
Figure 1. A schematic representation of our approach for quantifying and visualizing phylogenetic signal in a phylogenomic data matrix.
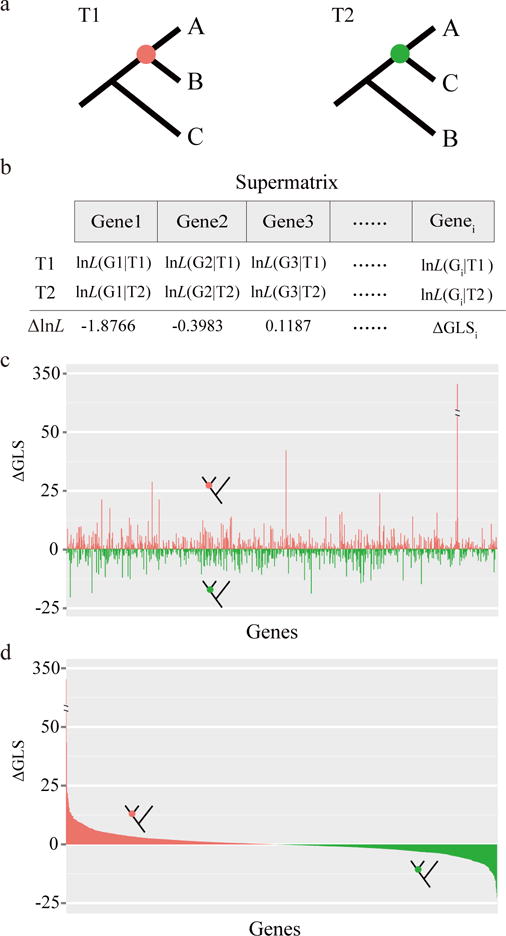
a, Two alternative phylogenetic hypotheses (T1, the unconstrained ML tree under concatenation; T2, the ML tree constrained to recover the T2 branch). b, Calculation of the difference in the gene-wise log-likelihood scores (ΔGLS) of T1 versus T2 for each gene in the data matrix. The difference in the site-wise log-likelihood scores, ΔSLS, of T1 versus T2 for each site in the data matrix is also calculated but is not shown here. The gene-wise phylogenetic signal (ΔGLS) for T1 versus T2 can be visualized either by arranging genes in the order of their placements in the data matrix (c) or in descending order of their ΔGLS values (d). Red bars denote genes supporting T1, whereas green bars denote genes supporting T2. The data for panels c and d are the actual values from the analysis of the Ascoideaceae branch in the fungal phylogenomic data matrix (Table 1). The schematic representation of our approach for quantifying and visualizing phylogenetic signal among three alternative phylogenetic hypotheses (T1, T2, and T3) is shown in Supplementary Fig. 1.