

Background
Serum amyloid A (SAA, 12 kDa) is a major acute-phase response plasma protein that has been evolutionarly conserved for 500 million years. SAA is a clinical marker of inflammation and a contributing factor to cardiovascular disease. SAA is best known as a causative agent of AA amyloidosis, a life-threatening complication of chronic inflammation and the major human systemic amyloidosis worldwide. In AA amyloidosis, N-terminal fragments of SAA (termed amyloid A, mainly residues 1–76) deposit as fibrils mainly in kidneys, causing renal damage. Recent failure of phase-III clinical trials of Kiacta, a small-molecule inhibitor of heparan sulfate binding to SAA, underscores the need for better understanding of the interactions between SAA with its ligands.
SAA modulates immune response, cholesterol homeostasis and cell signaling. Despite four decades of studies, normal functions of SAA remain elusive. In acute injury or infection, plasma levels of SAA increase up to 1000-fold reaching ~1 mg/ml. The advantage for survival of this dramatic increase is unclear; it is thought to result, in part, from the ability of SAA to mobilize cholesterol for cell repair. Most circulating SAA binds to plasma high-density lipoproteins (HDL, a. k. a. “good cholesterol”) and re-routs HDL cholesterol transport by interacting with cell receptors such as CD36 or LOX1. SAA also interacts with a wide array of other diverse ligands (cholesterol, retinol, heparan sulfate proteoglycans, metal ions, various proteins, etc.) Such binding promiscuity, which is a hallmark of this and other intrinsically disordered proteins, complicates functional studies of SAA often rendering them controversial. Our goal is to resolve this controversy and elucidate molecular underpinnings for normal functions of SAA and its pathologic misfolding in amyloid.
Methods
We used bioinformatics tools to perform amino acid sequence analysis of SAA family members [1] and combined the results with the atomic x-ray crystal structures of human SAA1.1 and murine SAA3 [2, 3]. This approach enabled us to identify a novel lipoprotein binding site in the SAA monomer whose shape explains, for the first time, the binding selectivity for HDL vis a vis larger lipoproteins. Analysis of the x-ray crystal structure of SAA suggests key determinants for the curvature of this binding site, and hence, for its binding selectivity. By combining high- and low-resolution structural studies of SAA in the crystalline form, on HDL and in solution, we proposed the conformation of SAA monomer bound to HDL surface. This conformation explains why HDL-bound SAA is protected from misfolding. Next, we re-evaluated the existing functional data on SAA to propose a novel function of SAA as an intrinsically disordered protein hub that mediates interactions among various proteins, lipids and proteoglycans on the dynamic HDL platform.
Results
Amino acid sequence analyses revealed that, out of four helices observed in the crystal structures of SAA, only two (h1 and h3) are amphipathic helices with a large hydrophobic moment and high membrane-binding affinity. We conclude that only h1 and h3 can bind HDL; this conclusion is strongly supported by previous experimental studies reporting cholesterol and HDL binding via h1 or retinol binding via h3. Notably, all predicted amyloidogenic segments in SAA are located in h1 and h3, suggesting that binding to HDL via h1 and h3 protects SAA from misfolding. Surface hydrophobicity distribution in the SAA monomer reveals a striking feature – an extended concave hydrophobic surface that has not been observed in any other HDL proteins and is apparently unique to SAA (Fig. 1). This exposed apolar surface explains why SAA is aggregated in solution and is largely unfolded at near-physiologic conditions in the absence of HDL.
Figure 1.
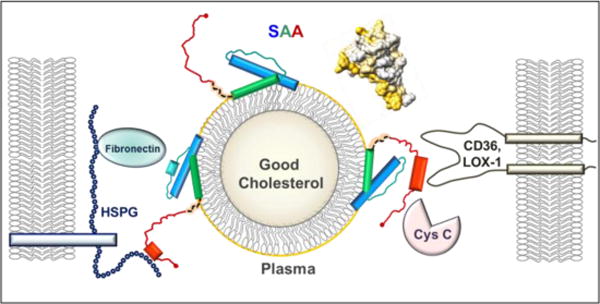
Structural model of SAA bound to an HDL particle. SAA monomer binds HDL via the concave apolar surface site (golden arc in the space-filling model, top right), which contains helices 1 and 3 (blue and green rectangles in the cartoon) from the N-domain (residues 1–69). Such a binding protects amyloidogenic segments (residues 2–9, 53–56 and 67–70) from initiating the misfolding. The dynamic C-domain, which is cleaved in AA amyloidosis, binds cell receptors (CD36, LOX1 etc.) and other ligands (HSPG, cystatin C, etc.) bridging them together on the dynamic HDL platform and mediating their interactions. When inflammation is resolved and SAA plasma levels drop, SAA is released from HDL and this dynamic network dissociates.
On the basis of these studies we propose the first structural model of the HDL-bound monomeric SAA wherein h1 and h3 from the N-domain (residue 1–69) form a concave hydrophobic surface that binds lipoproteins. The curvature of this surface is maintained through tight helical packing via the conserved GPGG motif (residues 48–51) that is unique to SAA, facilitating unusually close approach of h1 and h3 (3.6Å closest main chain separation). The GPGG motif, along with other conserved residues from h1 and h3 (e.g. A10, G13, A54, A55), locks the relative orientation of h1 and h3 as a socket wrench. This orientation determines the radius of curvature of the HDL binding site and confers the binding selectivity for HDL. Such a helical packing represents a novel structural motif that, to our knowledge, has not been reported for any other protein. A ~30-residue C-domain of SAA, connected to the N-domain via a flexible linker, binds various polar and charged ligands including cell receptors, bridging them together with HDL and thereby re-routing cholesterol transport for cell repair.
In our model, helices h1 and h3 are largely folded on HDL (blue and green rectangles in Figure 1) while the remainder of the SAA molecule is largely unfolded (teal and red), which accounts for the observed ~45% α-helical content of SAA on HDL. In comparison, SAA is 85% α-helical in the crystal structure and <20% α-helical in solution at 25°C in the absence of bound ligands. We propose that the C-domain (residues 70–104) can at least partially fold upon binding to various ligands, such as heparan sulfate or CD36 (red, Fig. 1). Our proposed model of SAA on HDL is supported by the SAA cleavage in the inter-domain linker that starts at residue 70 (orange, Fig. 1), to generate the 1–76 fragment that deposits in AA amyloidosis.
Discussion
In acute phase, the presence of multiple copies of SAA on one HDL particle suggests that different SAA molecules can bind different ligands and bridge them on the dynamic HDL platform (Fig. 1). This prompts us to propose that SAA acts as a molecular hub mediating dynamic interaction networks in immune response and inflammation. This action is consistent with the causative link between inflammation and atherosclerosis and may shed light on other functions of this enigmatic protein in health and disease. Future studies will experimentally test our model.
Footnotes
Declaration of interest
The authors report no conflict of interest.
References
- 1.Frame NM, Gursky O. Structure of serum amyloid A suggests a mechanism for lipoprotein binding and function as a hub in interaction networks. FEBS Letts. 2016;590:866–79. doi: 10.1002/1873-3468.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu J, Yu Y, Zhu I, Cheng Y, Sun PD. Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis. Proc Natl Acad Sci USA. 2014;111:5189–94. doi: 10.1073/pnas.1322357111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Derebe MG, Zlatkov CM, Gattu S, Ruhn KA, Vaishnava S, Diehl GE, MacMillan JB, et al. Serum amyloid A is a retinol binding protein that transports retinol during bacterial infection. eLife. 2014;3:e03206. doi: 10.7554/eLife.03206. [DOI] [PMC free article] [PubMed] [Google Scholar]