
Figure 1. Consanguineous family with multiple affected children presenting with a PME.
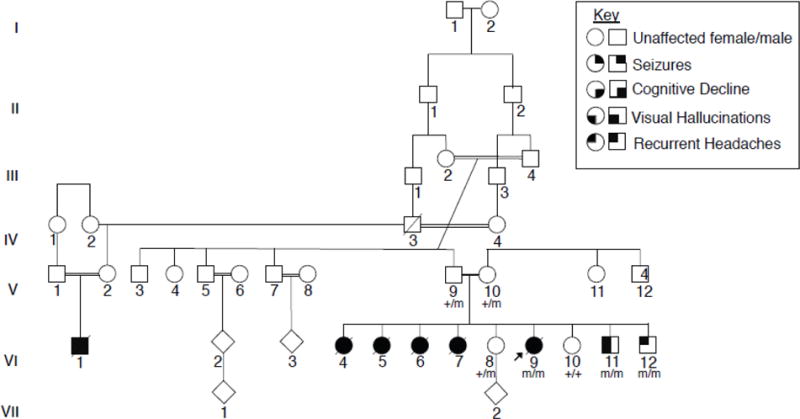
The proband (VI:9) was the sixth child born to a consanguineous couple of Iraqi descent, four older sisters, VI:4, VI:5, VI:6, and VI:7, were described as having a very similar clinical presentation. Affected children presented around the age of puberty with recurrent headaches, followed by visual hallucinations and seizures. Individuals eventually presented with cognitive decline and developed a progressive myoclonic epilepsy. Exome sequencing was completed for V:14, VI:8, VI:9., and VI:11. The proband was found to have a homozygous missense variant of unknown significance, GPR37L1: c.1047G>T (p.K349N). Sanger sequencing was completed for all available family members and confirmed that both parents are heterozygous for the variant, and the unaffected sisters are either heterozygous or wild type. The two younger brothers were found to be homozygous for the K349N variant. Each copy of the K349N mutant is labeled with an “m” in the pedigree shown here, as opposed to a “+” for the wild-type variant, in all individuals available for genetic testing.