

Summary
The IRE1/XBP1 signaling pathway is part of a cellular program that protects from endoplasmic reticulum (ER) stress, but also controls development and survival of immune cells. Loss of XBP1 in splenic type 1 conventional dendritic cells (cDC1s) results in phenotypic and functional alterations without affecting cell survival. However, in mucosal cDC1s, loss of XBP1 impaired survival in a tissue specific manner - while lung cDC1s die, intestinal cDC1s survive. This was not caused by differential activation of ER stress cell death regulators CHOP or JNK. Rather, cell fate was determined by a differential ability to shut down protein synthesis via a protective ATF4-dependent integrated stress response. In addition, regulated IRE1 dependent mRNA decay (RIDD) occurred mainly in intestinal cDC1s, and compound deficiency of IRE1 endonuclease led to cDC1 loss also in the intestine. Thus, mucosal DCs differentially mount ATF4 and IRE1 dependent adaptive mechanisms to survive in the face of ER stress.
Introduction
X-box Binding Protein-1 (XBP1) is a transcription factor activated by Inositol Requiring Enzyme-1 (IRE1), a transmembrane protein residing in the endoplasmic reticulum (ER) with both kinase and endonuclease activity. Upon activation, IRE1 oligomerizes and transphosphorylates neighbouring IRE1 molecules, stabilizing a conformational change and engaging its endonuclease activity. This process leads to the unusual splicing and religation of the mRNA encoding Xbp1, removing a 26 nucleotide long intron. The resulting frame shift bypasses an early stopcodon, allowing the translation of the full-length transcription factor XBP1s1–3.
The IRE1/XBP1 axis is part of a multipronged signaling cascade, dubbed the unfolded protein response (UPR), which aside of IRE1, comprises two other transmembrane sensors named activating transcription factor 6 (ATF6) and protein kinase RNA-like endoplasmic reticulum kinase (PERK)1. The UPR monitors the health state of the ER and maintains the fidelity of the cellular proteome. In this context, physiological and pathophysiological perturbations (e.g. viral infections, hypoxia or nutrient deprivation) can overwhelm the folding capacity of the ER, resulting into the accumulation of misfolded proteins, a condition referred to as ER stress. By increasing the folding capacity, enhancing ER associated degradation of misfolded proteins (ERAD) and inducing translation arrest, the UPR sensors cooperatively orchestrate the recovery of ER homeostasis (adaptive phase)4.
Nevertheless, in conditions of irremediable ER stress, the UPR can also execute apoptotic programs (terminal phase). A large set of studies has revealed multiple non-redundant apoptotic pathways originating from the UPR and there is growing evidence that all three of the UPR branches are capable of inducing cell death5. CHOP, a transcriptional regulator, is activated by PERK and is often regarded as the major culprit in ER stress-induced apoptosis pathways5. It executes cell death through the intrinsic as well as the extrinsic apoptotic pathway, enhances reactive oxygen species (ROS) production and activates cytoplasmic calcium pathways6,7. Through the upregulation of the phosphatase GADD34, the ATF4/CHOP branch modulates the levels of eIF2α phosphorylation, and this adaptive response allows an ER stressed-cell to resume protein synthesis. This is generally considered as a crucial checkpoint in determining cell death or survival during unresolved and chronic ER stress, as excessive client ER protein load might induce further ER stress when the cell has not yet fully adapted for this protein folding task8. Furthermore, under conditions of severe ER stress, IRE1 endonuclease activity can also result in cell death. In these conditions, the endonuclease substrate specificity of IRE1 broadens, and additional ER-localised RNAs are cleaved, in a process referred to as regulated IRE1 dependent degradation of RNA or RIDD9,10. Although this might again be an adaptive mechanism to reduce ER client proteins initially, the RIDD pathway has also been shown to trigger a proapoptotic response11–13.
Dendritic cells (DCs) are a heterogeneous group of immune cells that are present throughout the body. Their main function is to sample tissue-derived antigens, migrate to the draining lymph nodes (LN) and present antigen to naive T-cells, inducing proliferation and differentiation. Transcriptional profiling and ontogeny studies have allowed the distinction into plasmacytoid DCs (pDC) and the type 1 and type 2 conventional DCs (cDC1 and cDC2 respectively) lineages, each subtype having a specialized function14–18. We previously reported that IRE1-dependent splicing of XBP1 is a hallmark of splenic cDC1s, which occurs without activation of the additional branches of the UPR19. Furthermore, deletion of XBP1 in this cell subset resulted in disturbed ER architecture, decreased expression of the surface marker CD11c and a crippled ability to crosspresent dead cell-associated antigens, however without affecting cell survival in splenic DCs19.
In the present study, we have analyzed the function of XBP1 and the UPR in cDCs that reside in the mucosal immune system. We report that in contrast to the spleen, conditional deletion of XBP1 in CD11c+ cells resulted in a profound and cell intrinsic loss of lung cDC1s from tissue and draining lymph nodes. This was not due to a developmental defect of cDC1s, but to apoptosis of cells in absence of XBP1. Unexpectedly, cDC1s residing in the small intestine did not succumb to XBP1 deficiency. Cell survival was associated with reduction of protein translation through a stronger ATF4 dependent adaptive integrated stress response and stronger RIDD activation in intestinal cDC1 compared with lung cDC1s. As a consequence, loss of RIDD on top of XBP1 by compound deficiency of IRE1 and XBP1 caused cDC1 cell death also in the small intestine. We conclude that IRE1 endonuclease activity sets the threshold for cDC1 survival in conditions of chronic ER stress.
Results
Spontaneous IRE1 activation in cDC1s is tissue dependent
In contrast to most immune cells, dendritic cells (DCs) show high basal XBP1 splicing activity20,21. By making use of a mouse reporter strain that faithfully reports on IRE1 endonuclease activity (ERAI mice, for details see Material and Methods section and19,22), we have previously shown that within the spleen mainly cDC1s bear high levels of VenusFP protein19. Here, we questioned if this was a conserved feature among different tissues and probed the VenusFP signal in cDCs throughout several organs. To be able to compare DCs between variable tissues, we used a global staining protocol using CD11c, MHCII, XCR-1 and CD172α, carefully excluding macrophages (Fig 1A and18). As seen in the spleen, the use of XCR-1 and CD172α allowed bona fide identification of the peripheral cDC subsets in the examined organs; as confirmed by counterstaining with antibodies to CD103 and CD11b that match to cDC1 and cDC2 lineages respectively (Suppl Fig 1A). By applying this staining onto the different organs, we found that independently of the tissue of origin, DCs expressed higher levels of VenusFP compared to additional immune cells including B and T lymphocytes (Fig 1B,C). Within DC subtypes, cDC1s examined in different organs displayed the brightest VenusFP fluorescence, whereas, cDC2 throughout the tissues and draining nodes were much lower in VenusFP expression. A notable exception was noticed for lung derived cDC1s, which appeared to have low expression of VenusFP that was comparable to their CD24+ cDC223 counterparts (Fig 1B,C). We therefore additionally stained for IRE1 protein expression levels in various cDC1s, and found that differences in IRE1 endonuclease activity in ERAI mice closely correlated with differences in expression levels of IRE1. The highest levels were found in small intestinal cDC1s. Lung cDC1s showed the lowest level of IRE1, whereas splenic cDC1s had intermediate levels (Fig 1D). Thus, high activation of the endonuclease of IRE1 in steady state is a conserved feature among cDC1s, that is however strongly influenced by the tissue of residence.
Figure 1. Tissue specific regulation of the IRE1 endonuclease activity in cDCs.
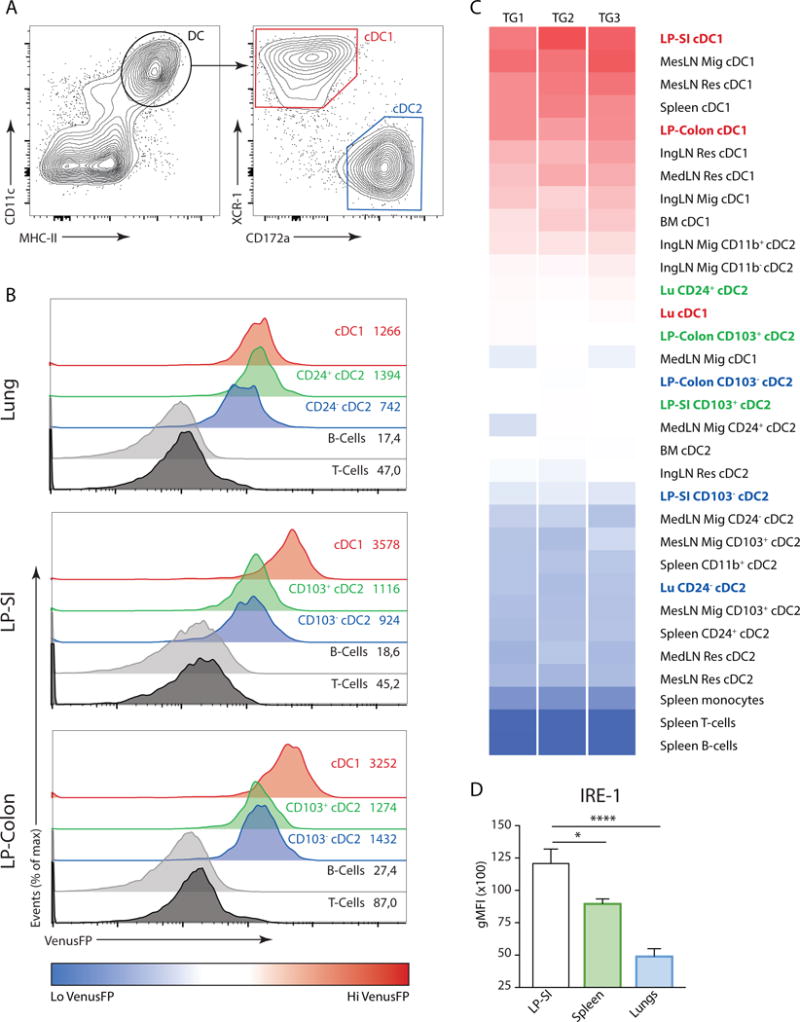
(A) Global gating strategy of cDCs, regardless of tissue origin. Graphs of spleen cDCs are shown. (B) ERAI VenusFP expression in lung (top), lamina propria of small intestine (LP-SI) (middle) and LP-colon (bottom histograms) in T-cells, B-cells and cDCs from ERAI Tg animals. Values depict geometrical mean fluorescence (gMFI) obtained from 1 representative sample. (C) Heat map analysis of ERAI VenusFP fluorescence in cDC1s and cDC2s derived from various organs. Splenic B-cells, T-cells and monocytes were used as immune cell controls for VenusFP levels. cDCs derived from lungs, small intestine and colon are highlighted. Data is representative of 2 independent experiments. (D) Fluorescence measured by flow cytometry of splenic, lung and LP-SI WT cDC1s stained with an antibody raised against IRE1. Bar graphs represent mean gMFI +/− S.E.M (n=4-6-6). Kruskal-Wallis test with Dunn’s multiple comparisons. Data is representative of 3 independent experiments.
XBP1 deletion affects mucosal cDCs differentially
To gain more insight in the function of the tissue specific regulation of XBP1 splicing, we used the Itgax-Cre (Cd11c-Cre) × Xbp1fl/fl mice, referred to as XBP1ΔDC mice19. We reported previously that cDC1s and cDC2s from XBP1ΔDC mice are present in normal cell numbers in the spleen19. Nevertheless, XBP1 deficient cDC1s express markedly low levels of the molecule CD11c19. This effect is attributed to the compensatory activation of RIDD, which is selectively observed in cDC1s from XBP1ΔDC mice, and that in turn mediates the degradation of the mRNA encoding for the integrin CD18, the obligate dimer partner of CD11c19. These findings precluded the use of CD11c as a universal marker for cDC identification in XBP1ΔDC mice. Therefore, we used a new gating strategy to define DC lineages independently of CD11c (18 and Suppl Fig 2A). Using this strategy, we confirmed that absence of XBP1 did not alter cell frequencies of splenic cDC1s and cDC2s (Fig 2A) yet selectively decreased CD11c expression in cDC1s (19 and Suppl Fig 2B).
Figure 2. Loss of XBP1 in CD11c+ cells disrupts the cDC compartment in peripheral organs.
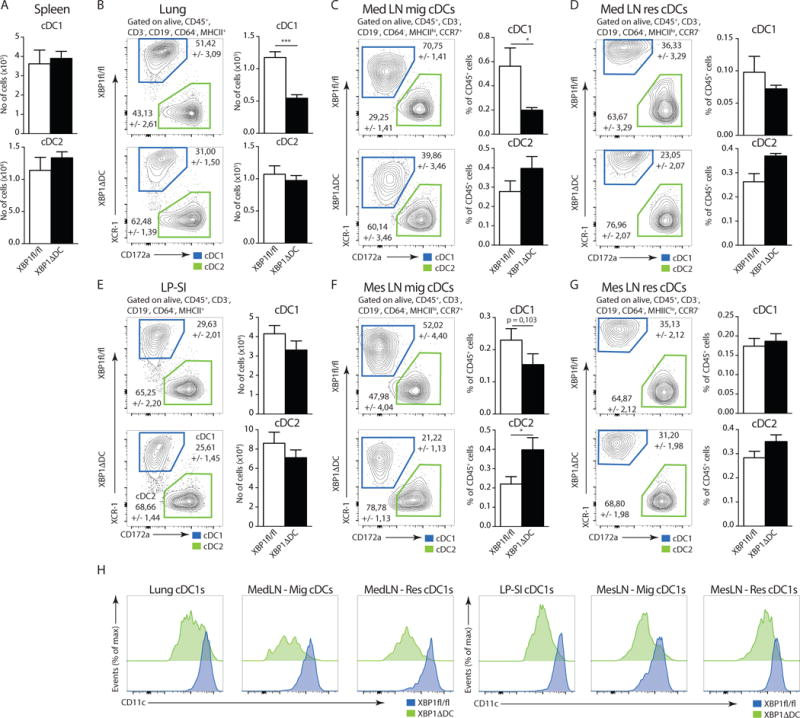
(A) Number (no) of cDC1s and cDC2s in the spleen of Xbp1fl/fl and XBP1ΔDC mice (n=6-8). (B) Distribution of cDC1s and cDC2s in lungs of Xbp1fl/fl and XBP1ΔDC mice. Bar graph depicts mean no of cells +/− S.E.M (n=6-7). P=0.0007, Mann-Whitney test. (C–D) Distribution of migratory (CCR7+) (C) and resident (CCR7−) (D) cDC1s and cDC2s in the mediastinal lymph node (MedLN) of Xbp1fl/fl and XBP1ΔDC mice. Bar graphs represent mean percentage +/− S.E.M. of migratory or resident cDCs relative to all CD45+ cells (n=11-14). P=0.0128, Mann-Whitney test. (E) Distribution of LP-SI cDC1s and cDC2s of Xbp1fl/fl and XBP1ΔDC mice. Bar graph depicts mean no (+/− S.E.M.) of cDC1s and cDC2s (n=6-8). (F–G) Distribution of migratory (F) and resident (G) cDC1s and cDC2s in the mesenteric lymph node (MesLN) of Xbp1fl/fl and XBP1ΔDC mice. Bar graphs represent mean percentage (+/− S.E.M.) of cDC1s and cDC2s relative to all CD45+ cells (n=11-14). P=0.0372, Mann-Whitney test. (H) CD11c expression in cDC1s from different organs of Xbp1fl/fl (blue) and XBP1ΔDC mice (green). Data is representative of 2 (C,D,F,G) or at least 3 independent experiments (A,B,E,H).
Next, we analyzed the lung and the lung draining mediastinal (Med) LNs of XBP1ΔDC mice and control littermates. Surprisingly, we found that in lungs from XBP1ΔDC mice, cDC1s were reduced by 50% approximately both in frequencies and cell numbers, while lung cDC2s remained unaffected (Fig 2B). Additional immune cell populations known to be targeted by the Cre recombinase were normally present in the lung of XBP1ΔDC mice, with the exception for a small but significant decrease in the number of alveolar macrophages (Suppl Fig 2C,D). The selective absence of lung cDC1s of XBP1ΔDC mice was not accounted by differences in the maturation state, as XBP1-deficient and -sufficient cDC1s from the lung displayed similar expression of MHCII and the costimulatory marker CD86 (Suppl Fig 2E). In the LNs, DCs can be subdivided into ‘lymph-node resident DCs’, which are cells that seed the organ directly from the blood and are closely related to splenic cDCs, and migratory DCs that traffic to the LN from non-lymphoid tissue15. These DC subtypes can be identified by differential expression of CCR7 and MHCII15. Similar to the lung, we found that migratory cDC1s from the lung-draining MedLN (identified as CCR7+, MHCIIhi) were strongly reduced in XBP1ΔDC mice. The frequencies of migratory cDC2s were slightly increased (Fig 2C). In addition, loss of XBP1 did not alter the frequencies of lymphoid-resident, non-migratory cDC1s and cDC2s (CCR7−, MHCIIlo) (Fig 2D), as previously noticed for splenic counterparts19. Altogether, these findings indicate that XBP1 selectively regulates the homeostasis of tissue-resident and migratory lung cDC1s but not cDC2s.
In contrast to the lung, we found that the cDC1 and cDC2 compartment of the lamina propria of the small intestine (LP-SI) remained largely unaltered in XBP1ΔDC mice (Fig 2E), with only a minor trend to fewer migratory cDC1s in the mesenteric (Mes) LN. We again found a significant increase in the frequency of migratory cDC2s (Fig 2F). Comparable to the spleen and MedLN, absence of XBP1 does not affect the frequency of resident cDCs (Fig 2G). Notably, both resident and tissue-derived cDC1 populations from lung and intestine of XBP1ΔDC mice demonstrated signs of XBP1 deletion, as reflected by selective reduction in the expression levels of CD11c, an effect never observed in cDC2s (Fig 2H and Suppl Fig 2B) and loss of Xbp1 exon 2 mRNA (Suppl Fig 2F). Altogether, these data indicate that XBP1 deficiency selectively affects the homeostasis of tissue-resident cDC1s in the lung but not in the intestine.
Absence of XBP1 causes cell-intrinsic loss of lung cDC1s
The development and survival of central and peripheral cDCs is regulated by multiple environmental cues including the cytokines FMS-related tyrosine kinase 3 ligand (Flt3L)24, that controls development of all cDCs, and granulocyte-macrophage colony-stimulating factor (GM-CSF), that controls peripheral cDC1s25. To address possible defects in the DC microenvironment accounting for the loss of lung cDC1s and migratory lung cDC1s, we generated bone marrow (BM) chimeric mice by injecting lethally irradiated CD45.1/2 mice with a mixture of BM cells of wild type (WT) CD45.1 and CD45.2 XBP1ΔDC origin (Fig 3A). CD45.2 XBP1ΔDC BM cells repopulated neutrophils and splenic DC populations as efficiently as the CD45.1 WT counterparts (Fig 3B). In contrast, cDC1s in the lung that originated from XBP1ΔDC BM precursors showed a pronounced defect compared to WT cells (Fig 3C). Along these lines, the ratios of the migratory cDC1 population in the MedLN were also reduced to a similar extent as in the lung. In the more stringent competitive setting of bone marrow chimerism, we also noted a small disadvantage for cDC2s of XBP1ΔDC in populating the lung, however this effect was no longer seen when migratory cDC2s were studied in the MedLN. Resident cDC1s and cDC2s cells were minimally affected by the absence of XBP1 (Fig 3C), indicating that XBP1 selectively regulates the homeostasis of lung-derived migratory cDC1s.
Figure 3. Cell intrinsic defects in the peripheral cDC1 compartment of XBP1ΔDC mice.
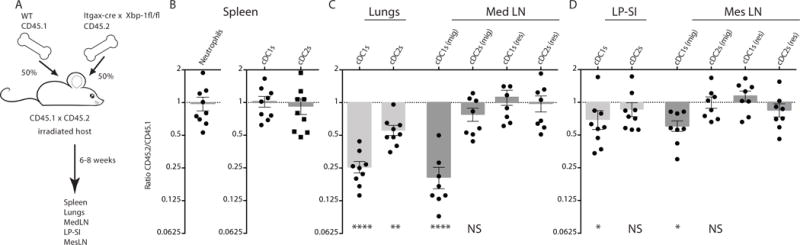
(A) Graphical representation of XBP1ΔDC chimeric mice setup. (B) Ratio of splenic CD45.2 XBP1ΔDC neutrophils, cDC1s and cDC2s over CD45.1 wild type counterparts in irradiated CD45.1/2 wild type hosts. (C) Ratio of lung and MedLN (both resident and migratory) CD45.2 XBP1ΔDC cDC1s and cDC2s over cDC1s and cDC2s derived from CD45.1 wild type origin. (D) Ratio of small intestine and MesLN (both resident and migratory) CD45.2 XBP1ΔDC cDC1s and cDC2s over cDC1s and cDC2s derived from CD45.1 wild type origin. Each symbol represents an individual mouse. Bar graph represents mean +/− S.E.M. (n=at least 8). Mann-Whitney test *: p<0.05, **: p<0.01, ****: p<0.001 Data is representative of 3 similar experiments.
On the other hand, the composition of the DC network in LP-SI and MesLN from the BM chimeras remained largely unaffected, with migratory and resident cDC1s and cDC2s being similarly represented between XBP1-sufficient and XBP1-deficient cells. We only found minor but statistically significant defects in the intestinal cDC1 population (Fig 3D). Therefore, we conclude that the selective loss of lung cDC1s in XBP1ΔDC mice is cell-intrinsic and cannot be explained by additional defects in the immune system.
XBP1 deficient cDC1s display normal development
XBP1 is an important transcription factor involved in the differentiation of highly secretory cells such as hepatocytes26, acinary cells27, Paneth cells28, plasma cells29, but also in non-secretory cells such as eosinophils30. To address whether XBP1 regulates DC development in-vivo, we monitored XBP1 activation in DC progenitor cells from the ERAI transgenic mice. A recent study reported that the presence of dedicated cDC1 and cDC2 precursors among pre-DC populations in BM and spleen can be identified through the use of Siglec-H and Ly6C31. Using this strategy, we found that only the direct precursors of cDC1s (identified as Siglec-H−, Ly6C−) in the spleen, but not in the BM expressed VenusFP (Fig 4A). In addition, we did not detect any VenusFP signal in the splenic precursors of cDC2s (Siglec-H−, Ly6C+) nor in the uncommitted pre-DC populations (Siglec-H+, Ly6C+/−) (Fig 4A and Suppl Fig 3A). cDC1s precursors in spleen also express CD2432 and flow cytometry analysis confirmed that the VenusFP signal is contained within the CD24high compartment, suggesting that IRE1-dependent XBP1 splicing defines the cDC1 lineage (Fig 4A). To assess the commitment of VenusFP+ precursors into cDC1 cells, splenic pre-DCs were sorted based on VenusFP expression and cultured ex-vivo. After 24 hours, VenusFP+ pre-DCs yielded mainly XCR1+ cDC1s whereas VenusFP− cells gave rise to Sirplα+ cDC2s (Fig 4B and data not shown). We were unable to detect any subpopulation of earlier progenitor cells in the BM expressing high levels of VenusFP (Fig 4A and C and Suppl Fig 3B), indicating that preferential activation of IRE1 in cDC1s is a late event in the commitment towards the cDC1 lineage.
Figure 4. XBP1 deficient cDC1s display normal development.
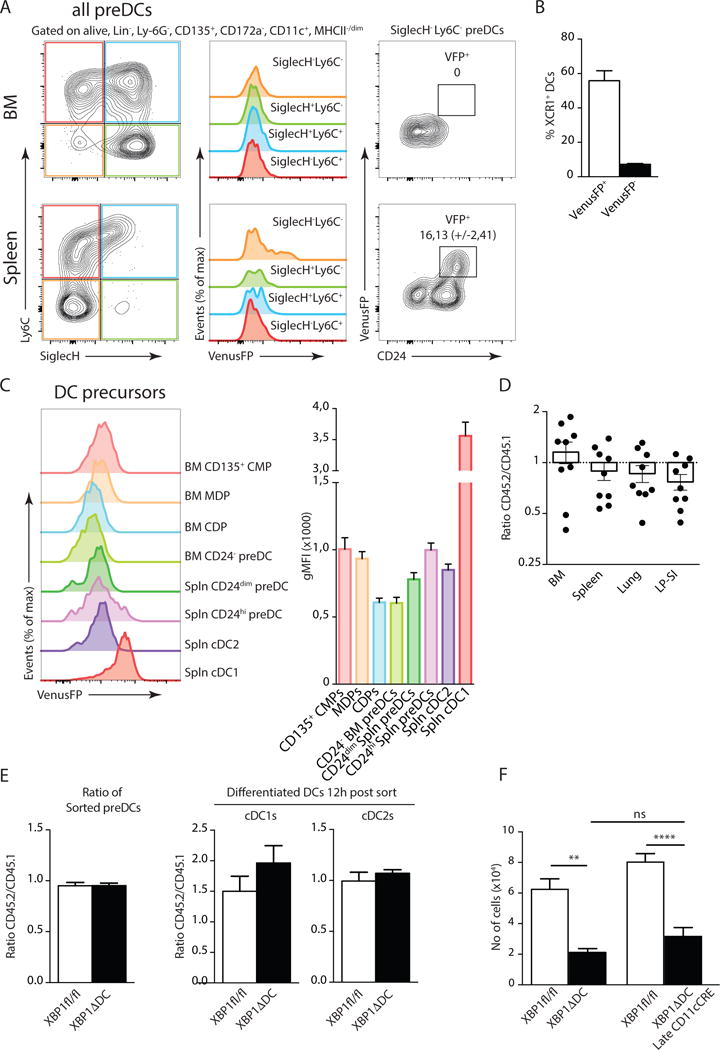
(A) VenusFP expression in BM and splenic pre-DC populations defined by SiglecH and Ly6C staining (left and middle plots). Expression of CD24 and VenusFP in SiglecH+ Ly6C+ pre-DCs (right plots). Numbers represent mean percentage of VenusFP+ pre-cDC1s (+/− S.E.M.). (n=3). (B) Percentage of XCR1+ cDCs relative to cDCs after 24h culture in-vitro of sorted ERAI VenusFP+ or VenusFP− pre-DCs in presence of Flt3L (n=4). (C) Expression of ERAI VenusFP in Flt3+ (CD135+) progenitor cells of cDCs. (CMP, Common Myeloid Progenitor, MDP, Macrophage Dendritic Cell Progenitor, CDP, Common DC progenitor). Bar graphs represent gMFI plus S.E.M. (n=8) (D) Ratio of bone marrow, spleen, lung or LP-SI CD45.2 XBP1ΔDC pre-DCs over CD45.1 wild type counterparts. Each symbol represents an individual mouse. Bar graph represents mean +/− S.E.M. (E) Ratio of CD45.2 Xbp1fl/fl or XBP1ΔDC pre-DCs over CD45.1 counterparts, directly after sort (left plot), or after overnight development into cDC1s or cDC2s in presence of Flt3L (right plot). (n=4-6) (F) No of lung cDC1s in Xbp1fl/fl and XBP1ΔDC and XBP1ΔDC-Late. Bar graphs represent mean +/− S.E.M. (n=at least 11 per group). Kruskal-Wallis test with Dunn’s multiple comparisons. Data is representative of 2 (B,C,D,E,F), or 4 (A) independent experiments.
We turned our attention to the CD45.1 WT/CD45.2 XBP1Δ BM chimeras previously established to address the contribution of XBP1 to the homeostasis of pre- DCs (Fig 3A). Data depicted in Fig 4D show similar frequencies of pre-DCs between WT and XBP1ΔDC cells in BM, spleen, lung and the LP-SI in BM chimeras, arguing against an effect of CD11c-mediated deletion of XBP1 at the pre-DC stage. Alternatively, DC precursors from XBP1ΔDC mice could be more committed to a cDC2 lineage. To address this, we sorted pre-DCs from CD45.1 WT and CD45.2 XBP1ΔDC mice and differentiated them in culture with Flt3L for 12 hours. At this point, up to 70% of the pre-DCs differentiated into CD11c+ MHCII+ cDCs that had efficiently recombined the Xbp1 allele (Suppl Fig 3C and data not shown). Data shows that CD45.1 WT and CD45.2 XBP1ΔDC preDCs gave rise to equal proportions of cDC1s or cDC2s (Fig 4E), indicating that XBP1 loss does not skew pre-DC differentiation. Furthermore, XBP1-deficient lung cDC1s expressed normal levels of key transcription factors required for DC differentiation15, as determined by real-time qPCR (RT-qPCR) (Suppl Fig 3D).
Finally, to formally exclude the possibility that the loss of lung cDC1s in the XBP1ΔDC mice is due to developmental defects, we made use of an alternative Cre line that specifically deletes the floxed gene in fully mature cDCs33. This line possesses a minimal CD11c promotor with an upstream enhancer-driving Cre and eGFP, separated by an internal ribosome entry site (IRES). Quantification of eGFP levels by flow cytometry confirmed selective expression of the promotor in terminally differentiated cDCs, while being absent in pre-DCs (Suppl Fig 3E). In line with the data obtained with XBP1ΔDC mice, the Xbp1fl/fl mice × Cd11c-Cre-eGFP mice (referred to as ‘XBP1ΔDC-Late’ mice) also display a profound loss in the absolute numbers of lung cDC1s, indicating that XBP1 deficiency affects the homeostasis of fully differentiated lung cDC1s (Fig 4F). Altogether, these data show that XBP1 loss does not affect cDC1 development.
Loss of XBP1 induces apoptotic cell death in lung cDC1s
Pre-DCs can still undergo proliferation before committing to a mature DC phenotype31,32,34. To assess possible replication defects we checked the expression of the proliferation marker Ki67, and found no decrease in expression in absence of XBP1 (Fig 5A). In contrast, XBP1 deficient lung cDC1s displayed higher expression of this proliferation marker (Fig 5A). This observation was further validated by the higher in-vivo incorporation of BrdU in XBP1 deficient lung cDC1s compared to their littermate controls (Fig 5B). These experiments show that XBP1 deficiency does not reduce the proliferative capacities of pre-DCs.
Figure 5. Absence of XBP1 cripples lung cDC1 survival.
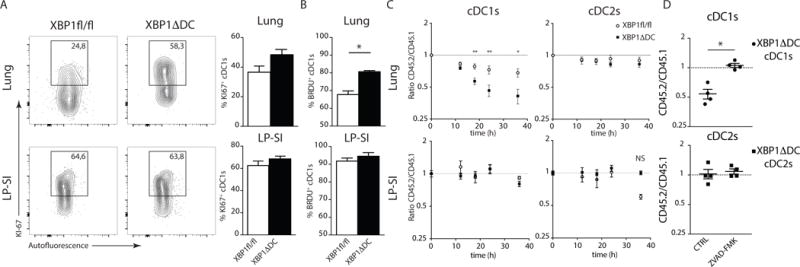
(A) Intracellular staining of Ki67 in lung or LP-SI derived cDC1s. Figure depicts percentage of Ki67+ cells in 1 representative animal. Bar graph represents mean +/− S.E.M. (n=5). (B) In-vivo BrdU incorporation after 3 days of exposition to BrdU in drJNKing water. Bar graphs represent mean +/− S.E.M. (n=4). P=0.0286(Lung), Mann-Whitney test. (C) Sorted lung or LP-SI CD45.2 Xbp1fl/fl and XBP1ΔDC cDC1s (left plot) or cDC2s (right plot) mixed with CD45.1 counterparts and cultured in medium without Flt3L. Ratios were monitored over time. Each symbol represents mean ratio +/− S.E.M. (Lung n=8, LP-SI n=7 or 3). Mann-Whitney test P(18h)=0.0026, P(24h)=0.0085, P(36h)=0.0117. (D) Sorted lung CD45.2 XBP1ΔDCs cDC1 (top) and cDC2 (bottom) were mixed with CD45.1 counterparts and treated overnight with zVAD-fmk or vehicle. Ratio of Ann-V− cells is given. Each symbol represents 1 mouse. Mean ratio +/− S.E.M. is given (n=4). For additional details see (Suppl Fig 4A). Data is representative of 1 (A,B), 2 (D) or 4 (C) independent experiments.
The increased turnover of XBP1-deficient cDC1s could be indicative of a compensatory mechanism to replenish dying cDC1s. To address whether XBP1ΔDC cDC1s are more prone to cell death, we performed an ex-vivo survival assay in which cDCs were sorted from the lung or intestine of CD45.2 XBP1ΔDC or CD45.2 littermate control mice and were mixed in a 1:1 ratio with CD45.1 WT counterparts, and cultured in the absence of pro-survival cytokines such as GM-CSF35. The ratios of viable CD45.2 XBP1ΔDC/CD45.1 WT DCs and CD45.2 Xbp1fl/fl/CD45.1 WT DCs were compared over time. The proportion of lung cDC1s from XBP1ΔDC mice more rapidly declined over time compared to CD45.2 Xbp1fl/fl control littermates, whereas survival of lung cDC2s and cDCs from the intestine remained largely unaltered (Fig 5C). Probing the role of apoptosis, the use of Annexin V (AnnV) revealed a reduced proportion of alive lung cDC1s (AnnV negative) from XBP1ΔDC mice compared to lung cDC1s derived from Xbp1fl/fl control littermates. In contrast, survival was similar in WT vs XBP1ΔDC lung cDC2s and in the intestinal cDC populations (Fig 5D and Suppl Fig 4B). Interestingly, the addition of zVAD-FMK, a potent inhibitor of caspase-activity, was able to rescue CD45.2 XBP1ΔDC cDC1s from cell death after an overnight co-culture (Fig 5D and Suppl Fig 4A) indicating that caspase-mediated apoptosis is the main driver of cell death in cDC1s isolated from XBP1ΔDC mice. zVAD treatment did not significantly alter the ratios in lung cDC2s nor intestinal DC populations (Suppl Fig 4B).
These data indicate that XBP1-deficient lung cDC1s succumb to apoptosis, whereas intestinal cDC1s defy XBP1 loss.
ER stress induced death of cDC1 is CHOP and JNK independent
Irremediable ER stress ultimately results in the execution of apoptosis, and loss of XBP1 in some-but not all-cell types causes ER stress5,19. Therefore, we studied whether the differences in cell fate of lung versus intestinal cDC1s noticed in XBP1ΔDC mice were caused by a difference in the extent of ER stress. First, we checked the presence of large aggregates of tubular ER, a feature of XBP1 deficiency in splenic cDC1s19, in lung and intestine cDC1s. Immunostaining for KDEL-containing proteins revealed that these ER aggregates were present and comparable in both lung and intestine derived cDC1s from XBP1ΔDC mice (Fig 6A). These data were also confirmed by transmission electron microscopy (Suppl Fig 5A). On the other hand, cDC2s were largely devoid of these structures (Suppl Fig 5B). Interestingly, the transcriptional program generally associated with ER stress (exemplified by Hspa5 (coding for BiP) Herpud, Atf4, Ddit3 and Trib3) (Suppl Fig 5F) appeared similarly activated in cDC1s from both lung and intestine, suggesting that XBP1 deficiency triggers ER stress to a similar extent in both tissues (Fig 6B. Thus, both lung and intestine cDC1 from XBP1ΔDC mice displayed signs of ongoing ER stress.
Figure 6. XBP1 deficiency triggers ER stress in peripheral cDC1s but cell death is CHOP and JNK independent.
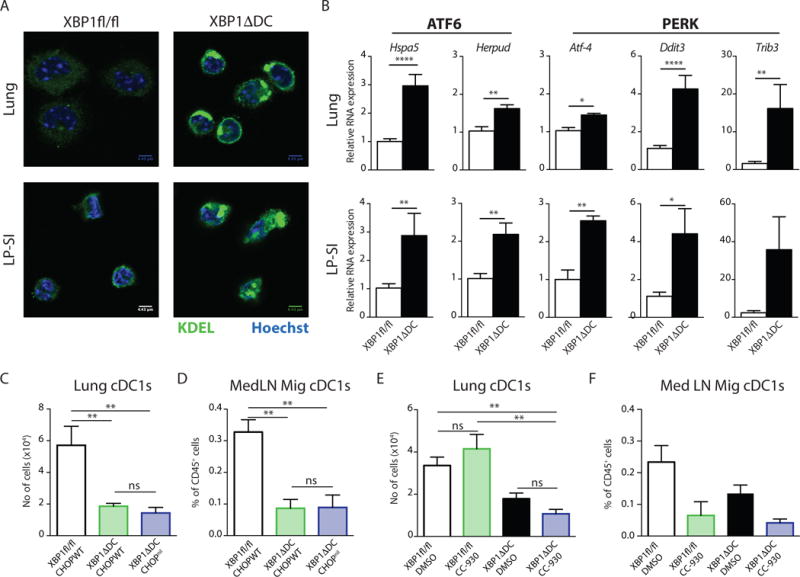
(A) Immunofluorescence of cDC1s, sorted by flow cytometry from lung and LP-SI, stained with an anti-KDEL antibody (ER-marker). Nuclei were stained with Hoechst. Scale bar: 4.43 μm. (B) Quantitative real-time PCR analysis of UPR target genes among total RNA prepared from sorted cDC1s of lung and LP-SI (n=6); results are normalized to housekeeping genes gapdh and ywhaz. Bar graphs depict mean +/− S.E.M. Unpaired Student T-test * : p<0.05, ** : p<0.01, **** : p<0.001. (C) No of lung cDC1s or (D) MedLN Mig cDC1s as percentage of CD45+ cells in Xbp1fl/fl, XBP1ΔDC and XBP1ΔDC CHOPnil mice. Bar graphs depict mean +/− S.E.M. (n=8-7-5). Kruskal-Wallis test with Dunn’s multiple comparisons. (E) No of lung cDC1s or (F) MedLN Mig cDC1s as percentage of CD45+ cells in XBP1fl/fl and XBP1ADC treated with CC-930 or vehicle. Bar graphs depict mean +/− S.E.M. (n=6-2-4-5).
Data is representative of 2 independent experiments (A–E) or 1 independent experiment (F).
The transcriptional regulator CHOP (encoded by Ddit3) is generally seen as an executor of ER stress-mediated cell death5. Considering the marked activation of Ddit3 mRNA upon XBP1 deletion (Fig 6B and reported in19), we tested whether CHOP mediated the apoptosis induced by XBP1 deficiency in the lung. For this, we crossed CHOP KO mice36 to XBP1ΔDC mice to generate double KO mice deficient for both XBP1 and CHOP in DCs (Suppl Fig 5C). Remarkably, CHOP deletion did not restore cDC1s numbers in the lung, nor in the migratory compartment of the MedLN (Fig 6C,D), suggesting that loss of lung cDC1s by XBP1 deficiency was not due to CHOP activation.
A second well established branch of ER stress-induced apoptosis is downstream of IRE1-mediated JNK activation28,37. We previously reported that loss of XBP-1 induces a reciprocal overexpression of IRE1 and hyperactivation of its kinase and endonuclease domains in DCs19. Immunoblotting confirmed enhanced JNK phosphorylation in XBP1Δ splenic cDC1s (Suppl Fig 5D). To explore whether JNK was mediating cell death of XBP1 deficient lung cDC1s, we treated the XBP1ΔDC mice with CC-930, a novel specific inhibitor of JNK phosphorylation (38 and validated in Suppl Fig 5E). As noticed in Fig 6E and F, CC-930 treatment was unable to rescue lung cDC1s and migratory counterparts in the MedLN. On the contrary, WT mice treated with CC-930 had remarkable fewer migratory MedLN cDC1s.
Together, these data indicated that XBP1 deficient cDC1s displayed a robust activation of the remaining UPR branches in absence of XBP1 but execution of apoptosis of lung cDC1s was not dependent on CHOP nor JNK activation (Suppl Fig 5F).
Protein translation and integrated stress response are distinctly regulated across tissue DCs
We found that despite similar expression of ER stress signature genes and ER remodeling (Fig 6A and B), XBP1-deficient lung cDC1s die, whereas intestinal cDC1s survive. We next hypothesized that discrete differences in the adaptation to chronic ER stress might explain the differential cell fate. PERK-mediated phosphorylation of eIF2α and the subsequent block in protein synthesis is a crucial adaptive mechanism for cell survival in conditions of chronic and severe ER stress, acting to reduce the amount of ER client protein load39. To investigate whether this pathway was differentially activated in lung versus intestinal cDC1s, we adopted a recently developed method in which a short pulse with O-propargyl puromycin (OP-Puro) can be used to measure translation of proteins in-vivo40. Subsequent fluorescent labeling of the incorporated OP-Puro into nascent polypeptides is a faithful read-out of translational rate, as shown by the inhibition of fluorescence in presence of translation inhibitors such as cycloheximide (40 and Suppl Fig 6A). Applying this method onto the previously established CD45.2-XBP1ΔDC/CD45.1-WT chimeric mice (Fig 3A) allowed us to monitor the effects of XBP1 deficiency on protein synthesis in a cell-intrinsic manner. We found that among the studied cell populations in spleen, lung, and intestine, cDC1s incorporate very high quantities of OP-Puro (Fig 7A and Suppl Fig 6B). XBP1 deficient cDC1s from the LP-SI efficiently reduced their translation rate to approximately 70% of the WT counterparts. In contrast, lung cDC1s displayed no difference in translation in absence of XBP1 (Fig 7A).
Figure 7. Tissue specific activation of the integrated stress response in cDC1s is not implicated in cell survival.
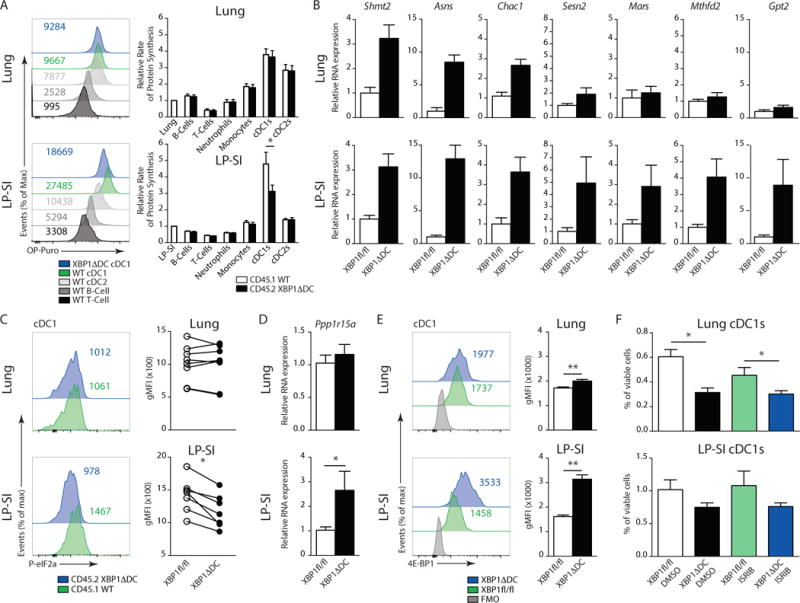
(A) In-vivo O-propargyl puromycin (OP-Puro) incorporation after intra-peritoneal injection into CD45.1 wild type/CD45.2 XBP1ΔDC BM chimeric mice. Histograms depict OP-Puro fluorescence in CD45.1 T-cells, B-cells and CD45.1 or XBP1ΔDC cDCs from lung (top) or LP-SI (bottom). Figures are from 1 representative sample. Bar graphs represent mean protein synthesis rate +/− S.E.M. (n=6). (B) RT-qPCR analysis of ATF4 target genes among total RNA prepared from sorted lung or LP-SI cDC1s from Xbp1fl/fl and XBP1ΔDC mice. RNA expression was normalized to household genes Gapdh and Ywhaz. Bar graphs show mean expression +/− S.E.M. XBP1ΔDClung (n=7-3) XBP1ΔDCLP-SI (6–4). (C) Intracellular staining of P-eIF2α in lung or LP-SI derived cDC1s. Histograms show fluorescence of 1 representative sample. Paired dot plots show gMFI of P-eIF2α of CD45.2 XBP1ΔDC cDC1s of matched CD45.1 WT controls. P=0.016, Wilcoxon matched pairs test. (D) RT-qPCR analysis of Gadd34 mRNA among total RNA prepared from sorted lung or LP-SI cDC1s from Xbp1fl/fl and XBP1ΔDC mice. RNA expression was normalized to household genes Gapdh and Ywhaz. Bar graphs show mean expression +/− S.E.M. XBP1ΔDClung (n=8-6) XBP1ΔDCLP-SI (7–6). (E) Intracellular staining of 4E-BP1 in lung or LP-SI derived cDC1s. Figures in histograms depict fluorescence of 1 representative sample. Bar graphs show mean fluorescence +/− S.E.M. P=0.0079, Mann-Whitney test (n=5). (F) No of lung cDC1s or LP-SI cDC1s as percentage of CD45+ cells in Xbp1fl/fl and XBP1ΔDC treated with ISRIB or vehicle. Bar graphs depict mean +/− S.E.M. (n=6 of n=4). Mann-Whitney test, * : p<0.05. Data is representative of 1 (B) or 2 (A,C,D,E,F) similar experiments.
These data demonstrate that cDC1s of the intestinal lamina propria turn down protein synthesis when facing ER stress. Several mechanisms could account for this, including eIF2α phosphorylation. Despite decreasing global protein translation, presence of phosphorylated eIF2α also leads to enhanced translation of a distinct set of mRNAs containing uORFs in their 5′ UTR, such as the activated transcription factor (ATF)4 (41 and Suppl Fig 6H). ATF4 acts as a transcriptional regulator and mounts a gene expression program known as the integrated stress response (ISR), that prepares cells for enhanced amino acid uptake, protein synthesis and folding capacity by inducing mRNAs for antioxidant response (Mtfhd2, Sesn2, Chac1) and amino acid metabolism (Mars, Asns, Shmt2, Gpt2) (41 and Suppl Fig 6H). We probed these members of the ISR and found that, in contrast to the lung, several of the tested genes were more induced in intestine cDC1s in the absence of XBP1 (Fig 7B). Unexpectedly, despite the presence of an ATF4 dependent gene signature, the phosphorylation status of eIF2α in XBP1 deficient LP-SI cDC1s was markedly reduced (Fig 7C and Suppl Fig 6C for validation of the P-eIF2α antibody). This correlated with an enhanced expression of the ATF4 target Gadd34 (Ppp1r15a) mRNA in LP-SI cDC1s (Fig 7D). This inducible phosphatase reduces P-eIF2α levels during ER stress and establishes a negative feedback loop, quickly restoring protein translation42,43,44. Lung cDC1s did not regulate phosphorylation of eIF2α (Fig 7C).
Translational control in DCs has been shown before to occur in an eIF2α-independent manner45, although the mechanism remains unclear. Puzzled by the sustained inhibition of translation in LP-SI cDC1s in absence of eIF2α phosphorylation, we decided to examine a second node of translational control mediated by 4E-BP146,47. 4E-BP1 is an ATF4 dependent gene that is known to be expressed in conditions of chronic ER stress48. It inhibits eIF4F assembly by competitively displacing eIF4G from eIF4E, thus promoting translation of select mRNAs containing IRES, at the expense of cap-dependent translation46. It is only active in the unphosphorylated state and is strictly regulated by direct phosphorylation as one of the main mTOR substrates49. We found that intestinal cDC1s lacking XBP1 mount higher levels of 4E-BP1 compared to lung cDC1s (Fig 7E). Phosphorylation, which inhibits 4E-BP1, was lower in intestinal cDC1s compared to lung cDC1s both in presence or absence of XBP1 (Suppl Fig 6D). The translation of the death associated protein DAP5 is promoted via an IRES46. Enhanced levels of intracellular DAP5 staining in absence of XBP1 confirmed the activation of IRES mediated translation (Suppl Fig 6E), suggesting that the 4E-BP1 node was responsible for sustained reduction in protein synthesis in cDC1s.
To examine the role of the integrated stress response as a potential adaptive survival pathway, we treated XBP1ΔDC mice and littermate controls with the inhibitor of the ISR, ISRIB, a drug that renders cells insensitive to the downstream effects of eIF2α phosphorylation such as ATF4 and 4E-BP1 induction50. Although in-vivo application of ISRIB efficiently blocked the ATF4 targets Ppp1r15a and 4E-BP1 (Suppl Fig 6F,G), suggestive of effective ISR inhibition, we did not observe reduced survival of intestinal cDC1s lacking XBP1 (Fig 7F). Thus, although intestinal cDC1s handle ER stress-induced by XBP-1 loss by mounting an ISR, at present we have insufficient evidence to indicate that this pathway protects from ER stress induced apoptosis (Suppl Fig 6H).
RIDD is differentially fine-tuned in XBP1 deficient cDC1s
Loss of XBP1 induces a reciprocal overexpression of IRE1 and hyperactivation of its kinase and endonuclease domains, potentially triggering regulated IRE-dependent mRNA decay (RIDD)9,19. Considering that RIDD was shown to promote cell death by inducing caspase 2 and thioredoxin interacting protein (TXNIP)51, we tested whether differential RIDD activation could explain the distinct cell fate of lung versus intestinal cDC1s lacking XBP1. First of all, we assessed whether the observed tissue specific differences in IRE1 activity in steady state (Fig 1C) were preserved in absence of XBP1. Therefore, we crossed ERAI mice with XBP1ΔDC mice, generating a transgenic mouse line that reports on IRE1 endonuclease activity in context of XBP1 deletion and concomitant IRE1 hyperactivation. VenusFP expression in these mice confirmed that XBP1 loss further increased the IRE1 endonuclease activity in cDC1s. Still, the differences in IRE1 activity levels between lung and intestinal cDC1s were preserved, with XBP1ΔDC cDC1s isolated from the lung displaying lower IRE1 endonuclease activity compared to small intestinal counterparts (Fig 8A). To validate induction of RIDD, we assessed the degradation of established RIDD mRNA targets Bloc1s1, Tapbp and Ergic3 in XBP1-deficient lung and intestinal cDC1s19. We observed a more pronounced degradation of these mRNAs species in cDC1s residing in the small intestine, confirming the results obtained by the ERAI reporter mice (Fig 8B). Thus, cDC1s from the small intestine and lung mount different degrees of RIDD, suggesting that RIDD may determine cell fate when XBP1 is lost.
Figure 8. IRE1 endonuclease activity is fine-tuned in an organ specific manner.
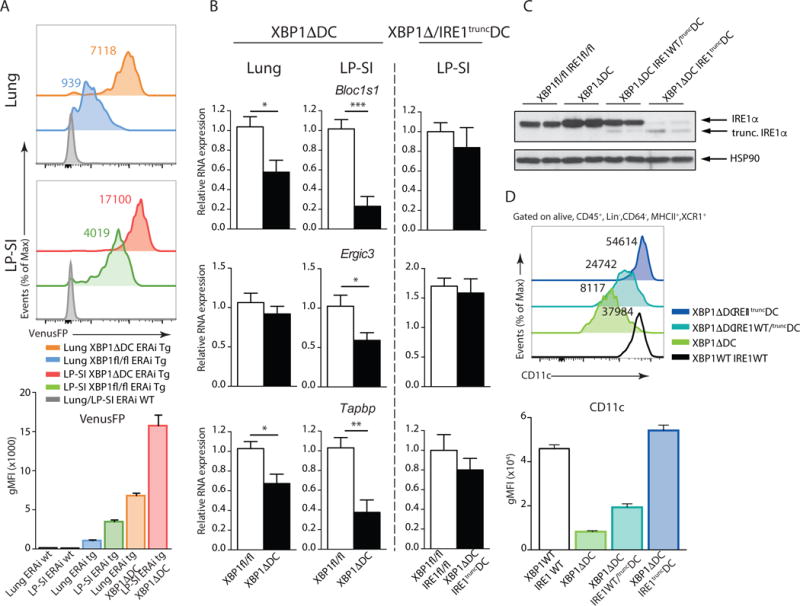
(A) ERAI VenusFP expression in lung (upper) and LP-SI (lower histograms) cDC1s from ERAI WT, ERAI Tg Xbp1fl/fl and ERAI Tg XBP1ΔDC mice. Bar graph depicts mean gMFI +/− S.E.M. (n=3-4-2). (B) RT-qPCR analysis of established RIDD targets among total RNA prepared from sorted lung or LP-SI cDC1s from Xbp1fl/fl, XBP1ΔDC, Xbp1fl/fl/Ire1fl/fl and XBP1ΔDC/IRE1truncDC mice. RNA expression was normalized to household genes Gapdh and Ywhaz. Bar graphs show mean expression +/− S.E.M. XBP1ΔDClung (n=12-6) XBP1ΔDCLP-SI (n=7-6) XBP1ΔDC/IRE1truncDCLP-SI (n=4-4). Unpaired Student T-test, P(Bloc1s1lung)=0.015, P(Bloc1s1lpsi)=0.0001, P(Tapbplung)=0.01, P(Tapbplp-si)=0.0019, P(Ergic3lpsi)=0.0461. (C) Immunoblot analysis of IRE1α levels in sorted splenic cDC1s of Xbp1fl/fl/Ire1fl/fl, XBP1ΔDC, XBP1ΔDC/IRE1WT/truncDC and XBP1ΔDC/IRE1truncDC mice. HSP-90 serves as a loading control. Each lane represents 1 mouse. (D) CD11c expression in Xbp1fl/fl/Ire1fl/fl, XBP1ΔDC, XBP1ΔDC/IRE1WT/truncDC and XBP1ΔDC/IRE1truncDC splenic cDC1s. Numbers in graph represent gMFI of CD11c of 1 representative sample. Bar graphs depict mean gMFI +/− S.E.M. Data is representative of 2 (A,B–D) or 1 (C) independent experiment.
RIDD activity promotes survival of XBP1 deficient intestinal cDC1s
To evaluate whether the differential induction of compensatory RIDD in lung versus intestinal DCs regulates cell survival of cDC1s, we turned into a genetic approach. We generated mice deficient in both XBP1 and RIDD by crossing XBP1ΔDC mice with Ern1fl/fl mice that bear loxP sites flanking exon 20 and 21 of the Ern1 gene52 (encoding IRE1 and referred to as “XBP1Δ/IRE1truncDC mice”). DCs from XBP1Δ/IRE1truncDC mice are deficient for XBP1 and harbor a truncated IRE1 isoform with preserved kinase function but crippled endonuclease activity53. Thus, we bred mice that were KO for XBP1 and either heterozygous or homozygous for the recombined IRE1 allele. IRE1 immunoblotting on sorted splenic cDC1s shows that the enzyme was upregulated in XBP1 KO cells19, whereas XBP1-KO cells bearing heterozygous deletion of IRE1 displayed intermediate expression of full size IRE1 and a faint band corresponding to the truncated IRE1 isoform (Fig 8C). In addition, cDC1s of double XBP1Δ/IRE1truncDC mice only displayed low levels of the truncated IRE1 (Fig 8C). To validate this strategy, we measured the expression levels of the RIDD targets Bloc1s1, Tapbp1 and Ergic3 mRNA in the double XBP1Δ/IRE1truncDC mice, which were no longer degraded in compound deficient intestinal cDC1s (Fig 8B). We also quantified surface expression levels of the molecule CD11c, which can be used as a surrogate marker of RIDD activity by flow cytometry (19 and Fig 8D). As expected, the levels of IRE1 inversely correlated with CD11c expression, as noticed by the reduced levels of the molecule in XBP1ΔDC mice, partially restored CD11c levels in XBP1ΔDC mice heterozygous for IRE1, and full restoration of CD11c expression in double XBP1Δ/IRE1truncDC mice (Fig 8D). These data confirmed that genetic targeting of IRE1 endonuclease in XBP1Δ/IRE1truncDC mice allowed for subtle titration of RIDD activation in-vivo (Fig 8D).
Subsequent analysis of the cDC compartment in lung and MedLN revealed that RIDD inhibition on top of XBP1 deficiency did not rescue lung cDC1s and MedLN cDC1s, arguing against a proapoptotic role for RIDD in DCs. On the contrary, the numbers of lung cDC1 were further decreased in mice with reduced or absent RIDD activity (Fig 9A–C). Remarkably, when RIDD was removed on top of XBP1 deficiency in the small intestine, cDC1s cell numbers declined, in a gene dosage dependent manner, as the effect was more marked in homozygous versus heterozygous XBP1Δ/IRE1truncDC mice. This defect was even more pronounced in the migratory cDC1 compartment of the MesLN (Fig 9D–F). In contrast to the cDC1s and much like the phenotype in the XBP1ΔDC mice, the effect on the cDC2 compartment was less pronounced, both in the lungs (Suppl Fig 7A–D) and small intestine (Suppl Fig 7E–H).
Figure 9. Organ specific RIDD activation orchestrates cDC1s survival in the face of chronic ER stress.
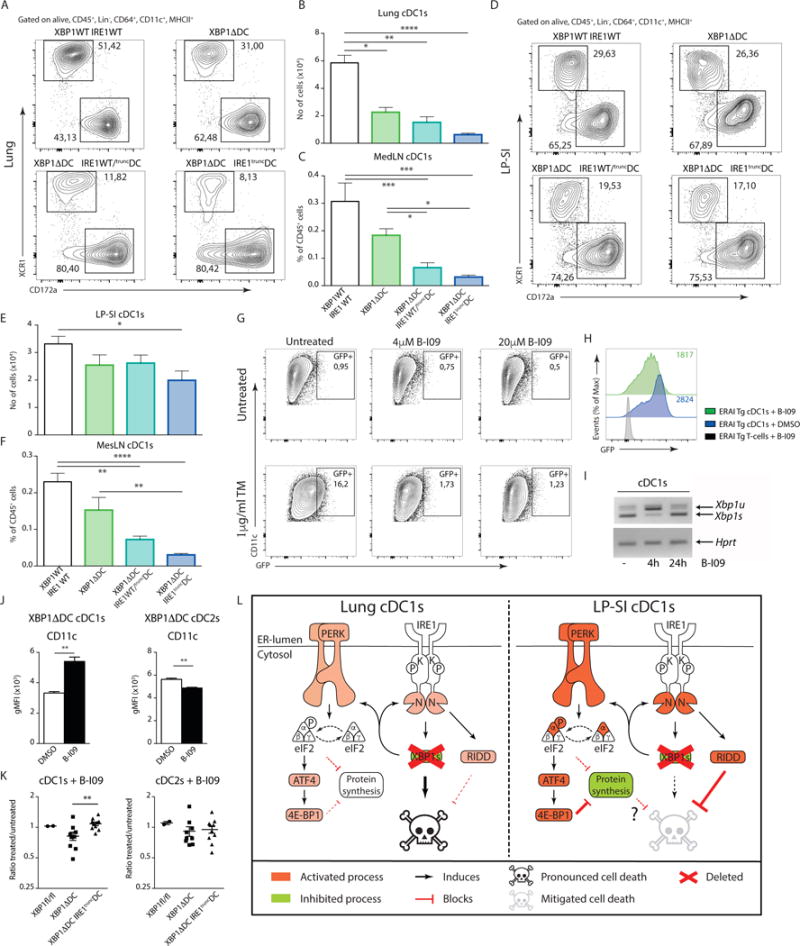
(A) Distribution of cDC1s and cDC2s in the lungs of given genotypes. Numbers are percentage of cDC1s and cDC2s relative to all cDCs from 1 representative sample. (B) Number of lung cDC1s in Xbp1fl/fl/Ire1fl/fl, XBP1ΔDC, XBP1ΔDC/IRE1WT/truncDC and XBP1ΔDC/IRE1truncDC mice. (C) Percentage of Mig cDC1s in the MedLN of Xbp1fl/fl/Ire1fl/fl, XBP1ΔDC, XBP1ΔDC/IRE1WT/truncDC and XBP1ΔDC/IRE1truncDC. (D) Distribution of cDC1s and cDC2s in the LP-SI of given genotypes. Numbers in graphs are percentage of cDC1s and cDC2s relative to all cDCs from 1 representative sample. (E) Number of LP-SI cDC1s in Xbp1fl/fl/Ire1fl/fl, XBP1ΔDC, XBP1ΔDC/IRE1WT/truncDC and XBP1ΔDC/IRE1truncDC mice. (F) Percentage of Mig cDC1s in the MesLN of Xbp1fl/fl/Ire1fl/fl XBP1ΔDC, XBP1ΔDC/IRE1WT/truncDC and XBP1ΔDC/IRE1truncDC. (A–F) Bar graphs represent mean +/− S.E.M. (n=at least 5). Kruskal-Wallis test Dunn’s multiple comparisons. (G) Venus FP expression in in-vitro cultured DCs from ERAi Tg bone marrow. Cells were treated with tunicamycin (TM) and increasing doses of the IRE1 inhibitor B-I09. Percentage of VenusFP+ cells are given. (H) VenusFP expression in ERAi transgenic splenic cDC1s upon in-vivo treatment with B-I09 or vehicle control. Geometrical mean fluorescence is given. (I) PCR analysis of unspliced Xbp1 mRNA (Xbp1u) and spliced Xbp1 mRNA (Xbp1s) in FACS sorted splenic cDC1s from wild-type C57BL/6 mice treated with B-I09 for 0, 4 or 24 hours. mRNA encoding Hprt serves as a loading control. (J) CD11c expression in XBP1 deficient MesLN cDC1s and cDC2s treated with B-I09 or vehicle control. Bar graphs represent mean +/− S.E.M. (n=5). Mann-Whitney test, ** : p<0.01. (K) Ratios of B-I09 treated over vehicle treated cDC1s and cDC2s derived of XBP1fl/fl, XBP1ΔDC and XBP1ΔDC/IRE1truncDC MesLNs. Each dot represents an individual mouse. Error bars depict S.E.M. Mann-Whitney test, ** : p<0.01. Data is representative of 2 (A–F,H,J,K) independent experiments or 1 (G,I) experiment. (L) Graphical summary of main events downstream of XBP1 deficiency in lung or intestinal cDC1s. Green represents inhibitory events, red equals activation. Loss of XBP1 in both lung and intestinal cDC1s activates both IRE1 and PERK. This is only modest in the lung and more pronounced in the LP-SI, resulting into a block in cell death in the LP-SI in a RIDD dependent manner. At this stage it is unclear whether the PERK mediated adaptive downregulation in protein synthesis in the LP-SI also contributes to cell survival.
Although the XBP1Δ/IRE1truncDC still contains a truncated version of IRE1, containing the kinase domain53, IRE1 expression levels in the double deficient mice are markedly lower, as can be noticed in Fig 8C. Therefore, we assessed the protective role of RIDD by a complementary approach, using an established chemical inhibitor of IRE1-dependent endonuclease activity with in-vivo applicability54. As can be seen in Fig 9G, tunicamycin treatment of in-vitro cultured ERAI transgenic DCs induced a strong increase in VenusFP. Concomitant treatment with B-I09 at different doses (5 and 20 μM) inhibited the IRE1 dependent VenusFP generation (Fig 9G). In-vivo application of B-I09 reduced the steady state VenusFP levels in ERAI transgenic splenic DCs (Fig 9H) and blocked the splicing of Xbp1 mRNA (Fig 9I). However, as can be observed in Fig 9I, the half-life of BI-09 is limited in-vivo and XBP1s levels are restored within 24 hours. Therefore, we turned to an ex-vivo approach to evaluate the effects of B-I09 on DC survival. MesLNs, containing migratory cDC1s from the LP-SI, were collected from XBP1fl/fl, XBP1ΔDC or XBP1Δ/IRE1truncDC mice. Single cell suspensions were cultured in presence of B-I09 or vehicle control. After 24 hours of culture, RIDD activity and survival of the DC populations was assessed. As seen in FIG 9J, RIDD measured by means of CD11c reduction, was efficiently blocked by B-I09 in XBP1 deficient cDC1. The opposite effect, albeit at lower levels, was noticed for the cDC2 population. B-I09 specifically induced the loss of XBP1Δ cDC1s, but not XBP1fl/fl or XBP1Δ/IRE1trunc cDC1, confirming previous findings that compound inhibition of IRE1 dependent endonuclease activity is detrimental for cDC1s in the face of chronic ER stress due to XBP1 loss. There was no effect of BI-09 on the survival of cDC2s in the three different genotypes (Fig 9K). Overall, these data suggest that IRE1-dependent RIDD activation alleviates the deleterious effects of XBP1 deficiency in cDC1s and protects against cell death (Suppl Fig 7I). This effect is particularly pronounced in the cDC1s of the small intestine, reflecting their high intrinsic IRE1 activity (Fig 9L).
Discussion
In recent years, our knowledge on the transcriptional pathways governing DC development and maintenance has expanded profoundly. cDC1s – essential for preserving immune tolerance and mounting antiviral and antitumour responses15 critically depend on the transcription factors Batf3, Irf8 and Id2 for their development55. Besides, additional factors have been identified that do not affect lymphoid tissue resident cDC1s homeostasis, but seem to have profound roles in the maintenance of peripheral tissue cDC1s, such as Csf2rb25, MycL56 and PI3Kγ57. Here we add XBP1, a major transcription factor of the UPR to the growing list of regulators involved in the homeostasis of tissue-resident cDC1s. Loss of XBP1 does not have any effect on the survival of splenic cDC1s, but appears a crucial regulator of peripheral tissue-resident cDC1s. This effect was particularly prominent in the lung compartment and mediastinal LN and was far less pronounced in the lamina propria of the small intestine, reminiscent to what has been described before for MycL56. It remains to be determined if the requirement for these additional factors reflects further functional specializations of peripheral tissue derived cDC1s.
Originally regarded as a linear and sequential developmental process, single cell RNA sequencing revealed an unexpected early developmental predisposition of DC progenitors for cDC1 subsets. Novel gating strategies allow for a clear distinction between all pre-DC subsets with pre-cDC1s corresponding to the Lys6c−Sig1ecH− population31. As revealed by Venus fluorescence expression in the ERAI reporter mice, pre-cDC1s can additionally be distinguished from pre-cDC2s by marked increase in IRE1-dependent XBP1 splicing. Despite this elevated expression level, XBP1 was not needed for the development of pre-DCs to cDC1s. On the contrary, it was required at the level of fully matured DCs to maintain their cellular fitness, since loss of XBP1 in cDC1s induced apoptosis. At first glance, these findings are in conflict with earlier reports claiming a role for XBP1 in the development of the whole DC compartment20,30. Importantly, the transgenic strains that were used in those studies had a more widespread deletion of XBP1 in the hematopoietic system. We believe that detrimental effects of XBP1 deletion in early progenitor cells lie at the heart of the observed discrepancies, since our ERAI data indicate that XBP1 is also active early on in BM precursors at the CMP level. Remarkably, our study also indicates that XBP1 is not necessary for terminal differentiation and survival of pDCs although functional analysis of this subset is still warranted (data not shown).
Both in the lung and in the intestine, cDC1s mounted a strong ER stress response the degree of which did not differ sufficiently to explain the differences in cell fate upon loss of XBP1. Chronic ER stress has been unequivocally linked with the induction of apoptosis, although the main mechanisms driving final demise of the cell remain as yet unclear5. Many studies in ER stress-induced apoptosis have focused on the role of the pro-apoptotic executer CHOP, the main culprit downstream of the PERK branch. In contrast to the expectations, CHOP was clearly not involved in lung cDC1 cell death in conditions of XBP-1 deficiency.
Several publications have demonstrated that adaptation to chronic ER stress is tightly coupled with translational control, to avoid excessive protein loading of the ER39,48,58. We found that only intestinal XBP1 deficient cDC1s reduced their protein translation rate in a P-eIF2α-independent, but 4E-BP1-dependent manner, in line with earlier findings45. 4E-BP1, activated by ATF4 as part of the integrated stress response (ISR), was much more expressed in intestinal cDC1s compared with lung cDC1s. 4E-BP1 blocks cap-dependent translation by displacing eIF4E from eIF4G, and in this way interferes with eIF4F assembly59. It has been suggested that 4E-BP1 is particularly important for the prolonged translational suppression in later stages of the UPR, when eIF2α-mediated control is turned off by upregulation of the GADD34-negative feedback loop43,47. This fits with our observations in the XBP1 deficient cDC1s of the small intestine where low levels of P-eIF2α correlated with high levels of GADD34 and induction of 4E-BP1. In pancreatic β-cells, 4E-BP1 is part of an adaptive modus that protects from chronic ER stress-induced cell death48. However, in intestinal cDC1 cells, inhibition of the whole ATF4 response by the chemical compound ISRIB did not lead to cell death. This indicates that the adaptive responses and translational nodes controlled at the level of ATF4 are likely to be very complex and/or are not easy to manipulate in vivo with current tools.
The IRE1 branch of the UPR has also been reported to mediate apoptosis in chronic ER stressed cells both in a kinase and endonuclease dependent manner. XBP1-deficient Paneth cells die due to overactivation of JNK and NF-κB by IRE1 kinase activity, and lowering IRE1 levels protects them from cell death28. In our hands however, higher steady state levels of IRE1 in intestinal cDC1s appeared to correlate with a protective phenotype in cDC1s lacking XBP1, while lower steady state levels of IRE1 activity in the lung correlated with cDC1s loss. This was not linked to JNK or NF-κB signaling (data not shown), but rather to a differential capacity to engage RIDD9,10. Loss of RIDD on top of XBP1 deficiency caused intestinal cDC1 demise suggesting that a DC intrinsic factor - IRE1-dependent RIDD activity - rather than local tissue micro-environmental factors, determined the balance between life and death in XBP1 deficient cDC1s.
Up to now, the physiological role of RIDD remains highly speculative. Revealed mostly in conditions of chronic ER stress or in conditions of XBP1 loss, RIDD has been proposed to serve as a way to lower mRNA abundance and hence protein folding in the ER9. This implies a random unbiased degradation of ER-localized mRNAs, though this is not what has been observed so far in mammalian cells, where RIDD appears to be a highly selective process60. Additionally, it has been claimed that the hyperactive state of IRE1 induces a pro-apoptotic pathway in conditions of irremediable ER stress. This depends on the selective degradation of particular miRNAs that leads to an induction of caspase-2 and/or TXNIP to cause caspase-2 or caspase-1 mediated cell death11,12. While this might be the case for embryonic fibroblasts, insulinoma, or kidney cell lines, we did not detect elevated levels of Casp2 or Txnip mRNA in XBP1ΔcDC1s, illustrating the existence of cell type-specific RIDD targets (data not shown). More recently, a pro-survival role of IRE1-dependent RIDD activation has been reported, in which IRE1 degrades Tnfrsf10b mRNA, a proapoptotic target molecule that transcribed upon ER stress in a PERK and CHOP-dependent pathway61. Similarly, XBP1 deficient hepatocytes degrade Ppp1r15b mRNA encoding for a phosphatase implicated in eIF2a dephosphorylation and hence important in the regulation of protein translation via RIDD62. Irrespective of the mechanism, IRE1-dependent RIDD activation in DCs has a prosurvival role, underscoring the tissue-specific functions of RIDD.
In general, it appears that UPR signaling pathways have different outcomes in cDC1s compared to non-immune cells. Despite the presence of XBP1 levels at basal stage, a typical XBP1s gene signature could not be observed in DCs (19 and data not shown). Rather, IRE1/XBP1s might trigger as yet poorly understood cell type specific gene programs63–65 that in DCs could be linked to their antigen presentation capacity66, type I IFN production67,68 or overall survival (this study).
Finally, we believe that our observations are relevant not only for understanding DC survival but also for better understanding the cellular specialization of different tissue DCs. All XBP1 KO cDC1s - but not cDC2s−, both in lung, spleen and intestine, show a marked aggregated ER that seems to invade the whole cytosol (see also 3D-EM movies19). We hypothesize that this aberrant ER and massive accumulation of lipid membranes might be the result of a defect in lipid handling due to XBP1 absence. This might be particularly relevant for crosspresenting cDC1s, as apoptotic cell uptake continues as part of their normal cellular program. Defects in XBP1 indeed cause lipotoxic stress, as reflected by induction of markers like Trib3 in cDC1s. It is tempting to speculate that in the small intestine, where apoptotic cell turnover and concomitant lipotoxic stress is expected to be high, DCs have developed more elaborate adaptive strategies to cope better with these sources of cellular stress.
Material and methods
Mice and generation of BM chimeras
Xbp1fl/fl (B6.129s6/SvEvTac-XBP-1tm2Glm)69, XBP1ΔDC (B6.129s6/SvEvTac-XBP-1tm2Glm × B6.Tgitgax-cre1-1Reiz)70, CD45.1 (B6.SJL-Ptprca Pep3b/BoyJ)71, ERAI (B6.TGpCAX-F-XBP-1DBD-venus/J)22, XBP1ΔDC-Late (B6.129s6/SvEvTac-XBP-1tm2Glm × B6.TgItgax-cre,-EGFP4097Ach/J)33, XBP1ΔDC/Chopnull (B6.129s6/SvEvTac-XBP-1tm2Glm × B6.TgItgax-cre1-1Reiz × B6;129P2/OlaHsd-Ddit3tm1Mori)36, ERAI/Xbp-1ΔDC, Xbp1fl/fl × Ern1afl/fl (B6.129s6/SvEvTac-XBP-1tm2Glm × B6;129S4-Ern1tm2.1Tiw)52, XBP1Δ/IRE1truncDC (B6;129S4-Ern1tm2.1Tiw × B6.129s6/SvEvTac-XBP-1tm2Glm × B6.TgItgax-cre1-1Reiz) mice were bred at Ghent University in specific pathogen-free conditions.
To generate radiation chimeras in C57BL/6 CD45.1/CD45.2 hosts, the bone marrow of CD45.1 and XBP-1ΔDC was used. In short, 12 hours after lethal irradiation (8 Gray), CD45.1/2 mice were injected intravenously with 2 × 106 bone marrow cells of a 1:1 mix derived from CD45.1 and CD45.2 XBP-1ΔDC mice. Mice were used for experiments at least 8 weeks after reconstitution.
All mice, except the XBP-1ΔDC-Chopnil (F3 offspring of XBP1ΔDC and Chopnull were used for experiments), were backcrossed at least 5 times to C57BL/6 background. All animal experiments were performed in accordance with institutional guidelines for animal care of the VIB site Ghent-Ghent University Faculty of Sciences. Litters with mice of both sexes at 6–14 weeks of age were used for experiments.
Preparation of cell suspensions
In the case of lung and spleen, organs were digested at 37°C for 30–45 min in RPMI 1640 (61870036; Gibco) containing Liberase TM (0.02 mg/ml; Roche) and Dnase I recombinant (0.01 U/μl; Roche) after mincing. The obtained cell suspension was cleared of red cells by osmotic lysis. In the case of small intestine, epithelial cells were removed by serial washes at 37°C with Hank’s balanced salt solution w/o Ca2+ and Mg2+ (HBSS; Gibco) containing 10% fetal calf serum (FCS; BodJNKo) and 2 mM EDTA (51234; Lonza). The lamina propria is subsequently digested at 37°C for 15–20 min in RPMI 1640 supplemented with 10% FCS, Collagenase VIII (0.66–1 μg/ml; Sigma) and Dnase I recombinant (0.01 U/μl). Single cell suspensions of bone marrow are obtained by crushing of tibia and femur and subsequent lysis of red blood cells.
Flow cytometry and cell sorting
Monoclonal antibodies labeled with fluorochromes or biotin recognizing following surface markers were used: CD3 (145-2C11; Tonbo), CD19 (1D3; Tonbo), CD45R (RA3-6B2; BD-Pharmingen), TER119 (TER119; eBioscience), Ly6G (1A8; BD-Pharmingen), CD64 (X54-5/7.1; BD Pharmingen), CD11c (N418; eBioscience), MHCII (M5/114.15.2; eBioscience), CD135 (A2F10; eBioscience), CD26 (H194-112; Biolegend), CD172a (P84; eBioscience), XCR1 (ZET; Biolegend), CD103 (2E7; eBioscience), CD86 (PO3; Biolegend), CD40 (3/23; BD Pharmingen), MHCI H-2b/d/p/q/w16 (ER-HR52; Abd Serotec), CD45 (30-F11; eBioscience), CD45.1 (A20; eBioscience), CD45.2 (104; eBioscience), SiglecH (eBio440c; eBioscience), Ly6C (HK1.4; eBioscience), CD115 (AFS98; eBioscience), CD117 (2B8; eBioscience), CD11b (M1/70; BD Pharmingen), CD24 (M1/69; BD Pharmingen), mPDCA1 (120g8; in-house manufactured). Viable cells were discriminated by the use of Fixable Viability dye eFluor 506 or 786 (eBioscience). Acquisition and analysis of labeled cell suspensions was performed with a LSR Fortessa (BD biosciences) and subsequent analysis of data with FlowJo10 software (FlowJo, LLC).
Cell sorting was performed on a FACS ARIAII and III (BD biosciences). In the case of the spleen and bone marrow, the single cell suspension was enriched prior to cell sorting by depletion of CD3+, CD19+, Ly6G+, NK1.1+, TER119+ and CD45R+ cells using fluorescein isothiocyanate labelled monoclonal antibodies recognizing CD3, CD19, Ly6G, NK1.1, TER119 and CD45R and anti-fluorescein isothiocyanate microbeads (130-048-701; Miltenyi Biotec).
Intracellular staining for IRE1, P-eIF2α, 4E-BP1, p4E-BP1 and DAP5
Cell suspensions were, after extracellular staining with antibody cocktail for surface markers, fixed at room temperature during 30 min and subsequently permeabilized with the FoxP3 staining kit (00-5523-00; eBioscience). Cells were labelled 45 min on RT or ice with the respective antibodies raised against IRE1 (H190; Santa Cruz), P-eIF2α (D9G8, Cell Signaling), 4E-BP1 (53H11, Cell Signaling), p4E-BP1 (236B4, Cell Signaling) and DAP5 (D88B6, Cell Signaling). After washing, the cells are incubated 30 min on ice with an AlexaFluor-647 labeled goat anti-rabbit antibody (A2144; Invitrogen) or in the case of P-eIF2α with a BV786 fluorochrome conjugated to streptavidin (563858, BD Pharmingen).
Flt3L supplemented cell cultures
Cells were cultured in RPMI 1640 medium supplemented with GlutaMAX, 2-mercaptoethanol, gentamycin (all from Gibco), 10% FCS and 250 ng/ml recombinant Flt3L (produced in-house).
Reagents
zVAD-fmk (N-1510.0005; Bachem) was resuspended in DMSO and used at a final concentration of 250 μM. ISRIB (SML0843-5MG; Sigma) was dissolved in DMSO (1 mg/ml), mixed 1:1 in PEG400 (91893-250ML-F; Sigma) and injected IP twice daily during 3 days at 2.5 mg/kg. CC-930 (HY-15495; MedChemExpress) was dissolved in DMSO at a concentration of 50 mg/ml, a 12.5% solution was made in PEG400 and injected IP daily (50mg/kg) during 5 days. Tunicamycin (T7765; Sigma) was dissolved in DMSO to a 10 mg/ml stock concentration and used at a concentration of 1 μg/ml in-vitro. Cycloheximide was dissolved in DMSO (10 mg/ml) and used at a concentration of 5μg/ml in-vitro. B-I09 (donated by Andrew Hu) was dissolved at a concentration of 80mM in DMSO and used in-vitro at 20 μM, unless stated otherwise. In-vivo, B-I09 was injected IP at a final concentration of 0.625 mg/25g mouse.
Measurement of protein synthesis
O-Propargyl Puromycin (NU-931-5; Jena Bioscience) was dissolved in DMSO, further diluted in PBS (10 mg/ml) and injected IP (50 mg/kg mouse weight). 1 hour after injection mice were euthanized by cervical dislocation and the organs of interest were collected. 7 × 106 cells were stained with mixtures of antibodies directed against cell surface markers. Each staining lasted approximately 30′ and was performed on ice protected from direct light. Next, cells were fixed and permeabilized using the FoxP3 Fixation/Permeabilization kit (00-5521-00; eBioscience). For OP-Puro labeling, Azide-AF647 was chemically linked to OP-Puro through a copper-catalyzed azide-alkyne cycloaddition. In short, 2.5 μM azide-AF647 (A10277; Invitrogen) is dissolved in the Click-iT Cell Reaction Buffer (C10269; Invitrogen) containing 400 μM CuSO4. Immediately after preparation, cells are incubated with this mixture on room temperature. After 10′ incubation, the reaction is quenched by addition of PBS supplemented with 5% heat-inactivated fetal calf serum and 5 mM EDTA. Cells are washed twice to remove unbound azide-AF647 (Adapted from40).
Immunoblotting
Sorted dendritic cells (2 × 105) were spun at 400g for 7 min. After sorting, the supernatant was removed, the pellet resuspended in ice cold PBS and transferred to a 1.5ml reaction tube. After a next round of centrifugation (400g, 7 min), the pellet was pipetted dry and resuspended in 30 μl of E1A buffer (1% NP40, 20 mM HEPES, pH 7.9, 250 mM NaCl, 1 mM EDTA) complemented with Complete-ULTRA (05 892 970 001; Roche) and PhosSTOP (04 906 837 001; Roche). Lysates were snapfrozen in liquid nitrogen and stored at −80°C until further use. Prior to SDS-PAGE, samples were spun at 12000g to remove insoluble material and were resuspended in 10 μl of loading dye. After wet transfer to polyvinyldifluoride membrane (Immobilon; Millipore), proteins were analyzed by immunoblotting and visualized by chemiluminescence (NEL 104001EA; Perkin Elmer). Antibodies used recognize IRE1 (14C10; Cell Signaling) and HSP-90 (H114; Santa Cruz), phospho-JNK (9251s; Cell Signaling), JNK (9258s; Cell Signaling) and β-Tubulin (ab21058; Abcam).
RNA isolation, qRT-PCR and primers
Dendritic cells (2–3.5 × 104) were sorted directly into RLT Plus-buffer (1048449; Qiagen) and stored at −80°C until further processing. RNA was obtained using the RNEasy Plus Micro Kit (74034; QIAGEN) following manufacturer’ s instructions. Integrity and concentration of RNA was assessed using Agilent RNA Pico chips and Bioanalyzer (5067-1513; Agilent). Complementary DNA was amplified using the Ovation PicoSL WTA System V2 kit (3312-24; NuGEN) according to manufacturer’s guidelines. Pollutants were removed using the MinElute PCR purification kit (28204; Qiagen) prior to SybrGreen based quantitative Real-Time PCR (Lightcycler 480, Roche).
Primers for qRT-PCR used in this study are enlisted as follows: XBP-1s forward 5-CAGCAAGTGGTGGATTTGG-3, XBP-1s reverse 5-CGTGAGTTTTCTCCCGTAAAAG-3, Herpud forward 5-AGCAGCCGGACAACTCTAAT-3, Herpud reverse 5-CTTGGAAAGTCTGCTGGACA-3, Hspa5 forward 5-CCTGCGTCGGTGTGTTCAAG-3, Hspa5 reverse 5-AAGGGTCATTCCAAGTGCG-3, ATF4 forward 5-CTCTTGACCACGTTGGATGAC-3, ATF4 reverse 5-CAACTTCACTGCCTAGCTCTAAA-3, Ddit3 forward 5-AAGCCTGGTATGAGGATCTGC-3, Ddit3 reverse 5-TTCCTGGGGATGAGATATAGGTG-3, Ppplrl5a forward 5-GCCTGCAAGGGGCTGATAAG-3, Ppp1r15a reverse 5-TTTGTATCCCGGAGCTATGGA-3, Trib3 forward 5-GCAAAGCGGCTGATGTCTG-3, Trib3 reverse 5-AGAGTCGTGGAATGGGTATCTG-3, Bloc1s1 forward 5-CACCCAGCCAGACTCGAC-3, Bloc1s1 reverse 5-GCAGCGATAGCTTCTCTCCTC-3, Tapbp forward 5-ACCATTCCCAGGAACTCAAA-3, Tapbp reverse 5-GAGAAGAAGGCTGTTGTTCTGG-3, Erp44 forward 5-GACACAGCCCCAGGAGAG-3, Erp44 reverse 5-TCATCTCGATCCCTCAATAAAGTA-3, Batf3 forward 5-AGAAGGCTGACAAGCTCCAC-3, Batf3 reverse 5-CTGCGCAGCACAGAGTTC-3, Spi1 forward 5-GGGATCTGACCAACCTGGA-3, Spi1 reverse 5-AACCAAGTCATCCGATGGAG-3, Irf8 forward 5-GAGCCAGATCCTCCCTGACT-3, Irf8 reverse 5-GGCATATCCGGTCACCAGT-3, Irf4 forward 5-GGAGTTTCCAGACCCTCAGA-3, Irf4 reverse 5-CTGGCTAGCAGAGGTTCCAC-3, Id2 forward 5-ACTATCGTCAGCCTGCATCA-3, Id2 reverse 5-AGCTCAGAAGGGAATTCAGATG-3, Gapdh forward 5-ACAAAATGGTGAAGGTCGGTG-3, Gapdh reverse 5-TGGCAACAATCTCCACTTTGC-3, Ywhaz forward 5-CTCTTGGCAGCTAATGGGCTT-3, Ywhaz reverse 5-GGAGGTGGCTGAGGATGGA-3. Sesn2 forward 5-TGCTGCTGCATACGGATGG-3, Sesn2 reverse 5-GGAACCCACCAGGTAAGAACA-3, Shmt2 forward 5-TGGCAAGAGATACTACGGAGG-3, Shmt2 reverse 5-GCAGGTCCAACCCCATGAT-3, Mthfd2 forward 5-AGTGCGAAATGAAGCCGTTG-3, Mthfd2 reverse 5-GACTGGCGGGATTGTCACC-3, Gpt2 forward 5-AACCATTCACTGAGGTAATCCGA-3, Gpt2 reverse 5-GGGCTGTTTAGTAGGTTTGGGTA-3, Chac1 forward 5-ACTTATTGCGTTTGGCAGACT-3, Chac1 reverse 5-TTCCTACGGCGTCCACAATG-3, Asns forward 5-GCAGTGTCTGAGTGCGATGAA-3, Asns reverse 5-TCTTATCGGCTGCATTCCAAAC-3, Mars forward 5-TGTCTAGTGGTTCAAGGCAAGA-3, Mars reverse 5-ACAGTTTTGACGACTCAAGCTG-3. mRNA expression was analyzed using qbase+ software version 2.6 (Biogazelle).
Immunocytochemistry
Dendritic cells (6–8 × 104) were seeded on fibronectin-coated Ibidi 8 well chambers and allowed to adhere for 1 hour at 37°C. Cells were rinsed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton-X-100, blocked with 0.2% normal donkey serum and stained overnight with KDEL, an ER marker (ADI-SPA-827-D; Enzo Life Sciences). The next day, cells were thoroughly washed, stained with an AlexaFluor488 (AF488) labelled donkey anti-mouse secondary antibody (A21202; Invitrogen) and Hoechst and mounted for microscopy. Confocal images were captured with a Zeiss LSM780 confocal microscope (Zeiss, Zaventem, Belgium). Images were taken by using a 63× Pln Apo/1.4 oil objective. Hoechst was excited with a Ti:Sa Mai Tai Laser at 790 nm and detected with a spectral bandwidth of 414–524 nm in front of the photomultiplier tube (PMT). AF488 was excited using the 488 line of a Multi Argon laser and detected with a spectral bandwidth of 495–579 nm in front of the PMT. The pinhole was set at 1 Airy Unit and 2x frame averaging was applied to reduce noise.
Electron microscopy
Sorted lung and intestinal DCs were fixed in 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M NaCacodylate buffer, pH 7.2 and centrifuged at 1500 rpm. Low melting point-agarose was used to keep the cells concentrated for further processing. Cells were fixed for 4 hours at RT followed by fixation O/N at 4°C after replacing with fresh fixative. After washing in buffer, they were post-fixed in 1% OsO4 with 1.5% K3Fe(CN)6 in 0.1 M NaCacodylate buffer at room temperature for 1 hour. After washing, cells were subsequently dehydrated through a graded ethanol series, including a bulk staining with 1% uranyl acetate at the 50% ethanol step followed by embedding in Spurr’s resin.
Ultrathin sections of a gold interference color were cut using an ultra microtome (Leica EM UC6), followed by a post-staining in a Leica EM AC20 for 40 min in uranyl acetate at 20°C and for 10 min in lead stain at 20°C. Sections were collected on Formvar-coated copper slot grids. Grids were viewed with a JEM 1400plus transmission electron microscope (JEOL, Tokyo, Japan) operating at 60 kV.
Statistical analysis
Data sets were analyzed using the non-parametric test Mann-Whitney U test to compare 2 populations. In the case of more than 2 populations the Kruskal-Wallis test combined with the Dunn’s test to correct for multiple comparisons was applied. Statistical significance for pairwise comparisons was tested with the Wilcoxon matched pairs test. All tests were performed as two-sided. Results with a P value of 0.05 or less were considered significant. Mean values, standard error of the mean and statistics were calculated with Prism6 (GraphPad software). No criteria of inclusion or exclusion of data were used in this study. Experiments were performed without prior randomization of the animals and without blinding.
Supplementary Material
Highlights.
-
-
IRE1 endonuclease reporter activity identifies cDC1 committed preDCs
-
-
Loss of XBP1 affects cDC1 cell survival in a tissue specific manner
-
-
Adaptive stress responses are differentially hardwired in lung versus intestinal cDCs
-
-
Regulated IRE1 dependent mRNA decay is antiapoptotic in DCs
-
-
CHOP does not control ER stress mediated cell death of DCs
Acknowledgments
B.N.L. is a recipient of an ERC Consolidator grant. B.N.L and S.J. are holders of several FWO program grants. B.N.L and S.J. are recipient of a UGent MRP grant (Group-ID). F.O. was recipient of a Marie Curie and FEBS grant grant and is currently funded by a FONDECYT No 1161212 grant. We would like to thank Laurie Glimcher (Dana-Farber Cancer Institute, MA, US) and Boris Reizis (Columbia University, NY, US) for providing us with the Xbp1fl/fl and the Itgax-Cre mice respectively. The Zeiss LSM780 with FLIM module was acquired through a grant from Minister Ingrid Lieten to the VIB Bio Imaging Core.
Abbreviations
- ATF6
activating transcription factor 6
- BM
bone marrow
- DC
dendritic cell
- cDC
conventional DC
- cDC1
conventional DC type 1 (XCR-1+ DC)
- cDC2
conventional DC type 2 (Sirp1α+ DC)
- eIF2α
eukaryotic initiation factor 2 α
- ER
endoplasmic reticulum
- ERAI
ER stress-activated indicator
- Flt3L
FMS-related tyrosine kinase 3 ligand
- FP
fluorescent protein
- GM-CSF
granulocyte macrophage colony-stimulating factor
- IRES
internal ribosome entry site
- IRE1
inositol-requiring enzyme 1
- JNK
c-Jun N-terminal kinase
- KO
Knock-out
- LP-SI
lamina propria of small intestine
- MedLN
mediastinal lymph node
- MesLN
mesenteric lymph node
- MHC class I
major histocompatibility class I
- pDC
plasmacytoid DC
- PERK
protein kinase R-like ER kinase
- RIDD
regulated IRE1-dependent decay
- ROS
reactive oxygen species
- UPR
unfolded protein response
- XBP1s
spliced XBP1
- XBP1u
unspliced XBP1
Footnotes
Author contributions
S.J.T., F.O., B.N.L and S.J. designed the research; S.J.T., F.O., L.V., J.V. and N.V. did the experiments; S.J.T. and F.O. analyzed the results; G.V.I. helped with all the FACS experiments; E.P. helped with microscopy analysis; T.I., A.H. and J.D.V. provided critical reagents for the study; S.J.T., F.O., B.N.L. and S.J. wrote the manuscript.
References
- 1.Janssens S, Pulendran B, Lambrecht BN. Emerging functions of the unfolded protein response in immunity. Nat Immunol. 2014;15:910–919. doi: 10.1038/ni.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calfon M, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 4.Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17:829–838. doi: 10.1038/ncb3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marciniak SJ, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 10.Hollien J, et al. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lerner AG, et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Upton JP, et al. IRE1α Cleaves Select microRNAs during ER Stress to Derepress Translation of Proapoptotic Caspase-2. Science. 2012 doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh R, et al. Allosteric Inhibition of the IRE1a RNase Preserves Cell Viability and Function during Endoplasmic Reticulum Stress. Cell. 2014;158:534–548. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hildner K, et al. Batf3 Deficiency Reveals a Critical Role for CD8 + Dendritic Cells in Cytotoxic T Cell Immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plantinga M, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. 2013;38:322–335. doi: 10.1016/j.immuni.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 17.Persson EK, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity. 2013;38:958–969. doi: 10.1016/j.immuni.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Guilliams M, et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity. 2016;45:669–684. doi: 10.1016/j.immuni.2016.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osorio F, et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α(+) dendritic cells. Nat Immunol. 2014 doi: 10.1038/ni.2808. [DOI] [PubMed] [Google Scholar]
- 20.Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. 2007;204:2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cubillos-Ruiz JR, et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell. 2015:1–13. doi: 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwawaki T, Akai R, Kohno K, Miura M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med. 2004;10:98–102. doi: 10.1038/nm970. [DOI] [PubMed] [Google Scholar]
- 23.Bajana S, Turner S, Paul J, Ainsua-Enrich E, Kovats S. IRF4 and IRF8 Act in CD11c+ Cells To Regulate Terminal Differentiation of Lung Tissue Dendritic Cells. The Journal of Immunology. 2016;196:1666–1677. doi: 10.4049/jimmunol.1501870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waskow C, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. 2008;9:676–683. doi: 10.1038/ni.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greter M, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36:1031–1046. doi: 10.1016/j.immuni.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reimold AM, et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–157. [PMC free article] [PubMed] [Google Scholar]
- 27.Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adolph TE, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–276. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reimold AM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 30.Bettigole SE, et al. The transcription factor XBP1 is selectively required for eosinophil differentiation. Nat Immunol. 2015;16:829–837. doi: 10.1038/ni.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlitzer A, et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol. 2015;16:718–728. doi: 10.1038/ni.3200. [DOI] [PubMed] [Google Scholar]
- 32.Naik SH, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. 2007;8:1217–1226. doi: 10.1038/ni1522. [DOI] [PubMed] [Google Scholar]
- 33.Stranges PB, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott CL, et al. CCR2(+)CD103(−) intestinal dendritic cells develop from DC-committed precursors and induce interleukin-17 production by T cells. Mucosal Immunol. 2014 doi: 10.1038/mi.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vremec D, et al. Maintaining dendritic cell viability in culture. Mol Immunol. 2015;63:264–267. doi: 10.1016/j.molimm.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Oyadomari S, et al. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA. 2001;98:10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 38.Plantevin Krenitsky V, et al. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg Med Chem Lett. 2012;22:1433–1438. doi: 10.1016/j.bmcl.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 39.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 40.Signer RAJ, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. 2014;509:49–54. doi: 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 42.Kojima E, et al. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J. 2003;17:1573–1575. doi: 10.1096/fj.02-1184fje. [DOI] [PubMed] [Google Scholar]
- 43.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harding HP, et al. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proceedings of the National Academy of Sciences. 2009;106:1832–1837. doi: 10.1073/pnas.0809632106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clavarino G, et al. Induction of GADD34 is necessary for dsRNA-dependent interferon-β production and participates in the control of Chikungunya virus infection. PLoS Pathog. 2012;8:e1002708. doi: 10.1371/journal.ppat.1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 47.Preston AM, Hendershot LM. Examination of a second node of translational control in the unfolded protein response. J Cell Sci. 2013;126:4253–4261. doi: 10.1242/jcs.130336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamaguchi S, et al. ATF4-Mediated Induction of 4E-BP1 Contributes to Pancreatic β Cell Survival under Endoplasmic Reticulum Stress. Cell Metab. 2008;7:269–276. doi: 10.1016/j.cmet.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 49.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 50.Sidrauski C, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife. 2013;2:e00498. doi: 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends in Biochemical Sciences. 2014 doi: 10.1016/j.tibs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 52.Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proceedings of the National Academy of Sciences. 2009;106:16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benhamron S, et al. Regulated IRE-1 dependent decay participates in curtailing immunoglobulin secretion from plasma cells. Eur J Immunol. 2013 doi: 10.1002/eji.201343953. [DOI] [PubMed] [Google Scholar]
- 54.Tang CHA, et al. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest. 2014;124:2585–2598. doi: 10.1172/JCI73448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Satpathy AT, Murphy KM, Kc W. Transcription factor networks in dendritic cell development. Semin Immunol. 2011 doi: 10.1016/j.smim.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wumesh KC, et al. L-Myc expression by dendritic cells is required for optimal T-cell priming. Nature. 2014;507:243–247. doi: 10.1038/nature12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nobs SP, et al. PI3-Kinase-g Has a Distinct and Essential Role in Lung-Specific Dendritic Cell Development. Immunity. 2015:1–17. doi: 10.1016/j.immuni.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 58.Scheuner D, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 59.Hershey JWB, Sonenberg N, Mathews MB. Principles of translational control: an overview. Cold Spring Harbor Perspectives in Biology. 2012;4 doi: 10.1101/cshperspect.a011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moore K, Hollien J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol Biol Cell. 2015;26:2873–2884. doi: 10.1091/mbc.E15-02-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu M, et al. Cell death. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345:98–101. doi: 10.1126/science.1254312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.So JS, Cho S, Min SH, Kimball SR, Lee A-H. IRE1α-dependent decay of CReP/Ppp1r15b mRNA increases eIF2α phosphorylation and suppresses protein synthesis. Mol Cell Biol. 2015 doi: 10.1128/MCB.00215-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Acosta-Alvear D, et al. XBP1 Controls Diverse Cell Type- and Condition-Specific Transcriptional Regulatory Networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 64.Zhou Y, et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat Med. 2011;17:356–365. doi: 10.1038/nm.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen X, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature. 2014 doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang Y, et al. Genetic Vaccines To Potentiate the Effective CD103+ Dendritic Cell-Mediated Cross-Priming of Antitumor Immunity. The Journal of Immunology. 2015;194:5937–5947. doi: 10.4049/jimmunol.1500089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith JA, et al. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu F, et al. ER stress and its regulator X-box-binding protein-1 enhance polylC-induced innate immune response in dendritic cells. Eur J Immunol. 2011;41:1086–1097. doi: 10.1002/eji.201040831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–1496. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8-dendritic cells in the spleen. J Exp Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shen FW, et al. Cloning of Ly-5 cDNA. Proc Natl Acad Sci USA. 1985;82:7360–7363. doi: 10.1073/pnas.82.21.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.