

Abstract
Cell cycle consists of different types of phases, transition from G1, S, G2, M. Inhibition of associated CDKs like CDK9/Cyclin T1 complex, which are indirectly involved in the Cell cycle progression in the form of transcription elongation, reduces diverse diseases such as Cardiac Hypertrophy, Alzheimer’s, Cancer, AIDS and Inflammation. Glide tool of the Schrodinger software has been used for performing Structure Based Virtual Screening and Docking against Drug Bank and MDPI database. The best hits were identified which go and bind in the active site of the target where ATP binds for the activity. The ADMET, MM-GBSA and DFT analysis is also done for the same. Compound 4-{4-[4-(3-aminopropoxy)phenyl]-1H-pyrazol-5-yl}-6-chlorobenzene-1,3-diol (DB08045) was found to be more potent, novel and selective as an inhibitor. Hopefully compound (DB08045) could be used as an anti-cancer agent for the treatment of life-threatening diseases.
Abbreviations: SBVS, structure based virtual screening; CTD, carboxy terminal domain; P-TEFB, positive transcription elongation factor B; CDK9, Cyclin Dependent Kinase 9; DFT, density functional theory; MW, molecular weight; HOMO, high occupied molecular orbital; LUMO, lowest unoccupied molecular orbital; ATP, adenosine triphosphate; MDPI, molecular diversity preservation international
Keywords: CDK, Drug Bank, Cancer, MDPI, Cell cycle, Potent
1. Introduction
Several diseases like Cancer and numerous Proliferative diseases occurred because of the alteration in the Cell cycle. The Cell cycle is controlled by the CDK family, which binds with the Cyclin unit for its activation (Morgan, 1997). CDK activation and regulation support cell division, changing the transition state and entry into the mitosis phase (Nigg, 1995, Vermeulen et al., 2003). Besides Cell cycle control some of the CDKs take part in the controlling of the transcription process (Chao et al., 2000). CDKs play different role such as CDK2 play an essential role in the apoptosis, CDK5 role in neuronal cell and CDK7, CDK8, CDK9 role in the transcription process (Gray et al., 1999). The transcription process is responsible for making mRNAs together with pre-initiation, initiation, elongation and termination. Among all the phases, the elongation phase is an extremely and vibrant step for regulating the transcription cycle (Sims et al., 2004, Loyer et al., 2005, Fuda et al., 2009). Processing of the elongation phase, the phosphorylation of the CDK substrate mechanism is crucial (Suryadinata et al., 2010). Protein kinases control the cellular signaling processes by performing catalyzed phospho transfer reaction (Johnson et al., 1998, Traxler and Furet, 1999). One of the CDKs is CDK9, which is a cdc2-like kinase involved in the transcription elongation phase with pairing of Cyclin units and making P-TEFB complex (CDK9/Cyclin T1) (Peng et al., 1998b). Cyclin K is also associated with the CDK9 and works as a CDK9 regulatory subunit that has been confirmed by the in vivo and in vitro studies and participates in the cellular control of the gene expression (Fu et al., 1999, Garriga and Grana, 2004). CDK9 is present in the two isoforms, one is CDK9 p42 and the other one is CDK9 p52, acting similarly. Both are present in the mammalian cells and show tissue specific expression pattern (Shore et al., 2003, Liu and Herrmann, 2005). The cdc-2 related kinase having motif Pro-Ile-Thr-Ala-Leu-Arg-Glu named PITALRE interacts with their associated Cyclin units (Grana et al., 1994). P-TEFB complex (CDK9/Cyclin T1) plays a significant role for transcription elongation (Zhu et al., 1997, Wei et al., 1998). Carboxy terminal domain is an important part of the Cyclin T1, deletion of that part decreases the kinase action and transcription activity around 10% (Zhu et al., 1997, Peng et al., 1998a, Peng et al., 1998b, Wei et al., 1998, Anand et al., 2007, Baumli et al., 2008). P-TEFB controls the transcription process with the activation of the CTD of the RNA polymerase II (Chao et al., 2000). Phosphorylation of the threonine residue in the CDK9 leads to the activation of the P-TEFB complex. Thus CDK9/Cyclin T1 is involved in the Apoptosis, Cell differentiation, Cell proliferation (Desai et al., 1992, De Falco and Giordano, 2002). CDK9/Cyclin T1 phosphorylates the Ser 2 sites of the RNA polymerase II for the elongation of the transcription process (Phatnani and Greenleaf, 2006, Egloff and Murphy, 2008, Krystof et al., 2010). ATP competitive inhibitor plays a crucial role for inquisitive the activity of CDK9 (Kamb, 1995, Lander et al., 2001). In Cell cycle numerous processes come about for the controlling of the CDK9 like Cyclin unit binding, phosphorylation and inhibitory INK4 subunits (Pavletich, 1999). CDK9 is the main target for Cancer prevention so that several inhibitors designed against the target and some of them show a promising binding affinity like DRB, which make Halogen bond with the CDK9 kinase hinge region (Baumli et al., 2010), 2-Anilino-4-(thiazol-5-yl) Pyrimidine (Wang et al., 2010), 4-Arylazo-3,5-diamino-1H-pyrazole (Krystof et al., 2006), Roscovitine, CR8 (Bettayeb et al., 2010, Berberich et al., 2011), Flavopiridol (Garriga and Grana, 2004, Schmerwitz et al., 2011), CAN-508 (Krystof et al., 2011). Flavopiridol has a wide specificity for CDK kinase inhibition. It blocks the transcription in the elongation phase and induces Apoptosis in the cell (Chao et al., 2000, Schmerwitz et al., 2011). Some of the Co-crystallized CDK9/Cyclin T1 inhibitors are available in the Protein Data Bank targeting CDK9 (Krystof et al., 2012): Flavopiridol (1), DRB (2), CAN-508 (3), 2-amino-4-heteroaryl-pyrimidine (4) (Fig. 1).
Figure 1.

List of co-crystallized CDK9/Cyclin T1 inhibitors found in the Protein Data Bank.
CDK kinase modulators are moreover promising for modulating the kinase activity via blocking Cell cycle progression (Senderowicz and Sausville, 2000). To date only 11 kinase inhibitors received US Food and Drug Administration approval as Cancer treatments (Zhang et al., 2009). Structure Based Drug Design for development of Cell cycle inhibitor is the right approach for the Cancer therapy (Davies et al., 2002, Dickson and Schwartz, 2009).
Hence, it is very important to develop and discovery some novel inhibitors, which can bind to the ATP binding site of the CDK9 kinase and reduce some of the life-threatening diseases like Cancer, AIDS, Cardiac Hypertrophy and several Proliferative diseases.
SBVS technique is very important to find out the drug compound very rapidly from the large library of the compounds in the Medicinal Chemistry Research (McInnes, 2007, Tuccinardi, 2009). Since the discovery of novel CDK9/Cyclin T1 (P-TEFB) complex inhibitor, orderly computational tools were used. Eight novel compounds were found from Drug Bank and three from MDPI database as CDK9 inhibitors using Structure Based Virtual Screening and Docking approach.
2. Materials and methods
2.1. Protein selection and preparation
CDK9/Cyclin T1 complex protein structure has been taken from the Protein Data Bank (PDB ID: 3BLQ). Before going to the docking procedure, the protein structure was prepared using the Protein preparation wizard of the Schrodinger software. A step by step procedure was used for the preparation. The protein structure was loaded into the Prep wizard where bond orders were assigned, after that hydrogen atoms were added. In the last stage a restrained minimization was performed with the RMSD cut off of 0.30 Å and OPLS-2005 force field (Baumli et al., 2008).
2.2. Ligand preparation
The small molecules of the ligand were retrieved from the Drug Bank and MDPI database. These molecules were prepared by using the Ligprep module of the Schrodinger software. The bond order and the bond angle were assigned, then minimization was done by using OPLS-2005 force field. The Epik option was used for keeping ligand in the correct protonation state (Banks et al., 2005).
2.3. Grid preparation
The Glide protocol was used for generating the grid of the protein structure. The ATP binding site was selected as centroid for the CDK9 protein and scaling factor was selected as 1.0 and the partial charge cutoff was selected as the 0.25 (Friesner et al., 2006).
2.4. Preparation of the reference compounds
The Reference compounds were retrieved from the co-crystallized structure of protein–ligand complexes from the Protein Data Bank, after that ligand was prepared by using the ligprep module of the Schrodinger software and docked with the protein CDK9 kinase and that docked score and already reported ligand were kept as a reference for finding the best drug compound from the Drug Bank and the MDPI database.
2.5. Virtual screening
High throughput virtual screening was performed against the CDK9/Cyclin T1 complex. A total number of 22,401 ligand compounds were taken from the MDPI database and 6858 ligand compounds were taken from the Drug Bank database. These compounds and the reference compounds were subjected to the Lipinski filtration and reactive functionality. The reference compounds were also incorporated in the database. Glide maestro protocol was used for the virtual screening using Schrodinger software. It performed docking of the drug compounds in the different phases like HTVS (High throughput virtual screening), SP (Standard-precision), XP (Extra-precision). The library contains a large number of compounds so that after performing HTVS screening remaining 10% was performed using the Standard-precision docking, and remaining 10% was performed using the Extra-precision docking. The virtual screening workflow is shown in Fig. 2.
Figure 2.

Structure Based Virtual Screening workflow.
2.6. Density functional theory analysis
LUMO and HOMO energy poses showed the region where small molecule accepts the electron during the complex formation and donates the electron during the complex formation. The gap energy is defined as the difference between HOMO and LUMO molecular orbital energy, that energy indicates the excitation energy showing the stability and reactivity of the compounds (Becke, 1993).
2.7. Drug like property analysis
The hits were selected for finding drug like properties. These properties follow Lipinski’s rule of five. The properties which have been considered are Molecular Weight (MW), Lipophilicity (log P), Hydrogen Bond Acceptor (HBA), Hydrogen Bond Donor (HBD), Human Oral Absorption (Lipinski et al., 2001).
2.8. MM-GBSA approach for drug-target binding energy estimation
Drug-Target binding energy was calculated by using Schrodinger software. This binding energy estimates the stability of the protein and the ligand complexes (Lyne et al., 2006).
3. Result and discussion
3.1. Database screening and docking against the target
Virtual screening and docking approach was used for finding the novel lead compound. Drug Bank database and MDPI database were selected for that. We found eight best lead compounds from Drug Bank database and three best lead compounds from MDPI database, which showed best binding affinity with the target protein.
Compound DB08045 was found to be more potent and selective, which interacts with the target protein active site residues. It makes H-bonding with residues (Cys106, Asp104, Lys48, Ile25, Asn154, Asp167) with docking score −14.072. The docking score is high and it covered the ATP binding site completely. It means this lead compound tightly docked in the active site of the target protein and showed good inhibition property. The docking score for DB08045, DB01204, DB07766, DB01772, and DB02594, DB07715, DB07163 and DB08133 has been identified (Table 1). The same docking and virtual screening procedure is used in MDPI database and three best hits are found. The docking score for MDPI14871, MDPI20746 and MDPI14352 has been shown in Table 2. The docking and Ligplot interaction diagram gives a better view of interaction between target and lead compounds (Figure 3, Figure 4).
Table 1.
2D structure of the selected inhibitors from Drug Bank database and reference compounds respectively with their docking scores.
| Drug Bank id | Compound structure | Mol. Wt. | Mol. formula | Docking score |
|---|---|---|---|---|
| DB08045 | 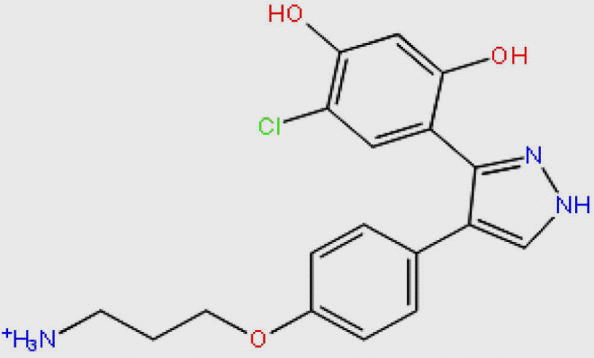 |
360.814 | C18H19ClN3O3+ | −14.072 |
| DB01204 | 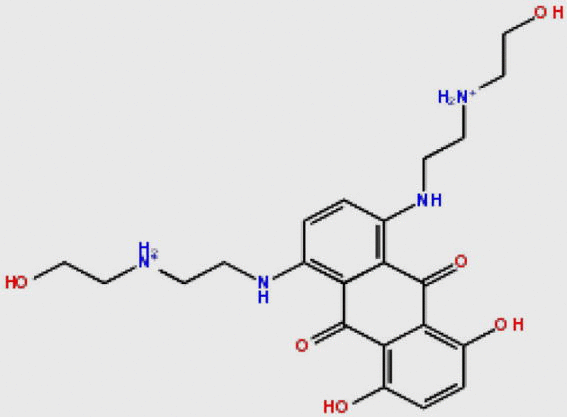 |
446.496 | C22H30N4O62+ | −13.456 |
| DB07766 | 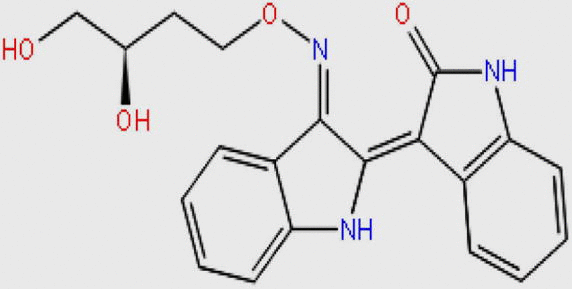 |
365.383 | C20H19N3O4 | −11.807 |
| DB01772 | 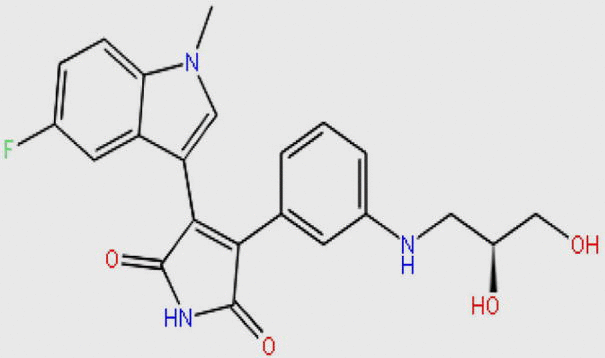 |
409.410 | C22H20FN3O4 | −11.505 |
| DB02594 | 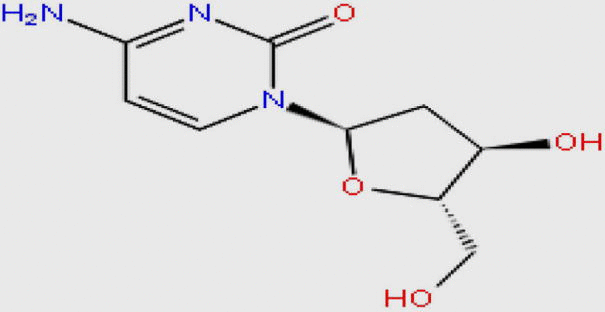 |
227.217 | C9H13N3O4 | −11.469 |
| DB07715 | 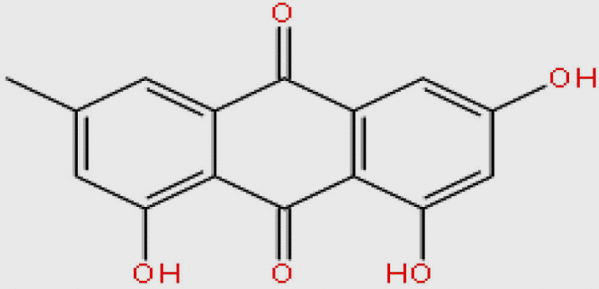 |
270.237 | C15H10O5 | −11.353 |
| DB07163 | 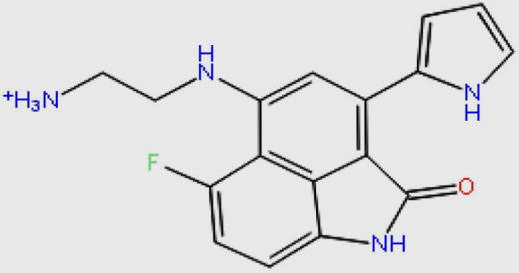 |
311.333 | C17H16FN4O+ | −10.826 |
| DB08133 | 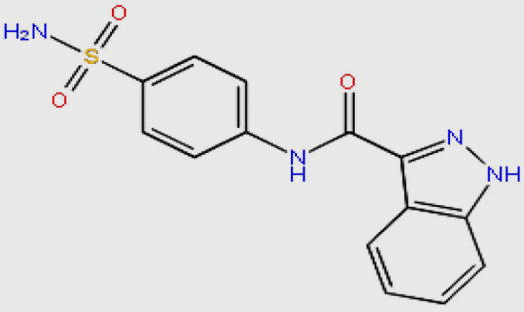 |
316.335 | C14H12N4O3S | −10.679 |
| 2-Amino-4-heteroaryl-pyrimidiner | – | – | – | −8.639 |
r = reference compound.
Table 2.
2D structure of the selected inhibitors from MDPI database respectively with their docking scores.
| MDPI id | Compound structure | Mol. Wt. | Mol. formula | Docking score |
|---|---|---|---|---|
| MDPI14871 | 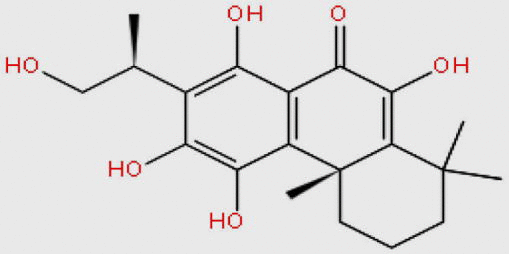 |
362.417 | C20H26O6 | −11.453 |
| MDPI20746 | 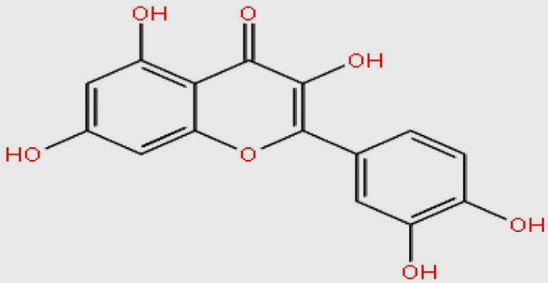 |
302.236 | C15H10O7 | −10.350 |
| MDPI14352 | 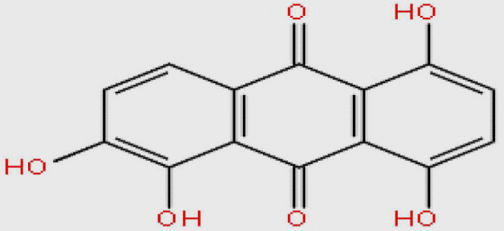 |
272.210 | C14H8O6 | −10.378 |
Figure 3.


Binding mode of the selected ligand in the active site of the CDK9/Cyclin T1 complex proposed by docking studies. (a) Binding mode of the ligand DB08045. (b) Ligplot Interaction diagram of the DB08045 ligand. (c) Binding mode of the ligand DB01204. (d) Binding mode of the ligand DB07766. (e) Binding mode of the ligand DB01772. (f) Binding mode of the ligand DB02594. (g) Binding mode of the ligand DB07715. (h) Binding mode of the ligand DB07163. (i) Binding mode of the ligand DB08133. The inhibitors have been shown in stick form and yellow dotted lines indicate the inhibitor protein H-bonding. The critical protein residues have been shown in gray color.
Figure 4.

Binding mode of the selected ligand in the active site of the CDK9/Cyclin T1 complex proposed by docking studies. (a) Binding mode of the ligand MDPI14871. (b) Binding mode of the ligand MDPI20746. (c) Binding mode of the ligand MDPI14352. The inhibitors have been shown in stick form and yellow dotted lines indicate the inhibitor protein H-bonding. The critical protein residues have been shown in gray color.
3.2. HOMO LUMO stability analysis of the screened compounds
Drug and protein interaction stability and contribution of selected inhibitors were analyzed using HOMO and LUMO for the Drug Bank database selected compounds (Table 3) and MDPI database selected compounds (Table 4). The orbital energy and gap energy between the two orbitals were estimated. Molecular reactivity was an analysis of the drug candidate (Figure 5, Figure 6). The lowest energy gap found for DB01772 is 0.10269 and the highest energy gap found for DB02594 is 0.18897 for Drug Bank selected compounds and 0.11703 and 0.14723 for MDPI selected compounds.
Table 3.
Orbital energy of the selected inhibitors of the Drug Bank database.
| S. No. | Compound name | HOMO energy | LUMO energy | HLG (eV) |
|---|---|---|---|---|
| 1 | DB08045 | −0.27511 | −0.16337 | 0.11174 |
| 2 | DB01204 | −0.35700 | −0.24697 | 0.11003 |
| 3 | DB07766 | −0.19160 | −0.08257 | 0.10903 |
| 4 | DB01772 | −0.19318 | −0.09049 | 0.10269 |
| 5 | DB02594 | −0.22590 | −0.03693 | 0.18897 |
| 6 | DB07715 | −0.23425 | −0.09745 | 0.1368 |
| 7 | DB07163 | −0.28094 | −0.16916 | 0.11178 |
| 8 | DB08133 | −0.23122 | −0.05651 | 0.17471 |
Table 4.
Orbital energy of the selected inhibitors of the MDPI database
| S. No. | Compound name | HOMO energy | LUMO energy | HLG (eV) |
|---|---|---|---|---|
| 1 | MDPI14871 | −0.19363 | −0.05277 | 0.14086 |
| 2 | MDPI20746 | −0.20836 | −0.06113 | 0.14723 |
| 3 | MDPI14352 | −0.21861 | −0.10158 | 0.11703 |
Figure 5.

Plot shows highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of Drug Bank screened selected compounds. The red color indicates positive electron density while blue color indicates negative electron density respectively.
Figure 6.

Plot shows highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of MDPI screened selected compounds. The red color indicates positive electron density while blue color indicates negative electron density respectively.
3.3. ADMET and drug-likeliness property analysis against screened compounds
These selected hits were evaluated for the physiochemical properties and drug likeliness properties. These selected hits followed Lipinski’s rule of five. Evidence for drug like characteristics, we selected various properties such as Molecular weight (MW), Total solvent accessible surface, Hydrogen bond acceptor (HBA), Hydrogen bond donor (HBD), Predicted aqueous solubility and Human oral absorption were assessed. The Total solvent accessible surface and Molecular weight have been shown to be a very good descriptor for characterizing drug absorption, including intestinal absorption and bioavailability. The chemical name of these eleven lead molecules with their corresponding Drug Bank id and MDPI database id has been given with their chemical properties. The chemical properties of the drug bank hits are illustrated in Table 5 and for MDPI hits are illustrated in Table 6.
Table 5.
Drug-like properties of the selected inhibitors from Drug Bank database.
| S. No. | Drug Bank ida | Mol. Wt.b | SASAc | Human oral absorptiond | HB donore | HB acceptorf | QP log Sg |
|---|---|---|---|---|---|---|---|
| 1 | DB08045 | 360.814 | 635.553 | 62.835 | 5 | 4.750 | −3.356 |
| 2 | DB01204 | 446.496 | 741.991 | 24.671 | 4 | 9.900 | −1.112 |
| 3 | DB07766 | 365.383 | 654.056 | 78.131 | 3 | 7.600 | −4.137 |
| 4 | DB01772 | 409.410 | 606.405 | 75.572 | 4 | 7.400 | −3.784 |
| 5 | DB02594 | 227.217 | 424.935 | 50.835 | 4 | 9.100 | −1.529 |
| 6 | DB07715 | 270.237 | 477.959 | 68.395 | 1 | 4.250 | −3.048 |
| 7 | DB07163 | 311.333 | 543.171 | 65.815 | 5 | 4.500 | −2.217 |
| 8 | DB08133 | 316.335 | 557.232 | 62.073 | 3 | 7.000 | −3.355 |
Drug Bank inhibitors id.
Molecular weight (acceptable range is: ⩽500).
Total solvent accessible surface area (acceptable range is: 300–1000).
Human oral absorption (acceptable range is: <25% less & >80% high).
Hydrogen bond donor (acceptable range is: ⩽5).
Hydrogen bond acceptor (acceptable range is: ⩽10).
Predicted aqueous solubility (acceptable range is: −6.5 to 0.5).
Table 6.
Drug-like properties of the selected inhibitors from MDPI database.
| S. No. | MDPI ida | Mol. Wt.b | SASAc | Human oral absorptiond | HB donore | HB acceptorf | QP log Sg |
|---|---|---|---|---|---|---|---|
| 1 | MDPI14871 | 362.417 | 588.370 | 75.441 | 4 | 5.700 | −3.573 |
| 2 | MDPI20746 | 302.236 | 531.625 | 53.717 | 4 | 5.250 | −3.096 |
| 3 | MDPI14352 | 272.210 | 454.633 | 61.573 | 1 | 4.000 | −2.565 |
MDPI database inhibitors id.
Molecular weight (acceptable range is: ⩽500).
Total solvent accessible surface area (acceptable range is: 300–1000).
Human oral absorption (acceptable range is: <25% less & >80% high).
Hydrogen bond donor (acceptable range is: ⩽5).
Hydrogen bond acceptor (acceptable range is: ⩽10).
Predicted aqueous solubility (acceptable range is: −6.5 to 0.5).
3.4. Drug-target binding energy analysis against screened compounds
Prime MM-GBSA (GB stands for Generalized Born) module of the Schrodinger software is very useful for finding out the protein–ligand binding affinity. The selected protein–ligand complexes were subjected to that module. It combines OPLS molecular mechanics energies (EMM), surface generalized born solvation model for polar solvation (GSGB), and a nonpolar solvation term (GNP). The total free energy of binding calculation is:
| (1) |
ΔGbind: total binding free energy of complex, Gcomplex: total energy of the complex, Gprotein: energy of the receptor without ligand, Gligand: energy of the unbound ligand.
| (2) |
The binding free energy estimated for Drug Bank compound complexes (Table 7) and for MDPI compound complexes is illustrated in Table 8.
Table 7.
Prime MM-GBSA energy calculation result of the selected inhibitors from Drug Bank database with the target.
| S. No. | Compound ida | ΔG bindb | Gevdwc | Gcould |
|---|---|---|---|---|
| 1 | DB08045 | −64.55 | −35.05 | −40.47 |
| 2 | DB01204 | −84.11 | −43.18 | −62.70 |
| 3 | DB07766 | −67.25 | −38.84 | −27.38 |
| 4 | DB01772 | −62.04 | −46.67 | −9.48 |
| 5 | DB02594 | −40.58 | −29.45 | −20.05 |
| 6 | DB07715 | −60.48 | −29.73 | −24.52 |
| 7 | DB07163 | −59.18 | −36.42 | −24.68 |
| 8 | DB08133 | −55.15 | −34.93 | 27.31 |
Inhibitors id from Drug Bank database.
Free binding energy.
Van der Waal energy.
Coulomb energy.
Table 8.
Prime MM-GBSA energy calculation result of the selected inhibitors from MDPI database with the target.
| S. No. | Compound ida | ΔG bindb | Gevdwc | Gcould |
|---|---|---|---|---|
| 1 | MDPI14871 | −74.98 | −36.46 | −20.06 |
| 2 | MDPI20746 | −60.64 | −32.07 | −27.92 |
| 3 | MDPI14352 | −56.10 | −30.04 | −21.35 |
Inhibitors id from MDPI database.
Free binding energy.
Van der Waal energy.
Coulomb energy.
3.5. Superimpose structure
The docked pose of Drug Bank hits and MDPI hits occupies the ATP binding site. It was superimposed on crystal structure (PDB ID: 3BLQ) that occupies this region. The binding pattern of Drug Bank hits and MDPI hits was found to be similar to that of the ATP binding site. The figure clearly indicates that the proposed binding pose is well occupied in the active site of CDK9/Cyclin T1 complex and has a binding pattern similar to that of ATP (Figure 7, Figure 8).
Figure 7.

Selected inhibitors after screened Drug Bank database has been superimposed in the ATP binding site of the target protein.
Figure 8.

Selected inhibitors after screened MDPI database has been superimposed in the ATP binding site of the target protein.
4. Conclusion
Virtual screening is a powerful technique which is widely used in the drug discovery as an initial phase in the medicinal chemistry research. The discovery of novel and potent inhibitors is reported in this present study. It has been clearly confirmed that the approach utilized in this study is successful in finding eleven potent CDK9/Cyclin T1 inhibitors from Drug Bank and MDPI databases. These inhibitors have shown drug-like properties upon the ADMET and Lipinski’s rule of five. Compound 4-{4-[4-(3-aminopropoxy) phenyl]-1H-pyrazol-5-yl}-6-chlorobenzene-1,3-diol (DB08045) was showing high binding affinity and docked perfectly within the binding pocket region forming interaction with Cys106, Asp104, Lys48, Ile25, Asn154, Asp167 with docking score −14.072. It is obvious that these hits could be potent and selective anticancer agent against Cyclin Dependent kinase/Cyclin T1 complex.
Acknowledgements
The Research work was carried out in the Department of Bioinformatics, Maulana Azad National Institute of Technology, India. Afzal Hussain is grateful to the University Grant Commission (UGC) fellowship for the financial assistance. The Authors also wish to acknowledge the Schrodinger team for providing software facility.
Footnotes
Peer review under responsibility of King Saud University.
References
- Anand K., Schulte A., Fujinaga K., Scheffzek K., Geyer M. Cyclin box structure of the P-TEFb subunit cyclin T1 derived from a fusion complex with EIAV tat. J. Mol. Biol. 2007;370(5):826–836. doi: 10.1016/j.jmb.2007.04.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks J.L., Beard H.S., Cao Y., Cho A.E., Damm W., Farid R., Felts A.K., Halgren T.A., Mainz D.T., Maple J.R., Murphy R., Philipp D.M., Repasky M.P., Zhang L.Y., Berne B.J., Friesner R.A., Gallicchio E., Levy R.M. Integrated modeling program, applied chemical theory (IMPACT) J. Comput. Chem. 2005;26(16):1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumli S., Lolli G., Lowe E.D., Troiani S., Rusconi L., Bullock A.N., Debreczeni J.E., Knapp S., Johnson L.N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008;27(13):1907–1918. doi: 10.1038/emboj.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumli S., Endicott J.A., Johnson L.N. Halogen bonds form the basis for selective P-TEFb inhibition by DRB. Chem. Biol. 2010;17(9):931–936. doi: 10.1016/j.chembiol.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Becke A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98(7):5648–5652. [Google Scholar]
- Berberich N., Uhl B., Joore J., Schmerwitz U.K., Mayer B.A., Reichel C.A., Krombach F., Zahler S., Vollmar A.M., Furst R. Roscovitine blocks leukocyte extravasation by inhibition of cyclin-dependent kinases 5 and 9. Br. J. Pharmacol. 2011;163(5):1086–1098. doi: 10.1111/j.1476-5381.2011.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettayeb K., Baunbaek D., Delehouze C., Loaec N., Hole A.J., Baumli S., Endicott J.A., Douc-Rasy S., Benard J., Oumata N., Galons H., Meijer L. CDK inhibitors roscovitine and CR8 trigger Mcl-1 down-regulation and apoptotic cell death in neuroblastoma cells. Genes Cancer. 2010;1(4):369–380. doi: 10.1177/1947601910369817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao S.H., Fujinaga K., Marion J.E., Taube R., Sausville E.A., Senderowicz A.M., Peterlin B.M., Price D.H. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 2000;275(37):28345–28348. doi: 10.1074/jbc.C000446200. [DOI] [PubMed] [Google Scholar]
- Davies T.G., Pratt D.J., Endicott J.A., Johnson L.N., Noble M.E. Structure-based design of cyclin-dependent kinase inhibitors. Pharmacol. Ther. 2002;93(2–3):125–133. doi: 10.1016/s0163-7258(02)00182-1. [DOI] [PubMed] [Google Scholar]
- De Falco G., Giordano A. CDK9: from basal transcription to cancer and AIDS. Cancer Biol. Ther. 2002;1(4):342–347. [PubMed] [Google Scholar]
- Desai D., Gu Y., Morgan D.O. Activation of human cyclin-dependent kinases in vitro. Mol. Biol. Cell. 1992;3(5):571–582. doi: 10.1091/mbc.3.5.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson M.A., Schwartz G.K. Development of cell-cycle inhibitors for cancer therapy. Curr. Oncol. 2009;16(2):36–43. doi: 10.3747/co.v16i2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff S., Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24(6):280–288. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Fu T.J., Peng J., Lee G., Price D.H., Flores O. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J. Biol. Chem. 1999;274(49):34527–34530. doi: 10.1074/jbc.274.49.34527. [DOI] [PubMed] [Google Scholar]
- Fuda N.J., Ardehali M.B., Lis J.T. Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature. 2009;461(7261):186–192. doi: 10.1038/nature08449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga J., Grana X. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene. 2004;337:15–23. doi: 10.1016/j.gene.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Grana X., De Luca A., Sang N., Fu Y., Claudio P.P., Rosenblatt J., Morgan D.O., Giordano A. PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retinoblastoma protein in vitro. Proc. Natl. Acad. Sci. U.S.A. 1994;91(9):3834–3838. doi: 10.1073/pnas.91.9.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray N., Detivaud L., Doerig C., Meijer L. ATP-site directed inhibitors of cyclin-dependent kinases. Curr. Med. Chem. 1999;6(9):859–875. [PubMed] [Google Scholar]
- Johnson L.N., Lowe E.D., Noble M.E., Owen D.J. The Eleventh Datta Lecture. The structural basis for substrate recognition and control by protein kinases. FEBS Lett. 1998;430(1–2):1–11. doi: 10.1016/s0014-5793(98)00606-1. [DOI] [PubMed] [Google Scholar]
- Kamb A. Cell-cycle regulators and cancer. Trends Genet. 1995;11(4):136–140. doi: 10.1016/s0168-9525(00)89027-7. [DOI] [PubMed] [Google Scholar]
- Krystof V., Cankar P., Frysova I., Slouka J., Kontopidis G., Dzubak P., Hajduch M., Srovnal J., de Azevedo W.F., Jr., Orsag M., Paprskarova M., Rolcik J., Latr A., Fischer P.M., Strnad M. 4-Arylazo-3,5-diamino-1H-pyrazole CDK inhibitors: SAR study, crystal structure in complex with CDK2, selectivity, and cellular effects. J. Med. Chem. 2006;49(22):6500–6509. doi: 10.1021/jm0605740. [DOI] [PubMed] [Google Scholar]
- Krystof V., Chamrad I., Jorda R., Kohoutek J. Pharmacological targeting of CDK9 in cardiac hypertrophy. Med. Res. Rev. 2010;30(4):646–666. doi: 10.1002/med.20172. [DOI] [PubMed] [Google Scholar]
- Krystof V., Rarova L., Liebl J., Zahler S., Jorda R., Voller J., Cankar P. The selective P-TEFb inhibitor CAN508 targets angiogenesis. Eur. J. Med. Chem. 2011;46(9):4289–4294. doi: 10.1016/j.ejmech.2011.06.035. [DOI] [PubMed] [Google Scholar]
- Krystof V., Baumli S., Furst R. Perspective of cyclin-dependent kinase 9 (CDK9) as a drug target. Curr. Pharm. Des. 2012;18(20):2883–2890. doi: 10.2174/138161212800672750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J.P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange-Thomann N., Stojanovic N., Subramanian A., Wyman D., Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J.C., Mungall A., Plumb R., Ross M., Shownkeen R., Sims S., Waterston R.H., Wilson R.K., Hillier L.W., McPherson J.D., Marra M.A., Mardis E.R., Fulton L.A., Chinwalla A.T., Pepin K.H., Gish W.R., Chissoe S.L., Wendl M.C., Delehaunty K.D., Miner T.L., Delehaunty A., Kramer J.B., Cook L.L., Fulton R.S., Johnson D.L., Minx P.J., Clifton S.W., Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J.F., Olsen A., Lucas S., Elkin C., Uberbacher E., Frazier M., Gibbs R.A., Muzny D.M., Scherer S.E., Bouck J.B., Sodergren E.J., Worley K.C., Rives C.M., Gorrell J.H., Metzker M.L., Naylor S.L., Kucherlapati R.S., Nelson D.L., Weinstock G.M., Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y., Taylor T., Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C., Wincker P., Smith D.R., Doucette-Stamm L., Rubenfield M., Weinstock K., Lee H.M., Dubois J., Rosenthal A., Platzer M., Nyakatura G., Taudien S., Rump A., Yang H., Yu J., Wang J., Huang G., Gu J., Hood L., Rowen L., Madan A., Qin S., Davis R.W., Federspiel N.A., Abola A.P., Proctor M.J., Myers R.M., Schmutz J., Dickson M., Grimwood J., Cox D.R., Olson M.V., Kaul R., Raymond C., Shimizu N., Kawasaki K., Minoshima S., Evans G.A., Athanasiou M., Schultz R., Roe B.A., Chen F., Pan H., Ramser J., Lehrach H., Reinhardt R., McCombie W.R., de la Bastide M., Dedhia N., Blocker H., Hornischer K., Nordsiek G., Agarwala R., Aravind L., Bailey J.A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D.G., Burge C.B., Cerutti L., Chen H.C., Church D., Clamp M., Copley R.R., Doerks T., Eddy S.R., Eichler E.E., Furey T.S., Galagan J., Gilbert J.G., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Johnson L.S., Jones T.A., Kasif S., Kaspryzk A., Kennedy S., Kent W.J., Kitts P., Koonin E.V., Korf I., Kulp D., Lancet D., Lowe T.M., McLysaght A., Mikkelsen T., Moran J.V., Mulder N., Pollara V.J., Ponting C.P., Schuler G., Schultz J., Slater G., Smit A.F., Stupka E., Szustakowski J., Thierry-Mieg D., Thierry-Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y.I., Wolfe K.H., Yang S.P., Yeh R.F., Collins F., Guyer M.S., Peterson J., Felsenfeld A., Wetterstrand K.A., Patrinos A., Morgan M.J., de Jong P., Catanese J.J., Osoegawa K., Shizuya H., Choi S., Chen Y.J. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Liu H., Herrmann C.H. Differential localization and expression of the Cdk9 42k and 55k isoforms. J. Cell. Physiol. 2005;203(1):251–260. doi: 10.1002/jcp.20224. [DOI] [PubMed] [Google Scholar]
- Loyer P., Trembley J.H., Katona R., Kidd V.J., Lahti J.M. Role of CDK/cyclin complexes in transcription and RNA splicing. Cell. Signal. 2005;17(9):1033–1051. doi: 10.1016/j.cellsig.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Lyne P.D., Lamb M.L., Saeh J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006;49(16):4805–4808. doi: 10.1021/jm060522a. [DOI] [PubMed] [Google Scholar]
- McInnes C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007;11(5):494–502. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Morgan D.O. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- Nigg E.A. Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. BioEssays. 1995;17(6):471–480. doi: 10.1002/bies.950170603. [DOI] [PubMed] [Google Scholar]
- Pavletich N.P. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J. Mol. Biol. 1999;287(5):821–828. doi: 10.1006/jmbi.1999.2640. [DOI] [PubMed] [Google Scholar]
- Peng J., Marshall N.F., Price D.H. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J. Biol. Chem. 1998;273(22):13855–13860. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]
- Peng J., Zhu Y., Milton J.T., Price D.H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998;12(5):755–762. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatnani H.P., Greenleaf A.L. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20(21):2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- Schmerwitz U.K., Sass G., Khandoga A.G., Joore J., Mayer B.A., Berberich N., Totzke F., Krombach F., Tiegs G., Zahler S., Vollmar A.M., Furst R. Flavopiridol protects against inflammation by attenuating leukocyte-endothelial interaction via inhibition of cyclin-dependent kinase 9. Arterioscler. Thromb. Vasc. Biol. 2011;31(2):280–288. doi: 10.1161/ATVBAHA.110.213934. [DOI] [PubMed] [Google Scholar]
- Senderowicz A.M., Sausville E.A. Preclinical and clinical development of cyclin-dependent kinase modulators. J. Natl Cancer Inst. 2000;92(5):376–387. doi: 10.1093/jnci/92.5.376. [DOI] [PubMed] [Google Scholar]
- Shore S.M., Byers S.A., Maury W., Price D.H. Identification of a novel isoform of Cdk9. Gene. 2003;307:175–182. doi: 10.1016/s0378-1119(03)00466-9. [DOI] [PubMed] [Google Scholar]
- Sims R.J., 3rd, Belotserkovskaya R., Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 2004;18(20):2437–2468. doi: 10.1101/gad.1235904. [DOI] [PubMed] [Google Scholar]
- Suryadinata R., Sadowski M., Sarcevic B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci. Rep. 2010;30(4):243–255. doi: 10.1042/BSR20090171. [DOI] [PubMed] [Google Scholar]
- Traxler P., Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999;82(2–3):195–206. doi: 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- Tuccinardi T. Docking-based virtual screening: recent developments. Comb. Chem. High Throughput Screen. 2009;12(3):303–314. doi: 10.2174/138620709787581666. [DOI] [PubMed] [Google Scholar]
- Vermeulen K., Van Bockstaele D.R., Berneman Z.N. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36(3):131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Griffiths G., Midgley C.A., Barnett A.L., Cooper M., Grabarek J., Ingram L., Jackson W., Kontopidis G., McClue S.J., McInnes C., McLachlan J., Meades C., Mezna M., Stuart I., Thomas M.P., Zheleva D.I., Lane D.P., Jackson R.C., Glover D.M., Blake D.G., Fischer P.M. Discovery and characterization of 2-anilino-4-(thiazol-5-yl)pyrimidine transcriptional CDK inhibitors as anticancer agents. Chem. Biol. 2010;17(10):1111–1121. doi: 10.1016/j.chembiol.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Wei P., Garber M.E., Fang S.M., Fischer W.H., Jones K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92(4):451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Zhang J., Yang P.L., Gray N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Pe’ery T., Peng J., Ramanathan Y., Marshall N., Marshall T., Amendt B., Mathews M.B., Price D.H. Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev. 1997;11(20):2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]