
Figure 11. Cardio protective role of oleanolic acid (OA) against doxorubicin (DOX) induced cardiotoxicity.
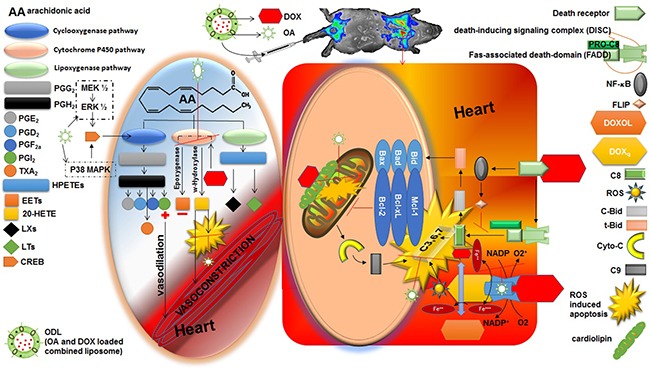
Right side square box illustrated the direct route of DOX induced myocardiotoxicity. The left side oval shape illustrated the indirect route of DOX induced myocardiotoxicity. Heart is the most susceptible organ to attack by DOX. In the presence of NADH, the quinone moiety of DOX molecule is transformed into a semiquinone moiety. The semiquinone moiety reacts with molecular oxygen to form a superoxide radical (O2-), and DOX molecule returns to its original quinone form. This redox cycling generates a huge amount of superoxide radical (O2-) and thus in turn produces “supernova” of ROS. DOX also interferes with iron used for normal metabolic reactions and produces ROS. Caspase activity can also be influenced by DOX via several routes. Moreover, DOX has high affinity for cardiolipin, a phospholipid abundant in myocardium located on mitochondria, and disturbs mitochondrium function. Aside by its direct role in cardiotoxicity, DOX also affect the heart by formation of P450-derived arachidonic acid metabolites via significantly increased CYP4A expression. DOX act on expoxygenase enzymes to reduce their activity. Hence, epoxyeicosatrienoic acids (EETs) to 20-Hydroxyeicosatetraenoic acid (20-HETE) equilibrium is disturbed that lead to increase level of 20-HETE in the myocardium. 20-HETE injured the cardiomyocytes via several routes 1) ROS induction; 2) activating nuclear factor-κB (NF- κB); 3) increased caspase-3 activity. Aside by this, 20-HETE causes vasoconstriction in blood vessels. OA has potential to reduce the stress full condition (ROS burden) by its radical scavenging properties. OA has antioxidant and inhibitory effect on NF- κB that is advantageous to balance toxic consequences of DOX. Moreover, increased formation of PGE2 and PGI2 (vasodilator prostanoids) by activation of cyclooxygenase (COX) balanced the vasoconstriction response of 20-HETE. The time and dose dependent PGI2 production is associated with the upregulation of COX by OA.