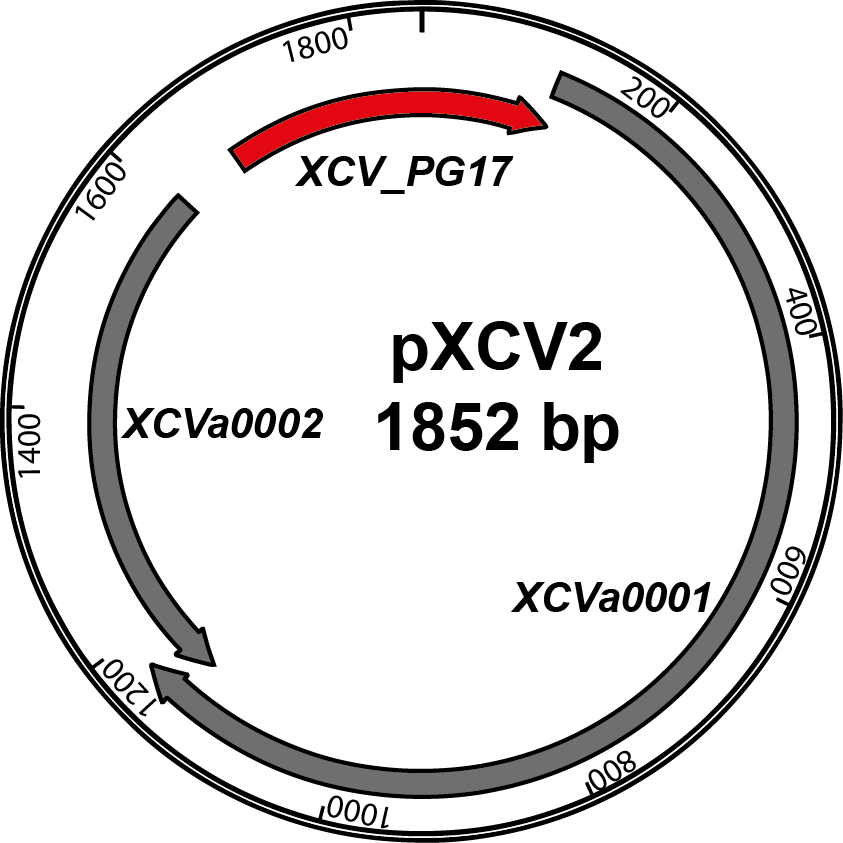
Intriguingly, our Xe proteomic approach identified 30 new genes encoding mostly small proteins with an average size of 191 amino acids (aa), ranging from 25 to 820 aa (Table 2). Among the small proteins is sX6, which was first assumed to act as sRNA but encodes a protein [21], which we could verify in our data. Xe harbours the 1852 bp plasmid pXCV2, which was thought to encode two protein-coding genes [26]. In this study, we detected peptides mapping to a third CDS which is located between position 1673 and 114 and encodes a protein of 60 aa (Additional file 5). Most new proteins have no annotated counterpart in other Xanthomonas genomes. However, 15 of the 30 protein-coding genes are conserved on the DNA level (Blast output: at least 80% coverage and 80% identity) suggesting that the corresponding proteins are also produced in other xanthomonads.
We predicted functional domains in the newly identified proteins. Interestingly, XCV_PG01, located between XCV0062 and XCV0063, encodes a putative serine/threonine phosphatase of the 2C family, which was previously overlooked. Furthermore, we identified a putative YecR-like lipoprotein, XCV_PG06, which was recently annotated in X. oryzicola [27]. A special case is XCV_PG30, which is encoded antisense to gcvP, predicted to encode a metal-dependent RNase. Both corresponding proteins are represented by peptides in this study. However, most new proteins lack known functional domains.
To validate the MS-data experimentally by an independent method, the expression of five new protein-coding genes was tested. An important criterion for the selected genes was the knowledge of the exact transcription start sites [21]. C-terminal c-Myc tagged expression constructs under the control of the native promoter were generated in pBRM-P and transformed into Xe 85–10. As shown in Fig. 5a, all tested new CDS expressed proteins of expected molecular mass. In case of XCV_PG02 there are five possible TSS (Fig. 5b). As the correct TSS cannot be deduced from the immunoblot it needs to be determined by alternative methods.
Fig. 5.

Validation of five new protein-coding genes. a Detection of the protein synthesis of new Xe proteins. Total protein extracts of Xe 85–10 containing pBRM-P (XCV_PG13), pBRM-P (XCV_PG17), pBRM-P (XCV_PG07), pBRM-P (XCV_PG02) or pBRM-P (XCV_PG06) grown in NYG were separated by 15% SDS PAGE and analyzed by immunoblotting using a c-Myc-specific antibody. b Gene organization of the XCV_PG02 locus. XCV0209 and XCV_PG02 are highlighted. Arrows represent possible translation start codons of XCV_PG02
The putative T6SS of Xe has no virulence function in standard virulence assays
Since the bacteria were grown in T3SS-inducing conditions, we expected to detect peptides corresponding to known virulence factors, i.e., T3SS components and T3E. Our MS-analysis identified 69% of T3E and 84% of structural and regulatory T3SS proteins (Table 3 and Additional file 6). Furthermore, 10 of 11 known Xps type II-secretion system (T2SS) components and 3 of 5 known substrates [28, 29] were detected (Table 3 and Additional file 6). Xe also encodes the Xcs T2SS, which in contrast to the Xps T2SS does not contribute to virulence [28]. No components of the Xcs T2SS were detected in our study. Various components of type IV-secretion systems (T4SS) [12] and type VI-secretion systems (T6SS) are encoded in Xe [30], but it is unknown if these putative secretion systems are functional in Xe. In order to identify a potential virulence function of putative T4SS and T6SS, we analyzed whether components were detected in our MS-data. Ten out of the 18 predicted components of the Vir-type T4SS were detected in our MS/MS-data, but no component of the Icm/Dot-type T4SS (Table 3 and Additional file 6). In addition, we analyzed two loci in the Xe genome, each encoding 15 conserved T6SS components (Fig. 6a). Only the T6SS component TssH/ClpV was detected in our MS-data (Table 3 and Additional file 6).
Table 3.
Summary of MS/MS-data on secretion systems
| Secretion system | # of detected / known proteins |
|---|---|
| Tat and Sec-dependent secretion | 15/19 |
| T1SS | 4/4 |
| T2SS – Xcs-type | 0/12 |
| T2SS – Xps-type | 10/11 |
| T2SS – substrates | 3/5 |
| T3SS | 21/25 |
| T3E | 25/36 |
| T4SS – vir-type | 10/18 |
| T4SS – icm-type | 0/15 |
| T5-autotransporter | 3/4 |
| T6SS – locus 1 | 0/16 |
| T6SS – locus 2 | 1/16 |
For detailed list see Additional file 6 (Excel file)
Fig. 6.
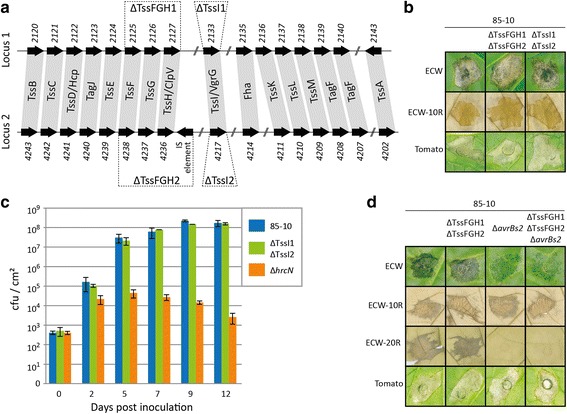
Deletion of conserved T6SS components has no effect on Xe virulence. a Schematic representation of the two genomic T6SS loci in Xe. Gene numbers and commonly used gene names of T6SS components identified in Xe 85–10 are given. Dashed lines mark genes deleted in this study. b Xe strains 85–10, 85-10ΔTssFGH1ΔTssFGH2 and 85-10ΔTssI1ΔTssI2 were inoculated into susceptible pepper plants (ECW), resistant pepper plants (ECW-10R) and susceptible tomato plants with an OD600 of 0.1. Phenotypes were documented 7 days post inoculation (7 dpi, ECW), 2 dpi (ECW-10R) and 8 dpi (tomato). c Xe strains 85–10, 85-10ΔTssI1ΔTssI2 and 85-10ΔhrcN (T3SS mutant) were inoculated in ECW plants with an OD600 of 4 × 10−5. Bacterial multiplication in leaves was monitored for 12 days. d Xe strains 85–10, 85-10ΔTssFGH1ΔTssFGH2, 85-10ΔavrBs2 and 85-10ΔTssFGH1ΔTssFGH2ΔavrBs2 were inoculated with an OD600 of 0.1 into leaves of pepper plants (ECW, ECW-10R, ECW-20R) and tomato plants. Phenotypes were documented 6 dpi (ECW), 2 dpi (ECW-10R, ECW-20R) and 9 dpi (tomato). Leaves of ECW-10R and ECW-20R plants were bleached in EtOH for better visualization of cell death reactions. Experiments were repeated twice with similar results
To test whether the putative T6SS contribute to virulence of Xe, deletion mutants were generated. On one hand, we deleted the TssI/VgrG-encoding gene of both loci (xcv2133 and xcv4217, termed TssI1 and TssI2) and on the other hand, TssF-, TssG- and TssH/ClpV-encoding genes of both loci (XCV2125-XCV2127, termed TssFGH1 and XCV4236-XCV4238, termed TssFGH2) were deleted (Fig. 6a). In characterized T6SS these components are essential for function [31]. The generated mutants, Xe 85-10ΔTssFGH1ΔTssFGH2 and Xe 85-10ΔTssI1ΔTssI2 were inoculated into pepper and tomato plants to test for virulence. The deletion mutants behaved like wild type, i.e., disease symptoms in susceptible plants and HR induction in resistant pepper plants (Fig. 6b). We also monitored the in planta growth of Xe 85-10ΔTssI1ΔTssI2 in comparison to Xe 85–10 in leaves of susceptible pepper plants; there were no significant differences (Fig. 6c). As a control, a strain without functional T3SS (Xe 85-10ΔhrcN) was used [32]. As expected, multiplication of Xe 85-10ΔhrcN was strongly reduced (Fig. 6c). Next, we additionally deleted avrBs2 in Xe 85-10ΔTssFGH1ΔTssFGH2. The T3E AvrBs2 is recognized in ECW-20R pepper plants [33] and is a conserved virulence factor in xanthomonads [34]. Deletion of avrBs2 renders Xe less virulent and helps to analyze subtle virulence effects when other genes are mutated. However, disease symptoms induced by Xe 85-10ΔTssFGH1ΔTssFGH2ΔavrBs2, in pepper ECW and tomato plants were comparable to those induced by Xe 85-10ΔavrBs2 (Fig. 6d). Taken together, deletion of conserved T6SS components did not affect virulence of Xe under the conditions tested.
Discussion
Because of its economical relevance, Xanthomonas spp. are currently subject of intense sequencing efforts and more and more genomes are available [35]. Here, we demonstrate the importance of proteogenomics for a better characterization of these important plant pathogens. Analyses of a large MS-spectra data set of Xe 85–10 and 85*, cultivated in different media, identified 30 new genes and 50 incorrectly annotated CDS. The number of inaccuracies in the Xe genome annotation [12] is comparable to previous proteogenomic studies of other bacteria, e.g., Yersina, Helicobacter, Mycobacterium, Rugeria and Deinococcus [25, 36–39]. These studies led to the refinement of 4–41 new and 5–73 falsely annotated genes and reached overall genome coverage of 31–80%. Thus, our study (55%) falls into the average genome coverage. It is expected that the coverage will increase with the number of conditions tested, because proteins might be exclusively synthetized under specific conditions or below the detection limit. As our lab focusses on the analysis of proteins important for the host-pathogen interaction, we chose respective conditions, i.e., XVM2, minimal medium A pH 7 (MA 7) and Xe strain 85*. Identified peptides corresponded to 25 (69%) known T3E and 21 (84%) gene products of the hrp-gene cluster (Additional file 6). Eleven known T3E were not detected, which might be due to a low abundance within the bacterial cell. Two detected T3E, XopD und XopAA, are longer than annotated. The original Xe annotation stipulates that these effectors have a size of 545 aa (XopD) and 616 aa (XopAA) respectively. The MS/MS-data showed that XopD and XopAA are 215 aa and 72 aa, respectively, longer. These results are consistent with published data [22, 40] and corroborate the idea that unusually long 5′ untranslated regions in Xe T3E mRNAs might hint to incorrectly annotated transcription start sites [21, 22, 41]. These findings are of special importance as the N-terminal regions of T3E usually harbor the T3SS-secretion and -translocation signals [42]. The knowledge of the exact TSS is crucial for further studies of T3E.
Genome annotation inaccuracies are often due to CDS which are present in a small number of organisms, so that the power of comparative genomics is limited. Validation of longer proteins and newly identified proteins requires additional experimental evidence. In contrast to previous studies [39], we made an effort to validate novel proteins by Western blot analysis, using expression constructs controlled by the corresponding native promoter. The combined use of MS- and transcriptome data can suggest the existence of new genes, but detection of RNA alone is no proof for the existence of a CDS.
The use of the native promoter is only feasible for genes with a known transcription start site. Based on the transcriptome data of Schmidtke et al. (2012) seven expression constructs were created, and the synthesis of proteins was demonstrated by Western blot. For DksA and XCV1265 we detected signals corresponding to proteins with higher molecular weight than previously annotated and confirmed the respective TSS using site-directed mutagenesis. The transcription of dksA starts within XCV2557 [21], encoded on the opposite strand (Fig. 4a). We propose that the previously annotated gene XCV2557 next to dksA, for which peptide data are missing, does not exist, as it greatly overlaps with the newly proposed annotation of dksA. As for dksA, Xe harbors many other transcription start sites internal of protein-coding regions which might be a hint for annotation mistakes.
Furthermore, we propose that the infC translation does not start with the annotated ATG, but with ATT. An ATT start codon was also found in a proteogenomic study of Deinococcus deserti [25]. Interestingly, the translation of infC in E. coli is also initiated at an ATT start codon. In E. coli, the ATT start codon is used for auto-regulation of translation [24].
Besides the T3SS other secretion systems might play a role in Xe virulence. Our MS-analysis detected 56% of the components of a putative Vir-type T4SS. By contrast, components of the Xcs T2SS and the putative IcmDot-type T4SS were not detectable. Besides Xe, putative T4SS are encoded in many other xanthomonads, e.g., X. axonopodis pv. citri [43], X. citri pv. citri [44] and X. campestris pv. campestris [45]. The function of these systems has only been studied in a few cases. The Vir-type T4SS of X. campestris pv. campestris does not contribute to bacterial virulence [46] and transcription of Vir-type T4SS components is downregulated during infection of host plants in X. citri pv. citri 306 [47]. In addition, the T4SS was shown to act against other Gram-negative bacteria in a contact-dependent manner [44, 48].
T6SS are encoded in many xanthomonads [30] and the genome of Xe 85–10 harbors two T6SS loci, each encoding 15 conserved T6SS components. It is not unusual that bacterial genomes harbor different T6SS loci. For example, Pseudomonas aeruginosa encodes three independent T6SS [49] and Burkholderia thailandensis five independent T6SS [50]. Only a single T6SS component was detected in our MS-approach, suggesting that both T6SS of Xe might play a role under different conditions. Since a function of a T6SS in xanthomonads is elusive, we generated mutants in putative T6SS genes in Xe. However, the deletion mutants revealed no obvious role of the putative T6SS in the interaction with plants. The genes we deleted are predicted to result in a loss of function [31]. The T6SS of Xe might target other bacterial species, as shown for T6SS of Vibrio cholerea [51, 52], Serratia marcescens [53], Salmonella Typhimurium [54] and P. aeruginosa [55]. To answer this question was out of scope of this study and has to await further studies.
Conclusions
Here, we describe that the well-annotated genome of Xe can be improved. Besides validation of “hypothetical” proteins, we discovered novel protein-coding genes and corrected the annotation of 50 genes. Proteins of particular biological interest, e.g., a serine/threonine phosphatase, putative secreted proteins and proteins containing domains of unknown functions were identified. Furthermore, the annotation of protein-coding genes which play a role in Xanthomonas virulence have been corrected, e.g., the T3SS-component HrpD6 and the T3SS-substrate XopAA. This proteogenomic analysis will improve annotations of Xanthomonadaceae genomes. Future studies of newly identified genes might unravel new virulence functions.
Methods
Bacterial strains and growth conditions
For bacterial strains, plasmids and oligonucleotides used in this study see Additional file 7. The Xe strains 85–10 [12, 56], 85* [20] and 85-10ΔsX13 [57] were grown in NYG [58], Minimal medium A pH 7 [59] and XVM2 [18], respectively, at 30 °C until OD600 of 0.5 (exponential), 0.8 (early stationary) or 1.2 (stationary). Plasmids were introduced into Xe by tri-parental conjugation, using pRK2013 as helper plasmid [60, 61]. Antibiotics were added to a final concentration of: gentamycin, 15 μg/ml; rifampicin, 100 μg/ml, 100 μg/ml spectinomycin.
Protein extraction and pre-separation
Cells were cracked in TE-buffer using three times French press. Cell debris and undissolved material were removed by centrifugation (15 min, 16,000×g, 4 °C). Protein concentrations were measured using the Bradford assay. 100 μg protein were precipitated over night with ice-cold acetone. Protein pellets were dissolved in 40 μl Laemmli-buffer, and 20 μl were subjected to 1-D-SDS PAGE (12% separation gel, 4% stacking gel). The gel was fixed overnight in 40% methanol and 10% acetic acid, and stained with colloidal Coomassie (20% Ethanol; 1.6% phosphoric acid; 8% ammonium sulfate; 0.08% Coomassie Brilliant Blue G-250).
LC-MS/MS-measurements and data analysis
Lanes of the protein gel were cut into 10 slices of equal size and proteins were digested in gel by trypsin. The eluted peptides were subjected to LC-MS/MS-analysis on a Proxeon nLC 1000 coupled to an Orbitrap Elite mass spectrometer. In-house self-packed columns (i.d. 100 μm, o.d. 360 μm, length 150 mm; packed with 1.7 μm Aeris XB-C18 reversed-phase material (Phenomenex, Torrance, CA, USA) were loaded, then desalted with 10 μl buffer A (0.1% (v/v) acetic acid) at a maximum pressure of 750 bar. For LC-MS/MS-analysis, peptides were eluted using a nonlinear 80 min gradient from 1 to 99% buffer B (0.1% (v/v) acetic acid in acetonitrile) at a constant flow rate of 300 nl/min. Spectra were recorded in an Orbitrap Velos (Thermo Fisher Scientific, Waltham, MA, USA) at a resolution of r = 30,000 with lockmass correction activated. After acquisition of the Full-MS-spectra, up to 20 dependent scans (MS/MS) were performed according to precursor intensity by collision-induced dissociation fragmentation (CID) in the linear ion trap.
Data were analyzed by Sorcerer Sequest against a six-frame translated database of the whole Xe genome (protein database containing 175,698 (21,627 ≤ 6 aa) entries). The following search parameters were used: enzyme type, trypsin (KR); peptide tolerance, 10 ppm; tolerance for fragment ions, 1 Da; b- and y-ion series; a maximum of two modifications per peptide was allowed. Peptide and protein identifications were accepted with a false discovery rate (FDR) of maximal 0.4%, requiring a minimum of at least two unique peptides for protein identification and quantification.
A second data set was generated using MS/MS-data obtained from a comparative proteome experiment. The tryptic digests obtained from the 1-D-SDS PAGE gel pieces were subjected to reversed phase column chromatography (Waters BEH 1.7 μm, 100 μm i. d. × 100 mm, Waters Corporation, Milford, MA, USA) operated on a nanoACQUITY-UPLC (Waters Corporation, Milford, MA, USA). Peptides were concentrated and desalted on a trapping column (Waters nanoACQUITY UPLC column, Symmetry C18, 5 μm, 180 μm × 20 mm, Waters Corporation, Milford, MA, USA) for 3 min at a flow rate of 1 ml/min with 99% buffer A (0.1% acetic acid). Subsequently, the peptides were eluted and separated using a non-linear 80-min gradient from 5 to 60% ACN in 0.1% acetic acid at a constant flow rate of 400 nl/min. MS and MS/MS-data were obtained using the LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a nanoelectrospray ion source. After a survey scan in the Orbitrap (r = 30,000) with the lockmass option enabled, MS/MS-data were recorded for the five most intensive precursor ions in the linear ion trap. Singly charged ions were not taken into account for MS/MS-analysis.
Data were analyzed by Sorcerer Sequest against the 6-frame database. The following search parameters were used: enzyme type, trypsin (KR); peptide tolerance, 10 ppm; tolerance for fragment ions, 1 Da; b- and y-ion series; a maximum of two modifications per peptide was allowed. Peptide and protein identifications were accepted with a false discovery rate below 1%, requiring a minimum of at least two unique peptides for protein identification and quantification.
Peptide mapping and visualization
Identified peptides were mapped to the Xe genome using TBlastN [62], perfect and full length sequence matches were used. With this setup the best fit for the peptide to the Xe-DNA sequence was selected. The peptides were visualized in Artemis genome browser [63]. GFF files can be found in Additional file 8.
Generation and mutation of expression constructs
For expression in Xe, protein coding sequences and the putative promoter region of XCV_PG02, XCV_PG06, XCV_PG07, XCV_PG13, XCV_PG17, dksA and XCV1265 were amplified from genomic DNA of Xe 85–10 by PCR using oligonucleotides listed in Table 3 and cloned into pBRM-P [64] by Golden Gate cloning [65]. pBRM-P encodes a c-Myc epitope which is fused to the 3′ end of the insert.
To mutate possible TSS, site-directed mutagenesis was employed. For this, pBRM-P (XCV1265) or pBRM-P (dksA) were used as a template and PCR amplified using oligonucleotides harboring the desired mutation (Additional file 7). Primers carried a 5′ phosphate for subsequent circulation of amplicons.
Protein analysis
To analyze the protein synthesis of XCV_PG02, XCV_PG06, XCV_PG07, XCV_PG13, XCV_PG17, DksA and XCV1265 Xe 85–10 bacteria with corresponding expression constructs were grown overnight in NYG medium until stationary phase. Protein extracts were analyzed by SDS-PAGE and immunoblotting using first an antibody specific for the c-Myc epitope (Santa Cruz Biotechnology, Dallas, TX, USA) and secondly, anti-GroEL (Enzo Life Sciences, Farmingdale, NY, USA). Secondary antibodies were horseradish peroxidase labeled anti-mouse or anti-rabbit antibodies (GE Healthcare, Chicago, IL, USA). Antibody reactions were visualized by enhanced chemiluminescence.
Generation of deletion mutants
To generate deletion mutants, regions of about 1 kb flanking of the deleted sequences were amplified by PCR and cloned into the suicide vectors pOGG2 via Golden Gate cloning or pOKI via classical cloning (Table 3). An IS-element is encoded subsequently before XCV4236 (TssH/ClpV), which was deleted together with XCV4236-XCV4238 (TssFGH2). pOGG2 derivatives or pOKI (avrBs2) were conjugated into Xe and mutants were selected by PCR.
Plant infection assays
Plants were grown in the greenhouse with 23 °C/25 °C day temperature (tomato/pepper) and 19 °C night temperature, 16 h of light and 40–60% humidity. For plant infection assays, Xe suspended in 10 mM MgCl2 were inoculated with a needleless syringe into leaves of the near-isogenic pepper (Capsicum annuum) cultivars ECW, ECW-10R or ECW-20R or tomato (Solanum lycopersicum) cultivar MoneyMaker [33, 66]. Pepper ECW is a commercial cultivar that has been used to introgress disease resistance genes and generate near-isogenic lines [33]. The tomato and pepper plants were grown as described before [56, 67].
Additional files
{kind=link}
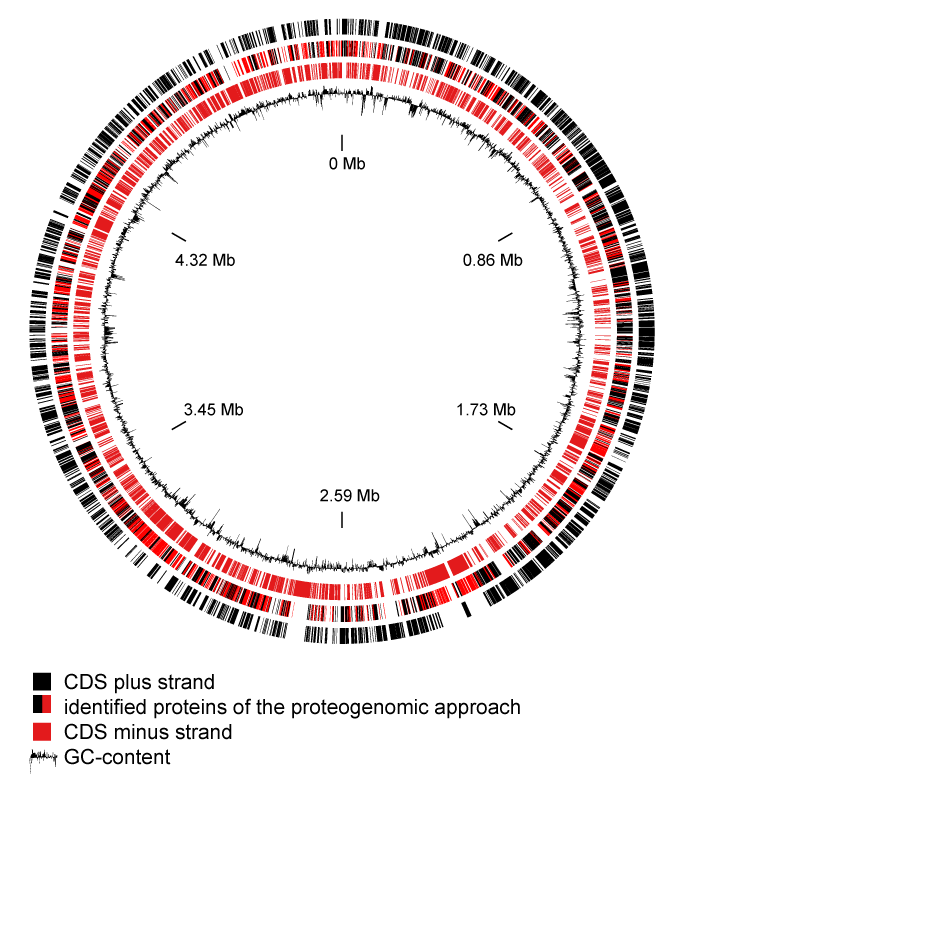
Proteogenomic identification of proteins in Xe 85–10. Overview of the Xe chromosome showing all annotated and MS-data based identified protein-coding genes. Black: annotated CDS plus strand, Red: annotated CDS minus strand, Black-Red: MS-data based identified CDS, Black serrates line: GC-content. (PNG 133 kb)
Identification of all detected proteins and their annotated function. List of all detected annotated proteins in MS-data and their predicted functions. (XLSX 693 kb)
Additional information to Tables 1 and 2 and all conditions and strains used in this study. The tables show additional information, e.g. predicted function, homology and transcription start site, of the new and falsely incorrectly annotated protein-coding genes and all conditions and strains used in this study. (XLSX 29 kb)
{kind=link}
Reannotation of dcp2, exbB2, flgG and infC. Multiple sequence alignment of dcp2, exbB2, flgG and infC homologs of Xe, X. axonopodis pv. citrumelo F1 (XacF1), X. oryzae pv. oryzae KACC10331 (Xoo), X. oryzae pv. oryzicola BLS256 (Xoc), X. fuscans subsp. aurantifolii ICPB 11122 (Xfa), X. perforans 91–118 (Xp). Green: experimentally detected by MS, underlined in red: annotated start codons, underlined in green: possible new start codon. (PNG 836 kb)
{kind=link}
pXCV2 carries a third CDS. Representation of pXCV2 plasmid of Xe 85–10. Grey arrows show position of annotated CDS and the red arrow indicates the position of the newly identified protein-coding CDS. (PNG 88 kb)
Detection of proteins which are components or substrates of (potential) secretion systems. +: specific peptide detected, −: no specific peptide detected in MS-data. (XLSX 15 kb)
Oligonucleotides, plasmids and strains used in this study. List of oligonucleotides, plasmids and strains used in this study. (DOCX 45 kb)
GFF annotation file. GFF annotation file for artemis genome browser. (TXT 147 kb)
Acknowledgements
We thank Alexander Schröder, Hannelore Espenhahn and Bianca Rosinsky for excellent technical assistance and Johannes Stuttmann and Cornelius Schmidtke for helpful discussions.
Funding
The work was supported by grands from the Deutsche Forschungsgemeinschaft as part of the priority program ‘Sensory and Regulatory RNAs in Prokaryotes (SPP 1258) and the Gottfried Wilhelm Leibniz-Preis to UB. The funding bodies were not involved in the planning and execution of the study.
Availability of data and materials
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [68, 69] partner repository with the dataset identifier PXD007140.
Abbreviations
- aa
Amino acid
- CDS
Coding sequence
- Da
Dalton
- ECW
Early cal wonder
- FDR
False discovery rate
- GFF
General feature format
- LC
Liquid chromatography
- MA
Mini medium A
- MS
Mass spectrometry
- sRNA
Small RNA
- T2SS
Type II secretion system
- T3E
Type III effector
- T3SS
Type III - secretion system
- T4SS
Type IV - secretion system
- T6SS
Type VI - secretion system
- TSS
Translation start site
- UTR
Untranslated region
Authors’ contributions
UA and UB planned experimental approach. AO and DB performed mass spectrometry of protein samples, delivered by UA. UA, together with BG, AO and DB, analyzed MS-data. UA interpreted MS-data and validated new and re-annotated proteins. NA characterized the T6SS. UA prepared the manuscript with contribution from NA and UB and all authors reviewed the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
There is no permission needed to work with the pepper and tomato plants used in this study. The plants are commercially used and were bred and published. There are no local guidelines restricting the use of the plants used in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4041-7) contains supplementary material, which is available to authorized users.
Contributor Information
Ulrike Abendroth, Email: ulrike.abendroth@gmx.net.
Norman Adlung, Email: norman.adlung@genetik.uni-halle.de.
Andreas Otto, Email: andreas.otto@uni-greifswald.de.
Benjamin Grüneisen, Email: benjamin.grueneisen@uk-halle.de.
Dörte Becher, Email: dbecher@uni-greifswald.de.
Ulla Bonas, Email: ulla.bonas@genetik.uni-halle.de.
References
- 1.Sanger F, Coulson AR, Friedmann T, Air GM, Barrell BG, Brown NL, et al. The nucleotide sequence of bacteriophage phiX174. J Mol Biol. 1978;125(2):225–246. doi: 10.1016/0022-2836(78)90346-7. [DOI] [PubMed] [Google Scholar]
- 2.Médigue C, Danchin A. Annotating bacterial genomes. Mod Genome Annotation. 2008:165–90.
- 3.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics. 2007;23(6):673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11(1):119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;33(Web Server issue):W451–W454. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larsen TS, Krogh A. EasyGene–a prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinform. 2003;4(1):21. doi: 10.1186/1471-2105-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salzberg SL, Delcher AL, Kasif S, White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998;26(2):544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29(12):2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renuse S, Chaerkady R, Pandey A. Proteogenomics. Proteomics. 2011;11(4):620–630. doi: 10.1002/pmic.201000615. [DOI] [PubMed] [Google Scholar]
- 10.Jones JB, Lacy GH, Bouzar H, Stall RE, Schaad NW. Reclassification of the xanthomonads associated with bacterial spot disease of tomato and pepper. Syst Appl Microbiol. 2004;27(6):755–762. doi: 10.1078/0723202042369884. [DOI] [PubMed] [Google Scholar]
- 11.Barak JD, Vancheva T, Lefeuvre P, Jones JB, Timilsina S, Minsavage GV, et al. Whole-genome sequences of Xanthomonas euvesicatoria strains clarify taxonomy and reveal a stepwise erosion of type 3 effectors. Front Plant Sci. 2016;71805 [DOI] [PMC free article] [PubMed]
- 12.Thieme F, Koebnik R, Bekel T, Berger C, Boch J, Büttner D, et al. Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. Vesicatoria revealed by the complete genome sequence. J Bacteriol. 2005;187(21):7254–7266. doi: 10.1128/JB.187.21.7254-7266.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leyns F, De Cleene M, Swings J-G, De Ley J. The host range of the genus Xanthomonas. Bot Rev. 1984;50(3):308–356. doi: 10.1007/BF02862635. [DOI] [Google Scholar]
- 14.Büttner D, Bonas U. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol Rev. 2010;34(2):107–133. doi: 10.1111/j.1574-6976.2009.00192.x. [DOI] [PubMed] [Google Scholar]
- 15.Bonas U, Schulte R, Fenselau S, Minsavage GV, Staskawicz BJ, Stall RE. Isolation of a gene cluster from Xanthomonas campestris pv. vesicatoria that determines pathogenicity and the hypersensitive response on pepper and tomato. Mol Plant-Microbe Interact. 1991;4(1):81–88. doi: 10.1094/MPMI-4-081. [DOI] [Google Scholar]
- 16.Fenselau S, Balbo I, Bonas U. Determinants of pathogenicity in Xanthomonas campestris pv. vesicatoria are related to proteins involved in secretion in bacterial pathogens of animals. Mol Plant-Microbe Interact. 1992;5(5):390–396. doi: 10.1094/MPMI-5-390. [DOI] [PubMed] [Google Scholar]
- 17.Bonas U, Van den Ackerveken G, Büttner D, Hahn K, Marois E, Nennstiel D, et al. How the bacterial plant pathogen Xanthomonas campestris pv. vesicatoria conquers the host. Mol Plant Pathol. 2000;1(1):73–76. doi: 10.1046/j.1364-3703.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 18.Wengelnik K, Marie C, Russel M, Bonas U. Expression and localization of HrpA1, a protein of Xanthomonas campestris pv. Vesicatoria essential for pathogenicity and induction ofthe hypersensitive reaction. J Bacteriol. 1996a;178(4):1061-9. [DOI] [PMC free article] [PubMed]
- 19.Wengelnik K, Van den Ackerveken G, Bonas U. HrpG, a key hrp regulatory protein of Xanthomonas campestris pv. vesicatoria is homologous to two-component response regulators. Mol Plant-Microbe Interact. 1996b;9(8):704-12. [DOI] [PubMed]
- 20.Wengelnik K, Rossier O, Bonas U. Mutations in the regulatory gene hrpG of Xanthomonas campestris pv. Vesicatoria result in constitutive expression of all hrp genes. J Bacteriol. 1999;181(21):6828–6831. doi: 10.1128/jb.181.21.6828-6831.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmidtke C, Findeiß S, Sharma CM, Kuhfuß J, Hoffmann S, Vogel J, et al. Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions. Nucleic Acids Res. 2012;40(5):2020–2031. doi: 10.1093/nar/gkr904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canonne J, Marino D, Noël LD, Arechaga I, Pichereaux C, Rossignol M, et al. Detection and functional characterization of a 215 amino acid N-terminal extension in the Xanthomonas type III effector XopD. PLoS One. 2010;5(12):e15773. doi: 10.1371/journal.pone.0015773. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Nielsen P, Krogh A. Large-scale prokaryotic gene prediction and comparison to genome annotation. Bioinformatics. 2005;21(24):4322–4329. doi: 10.1093/bioinformatics/bti701. [DOI] [PubMed] [Google Scholar]
- 24.Butler JS, Springer M, Grunberg-Manago M. AUU-to-AUG mutation in the initiator codon of the translation initiation factor IF3 abolishes translational autocontrol of its own gene (infC) in vivo. Proc Natl Acad Sci U S A. 1987;84(12):4022–4025. doi: 10.1073/pnas.84.12.4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baudet M, Ortet P, Gaillard JC, Fernandez B, Guérin P, Enjalbal C, et al. Proteomics-based refinement of Deinococcus deserti genome annotation reveals an unwonted use of non-canonical translation initiation codons. Mol Cell Proteomics. 2010;9(2):415–426. doi: 10.1074/mcp.M900359-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu L-T, Tseng Y-H. Characterization of the IncW cryptic plasmid pXV2 from Xanthomonas campestris pv. Vesicatoria. Plasmid. 2000;44(2):163–172. doi: 10.1006/plas.2000.1468. [DOI] [PubMed] [Google Scholar]
- 27.Niu XN, Wei ZQ, Zou HF, Xie GG, Wu F, Li KJ, et al. Complete sequence and detailed analysis of the first indigenous plasmid from Xanthomonas oryzae pv. oryzicola. BMC Microbiol. 2015;15:233. doi: 10.1186/s12866-015-0562-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szczesny R, Jordan M, Schramm C, Schulz S, Cogez V, Bonas U, et al. Functional characterization of the Xcs and Xps type II secretion systems from the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria. New Phytol. 2010;187(4):983–1002. doi: 10.1111/j.1469-8137.2010.03312.x. [DOI] [PubMed] [Google Scholar]
- 29.Solè M, Scheibner F, Hoffmeister AK, Hartmann N, Hause G, Rother A, et al. Xanthomonas campestris pv. vesicatoria secretes proteases and xylanases via the Xps-type II secretion system and outer membrane vesicles. J Bacteriol. 2015;197(17):2879–2893. doi: 10.1128/JB.00322-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I. Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genomics. 2009;10(1):104. [DOI] [PMC free article] [PubMed]
- 31.Cianfanelli FR, Monlezun L, Coulthurst SJ. Aim, load, fire: the type VI secretion system, a bacterial nanoweapon. Trends Microbiol. 2016;24(1):51–62. doi: 10.1016/j.tim.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Lorenz C, Büttner D. Functional characterization of the type III secretion ATPase HrcN from the plant pathogen Xanthomonas campestris pv. vesicatoria. J Bacteriol. 2009;191(5):1414–1428. doi: 10.1128/JB.01446-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minsavage G, Dahlbeck D, Whalen M, Kearney B, Bonas U, Staskawicz B, et al. Gene-for-gene relationships specifying disease resistance in Xanthomonas campestris pv. vesicatoria―pepper interactions. Mol Plant-Microbe Interact. 1990;3(1):41–47. doi: 10.1094/MPMI-3-041. [DOI] [Google Scholar]
- 34.Kearney B, Staskawicz BJ. Widespread distribution and fitness contribution of Xanthomonas campestris avirulence gene avrBs2. Nature. 1990;346:385–386. doi: 10.1038/346385a0. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz AR, Potnis N, Timilsina S, Wilson M, Patane J, Martins Jr. J, et al. Phylogenomics of Xanthomonas field strains infecting pepper and tomato reveals diversity in effector repertoires and identifies determinants of host specificity. Front Microbiol. 2015. doi:10.3389/fmicb.2015.00535. [DOI] [PMC free article] [PubMed]
- 36.Payne SH, Huang S-T, Pieper R. A proteogenomic update to Yersinia: enhancing genome annotation. BMC Genomics. 2010;11(1):460. doi: 10.1186/1471-2164-11-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Müller SA, Findeiß S, Pernitzsch SR, Wissenbach DK, Stadler PF, Hofacker IL, et al. Identification of new protein coding sequences and signal peptidase cleavage sites of Helicobacter pylori strain 26695 by proteogenomics. J Proteome. 2013;86:27–42. doi: 10.1016/j.jprot.2013.04.036. [DOI] [PubMed] [Google Scholar]
- 38.Kelkar DS, Kumar D, Kumar P, Balakrishnan L, Muthusamy B, Yadav AK, et al. Proteogenomic analysis of Mycobacterium tuberculosis by high resolution mass spectrometry. Mol Cell Proteomics. 2011;10(12):M111.011627. doi: 10.1074/mcp.M111.011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christie-Oleza JA, Miotello G, Armengaud J. High-throughput proteogenomics of Ruegeria pomeroyi: seeding a better genomic annotation for the whole marine Roseobacter clade. BMC Genomics. 2012;13:73. doi: 10.1186/1471-2164-13-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsuge S, Furutani A, Ikawa Y. Regulatory network of hrp gene expression in Xanthomonas oryzae pv. oryzae. J Gen Plant Pathol. 2014;80(4):303–313. doi: 10.1007/s10327-014-0525-3. [DOI] [Google Scholar]
- 41.Morales C, Posada J, Macneale E, Franklin D, Rivas I, Bravo M, et al. Functional analysis of the early chlorosis factor gene. Mol Plant-Microbe Interact. 2005;18(5):477–486. doi: 10.1094/MPMI-18-0477. [DOI] [PubMed] [Google Scholar]
- 42.Büttner D. Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol Mol Biol Rev. 2012;76(2):262–310. doi: 10.1128/MMBR.05017-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alegria MC, Souza DP, Andrade MO, Docena C, Khater L, Ramos CH, et al. Identification of new protein-protein interactions involving the products of the chromosome- and plasmid-encoded type IV secretion loci of the phytopathogen Xanthomonas axonopodis pv. Citri. J Bacteriol. 2005;187(7):2315–2325. doi: 10.1128/JB.187.7.2315-2325.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Souza DP, Andrade MO, Alvarez-Martinez CE, Arantes GM, Farah CS, Salinas RK. A component of the Xanthomonadaceae type IV secretion system combines a VirB7 motif with a N0 domain found in outer membrane transport proteins. PLoS Pathog. 2011;7(5):e1002031. doi: 10.1371/journal.ppat.1002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qian W, Jia Y, Ren S-X, He Y-Q, Feng J-X, Lu L-F, et al. Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 2005;15(6):757–767. doi: 10.1101/gr.3378705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He Y-Q, Zhang L, Jiang B-L, Zhang Z-C, Xu R-Q, Tang D-J, et al. Comparative and functional genomics reveals genetic diversity and determinants of host specificity among reference strains and a large collection of Chinese isolates of the phytopathogen Xanthomonas campestris pv. campestris. Genome Biol. 2007;8(10):R218. doi: 10.1186/gb-2007-8-10-r218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacob TR, Laia MLd, Moreira LM, Gonçalves JF, Carvalho FMdS, Ferro MIT, et al. Type IV secretion system is not involved in infection process in citrus. Int J Microbiol. 2014;2014:763575. [DOI] [PMC free article] [PubMed]
- 48.Souza DP, Oka GU, Alvarez-Martinez CE, Bisson-Filho AW, Dunger G, Hobeika L, et al. Bacterial killing via a type IV secretion system. Nat Commun. 2015;6:6453. doi: 10.1038/ncomms7453. [DOI] [PubMed] [Google Scholar]
- 49.Sana TG, Berni B, Bleves S. The T6SSs of Pseudomonas aeruginosa strain PAO1 and their effectors: beyond bacterial-cell targeting. Front Cell Infect Microbiol. 2016;6:61. doi: 10.3389/fcimb.2016.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwarz S, West TE, Boyer F, Chiang W-C, Carl MA, Hood RD, et al. Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 2010;6(8):e1001068. doi: 10.1371/journal.ppat.1001068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacIntyre DL, Miyata ST, Kitaoka M, Pukatzki S. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc Natl Acad Sci U S A. 2010;107(45):19520–19524. doi: 10.1073/pnas.1012931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fu Y, Waldor MK, Mekalanos JJ. Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe. 2013;14(6):652–663. doi: 10.1016/j.chom.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murdoch SL, Trunk K, English G, Fritsch MJ, Pourkarimi E, Coulthurst SJ. The opportunistic pathogen Serratia marcescens utilizes type VI secretion to target bacterial competitors. J Bacteriol. 2011;193(21):6057–6069. doi: 10.1128/JB.05671-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sana TG, Flaugnatti N, Lugo KA, Lam LH, Jacobson A, Baylot V, et al. Salmonella Typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. Proc Natl Acad Sci U S A. 2016;113(34):E5044–E5051. doi: 10.1073/pnas.1608858113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang F, Waterfield NR, Yang J, Yang G, Jin Q. A Pseudomonas aeruginosa type VI secretion phospholipase D effector targets both prokaryotic and eukaryotic cells. Cell Host Microbe. 2014;15(5):600–610. doi: 10.1016/j.chom.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 56.Bonas U, Stall RE, Staskawicz B. Genetic and structural characterization of the avirulence gene avrBs3 from Xanthomonas campestris pv. vesicatoria. Mol Gen Genet. 1989;218(1):127–136. doi: 10.1007/BF00330575. [DOI] [PubMed] [Google Scholar]
- 57.Schmidtke C, Abendroth U, Brock J, Serrania J, Becker A, Bonas U. Small RNA sX13: a multifaceted regulator of virulence in the plant pathogen Xanthomonas. PLoS Pathog. 2013;9(9):e1003626. doi: 10.1371/journal.ppat.1003626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daniels MJ, Barber CE, Turner PC, Sawczyc MK, Byrde RJ, Fielding AH. Cloning of genes involved in pathogenicity of Xanthomonas campestris pv. campestris using the broad host range cosmid pLAFR1. EMBO J. 1984;3(13):3323–3328. doi: 10.1002/j.1460-2075.1984.tb02298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ausubel F, Brent R, Kingston R, Moore D, Seidman J, Smith J, et al. Current Protocols in Molecular Biology. New York: John Wiley & Sons; 1996.
- 60.Backman K, Ptashne M, Gilbert W. Construction of plasmids carrying the cI gene of bacteriophage lambda. Proc Natl Acad Sci U S A. 1976;73(11):4174–4178. doi: 10.1073/pnas.73.11.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Figurski DH, Helinski DR. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A. 1979;76(4):1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 63.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 64.Lorenz C, Hausner J, Büttner D. HrcQ provides a docking site for early and late type III secretion substrates from Xanthomonas. PLoS One. 2012;7(11):e51063. doi: 10.1371/journal.pone.0051063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One. 2008;3(11):e3647. doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ballvora A, Pierre M, van den Ackerveken G, Schornack S, Rossier O, Ganal M, et al. Genetic mapping and functional analysis of the tomato Bs4 locus governing recognition of the Xanthomonas campestris pv. vesicatoria AvrBs4 protein. Mol Plant-Microbe Interact. 2001;14(5):629–638. doi: 10.1094/MPMI.2001.14.5.629. [DOI] [PubMed] [Google Scholar]
- 67.Marois E, Van den Ackerveken G, Bonas U. The Xanthomonas type III effector protein AvrBs3 modulates plant gene expression and induces cell hypertrophy in the susceptible host. Mol Plant-Microbe Interact. 2002;15(7):637–646. doi: 10.1094/MPMI.2002.15.7.637. [DOI] [PubMed] [Google Scholar]
- 68.Hermjakob H, Apweiler R. The proteomics identifications database (PRIDE) and the ProteomExchange consortium: making proteomics data accessible. Expert Rev Proteomics. 2006;3(1):1–3. doi: 10.1586/14789450.3.1.1. [DOI] [PubMed] [Google Scholar]
- 69.Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45(D1):D1100–D1106. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proteogenomic identification of proteins in Xe 85–10. Overview of the Xe chromosome showing all annotated and MS-data based identified protein-coding genes. Black: annotated CDS plus strand, Red: annotated CDS minus strand, Black-Red: MS-data based identified CDS, Black serrates line: GC-content. (PNG 133 kb)
Identification of all detected proteins and their annotated function. List of all detected annotated proteins in MS-data and their predicted functions. (XLSX 693 kb)
Additional information to Tables 1 and 2 and all conditions and strains used in this study. The tables show additional information, e.g. predicted function, homology and transcription start site, of the new and falsely incorrectly annotated protein-coding genes and all conditions and strains used in this study. (XLSX 29 kb)
Reannotation of dcp2, exbB2, flgG and infC. Multiple sequence alignment of dcp2, exbB2, flgG and infC homologs of Xe, X. axonopodis pv. citrumelo F1 (XacF1), X. oryzae pv. oryzae KACC10331 (Xoo), X. oryzae pv. oryzicola BLS256 (Xoc), X. fuscans subsp. aurantifolii ICPB 11122 (Xfa), X. perforans 91–118 (Xp). Green: experimentally detected by MS, underlined in red: annotated start codons, underlined in green: possible new start codon. (PNG 836 kb)
pXCV2 carries a third CDS. Representation of pXCV2 plasmid of Xe 85–10. Grey arrows show position of annotated CDS and the red arrow indicates the position of the newly identified protein-coding CDS. (PNG 88 kb)
Detection of proteins which are components or substrates of (potential) secretion systems. +: specific peptide detected, −: no specific peptide detected in MS-data. (XLSX 15 kb)
Oligonucleotides, plasmids and strains used in this study. List of oligonucleotides, plasmids and strains used in this study. (DOCX 45 kb)
GFF annotation file. GFF annotation file for artemis genome browser. (TXT 147 kb)
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [68, 69] partner repository with the dataset identifier PXD007140.