
Fig 6. Roles of nitric oxide (NO), endothelium-derived hyperpolarizing factors (EDHF), and calcium-activated K+ (KCa) channels in renal arcuate arteries.
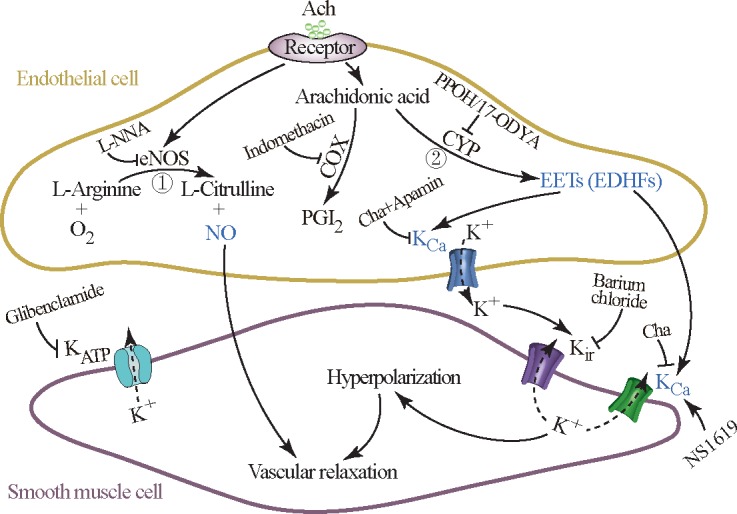
Acetylcholine (ACh) can induce endothelium-dependent vasodilation by the release of NO (①) and EDHF (②) from endothelial cells in renal arcuate arteries. NO is derived from endothelial NO synthase (eNOS), while prostacyclin (PGI2) and epoxyeicosatrienoic acids (EETs, as EDHFs) are generated from arachidonic acids by cyclooxygenase (COX), and cytochrome P-450 (CYP) epoxygenase, respectively. EETs facilitate the hyperpolarization of smooth muscle cells and vascular relaxation through K+ efflux mediated by the opening of KCa channels either in endothelial cells or in smooth muscle cells. The K+ released from KCa channels of endothelial cells into the subendothelial space (potential connective tissue space beneath the endothelium) subsequently actives inward rectifying K+ (Kir) channels on smooth muscle cells, while producing K+ efflux as well. In the present study, the contribution of NO and EDHF to endothelium vasodilation was impaired and the activity of KCa channels was downregulated in renal arcuate arteries of obese Zucker (OZ) rats at 20 weeks of age, but PGI2 had no effect on ACh-elicited vasodilation. Barium chloride, inhibitor of the Kir channel; Charybdotoxin (Cha)+Apamin, inhibitor of the KCa channel; Glibenclamide, inhibitor of the KATP channel; Indomethacin, inhibitor of COX; L-NNA, inhibitor of eNOS; NS1619, agonist of the KCa channel; and PPOH or 17-ODYA, inhibitors of CYP epoxygenase.