
Figure 5. Intron evolution.
(A) Rates of intron gain and loss per lineage, including extant genomes and ancestral reconstructed nodes. Diameter and color of circles denote the number of introns per kbp of coding sequence at each ancestral node. Bolder edges mark the lines of descent between the LECA and Metazoa/Ichthyophonida, which were characterized by continued high intron densities (see text). Red and green bars represent the inferred number of intron gains (green) and losses (red) in ancestral nodes. (B) Difference between intron site gains and losses in selected ancestors, including animals (left; from Metazoa to Unikonta/Amorphea) and unicellular holozoans (right). For each ancestor, we specify the variance-to-mean ratio of the inferred number of introns from 100 bootstrap replicates (higher values, denoted by lighter purple, indicate less reliable inferences; see Methods. The color code denotes modes of intron evolution: dominance of gains (green), losses (pink) and stasis (light gray). (C) Conservation of the NMD machinery and SR splicing factors in unicellular holozoans (up) and selected ancestors (down). Black dots indicate the presence of an ortholog, and empty dots partial conservation. For the NMD machinery, each column summarizes the presence of multiple gene families (number between brackets). † denotes the ancestral eukaryotic origin of TRA2 according to (Plass et al., 2008). Complete survey at the species and gene levels available as Figure 4—figure supplements 2 and 3. Figure 5—source data 1, 2 and 3.
DOI: http://dx.doi.org/10.7554/eLife.26036.018

Figure 5—figure supplement 1. Classification of intron sites by conservation in protein alignments, as used in (Csűrös and Miklós, 2006; Csurös, 2008).
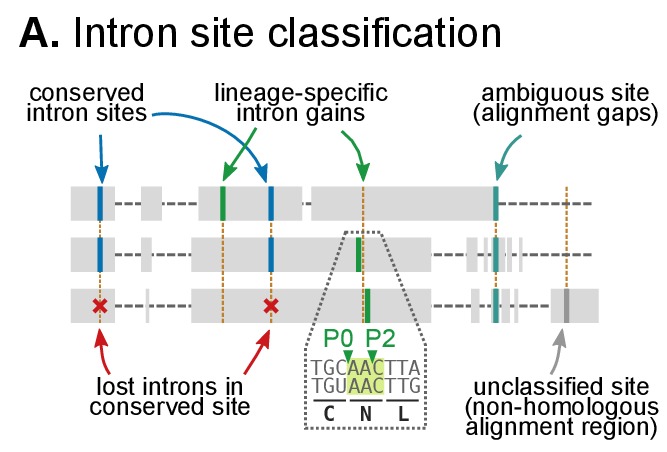
Figure 5—figure supplement 2. Phylogenetic distribution of the NMD machinery, SR splicing factors and RNA-binding domains in eukaryotes.

Figure 5—figure supplement 3. Phylogenetic analysis of (A) eIF4A3, (B) Smg5/6/7, and (C) eRF3, using Maximum likelihood in IQ-TREE (supports are SH-like approximate likelihood ratio test/UFBS, respectively); including Bayesian inference supports for the ortologous groups of interest (BPP statistical supports, in red).
