
Figure 1. Notch signaling is reactivated in the atrium with increased right-sided pressure and results in sinus bradycardia.
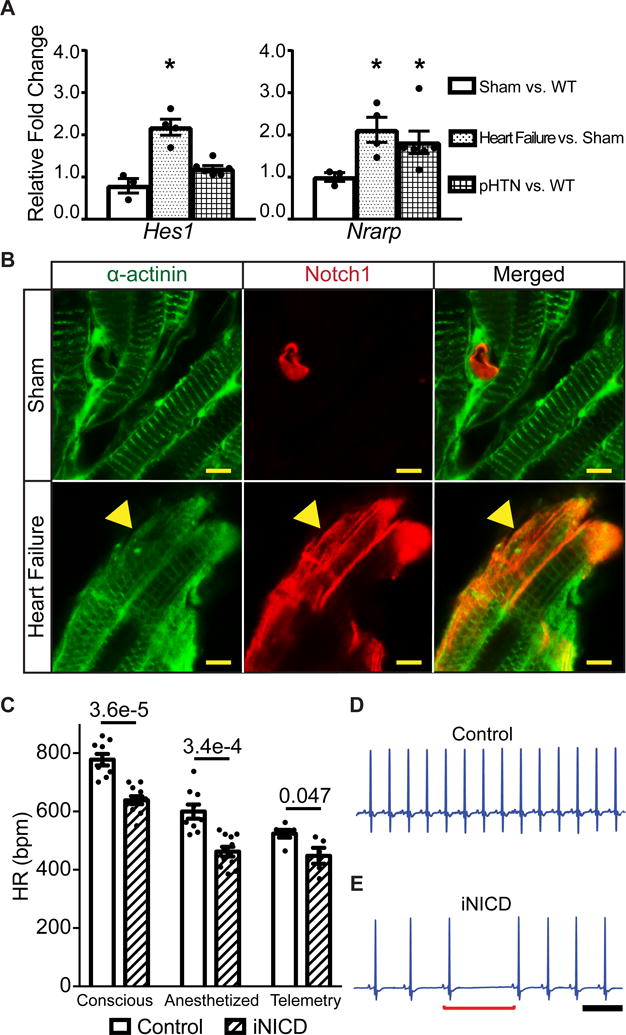
(A) We utilized a previously validated surgical approach that combines moderate transverse aortic constriction (TAC) and distal left anterior coronary ligation (MI) to produce a gradual and predictable progression of adverse left ventricular (LV) remodeling that leads to heart failure. Mice were sacrificed 4 weeks after surgery and RT-qPCR was performed on RA samples to assess expression of Notch target genes. Gene expression is represented as fold change in sham CD-1 RA versus non-instrumented CD-1 mice as a negative control, or TAC + MI mice versus shams (CD-1 control: n=2 females, n=1 male; sham: n=2 female, n=1 male; heart failure: n=3 females, n=1 male). In this heart failure model, the direct Notch targets Hes1 and Nrarp are significantly up-regulated. To test for Notch activation in a model of pulmonary hypertension, wild type CD-1 mice (n=3 females, n=3 males) were subjected to normoxic (21% O2) or hypoxic (10% O2) conditions for four weeks in an in vivo cabinet. Right atrial pressures are increased in chronic hypoxia when compared with normoxia (Supplemental Table II) and the direct Notch target Nrarp is significantly up-regulated in chronic hypoxic versus normoxic mice. All fold changes are relative to β-actin. Unpaired t test with Welch’s correction was performed between sham versus control, heart failure versus sham, and chronic hypoxia versus control normoxia for each gene and values of *P<0.05 were considered statistically significant. (B) Heart failure was induced via TAC + MI in NIP1::CreERT2; R26RtdTomato mice, where the intracellular domain of Notch1 was replaced with a complementary DNA encoding a 6× myc-tagged CreERT2 (6mtCreERT2). In this line, the intracellular domain of Notch1 was replaced with a complementary DNA encoding a 6× myc-tagged CreERT2 (6mtCreERT2). Binding of Notch ligands to the NIP1::CreERT2 triggers CreERT2 release from the membrane, but it only enters the nucleus in the presence of tamoxifen. While in the absence of tamoxifen no cells are labeled (data not shown), sham surgical animals treated with tamoxifen display active Notch1 signaling within PECAM-1+ (platelet and endothelial cell adhesion molecule-1) endothelial cells, as expected, and labeling of α-actinin+ atrial cardiomyocytes was not detected (Figure 1B, Supplemental Figure II). In contrast, induction of left ventricular remodeling together with administration of tamoxifen results in Notch1 activation (red) in many α-actinin+ atrial cardiomyocytes (green) (yellow arrowhead) as well as in PECAM-1+ endothelial cells (Supplemental Figure II). (C) Notch activation in adult mice significantly slows HR as assessed by conscious EKG, light anesthesia with isoflurane, and during conscious telemetric monitoring. Conscious and anesthetized EKGs were performed on mice that were administered doxycycline chow at 2 months of age to activate Notch for 3 weeks, followed by a 1–2 month washout period. Controls were either tetO_NICD (n=4 males) or αMHC-rtTA (n=1 female, n=4 males) mice fed doxycycline chow. Experimental iNICD mice were αMHC-rtTA; tetO_NICD (n=6 females, n=5 males). For telemetry studies, mice were between 3–5 months of age when doxycycline was administered. Controls were αMHC-rtTA (n=2 females, n=4 males) fed doxycycline, and experimental iNICD mice were αMHC-rtTA; tetO_NICD (n=2 females, n=3 males). The HR was slower in iNICD mice during both low and high activity periods (Supplemental Figure III). iNICD mice exhibit an increased frequency of pauses during telemetric monitoring, as demonstrated by HR variability in Poincaré plots (Supplemental Figure IV), and representative tracings from a control (D) and iNICD mouse (E) showing a sinus pause (underlined by red bracket) and slower HR in iNICD mice. Scale bar=500 ms. Statistics were performed using an unpaired t test with Welch’s correction. P values comparing control with iNICD mice are indicated for each condition, values of P<0.05 were considered statistically significant.