

ABSTRACT
Claudin-1 is a hepatitis C virus (HCV) coreceptor required for viral entry. Although extensive studies have focused on claudin-1 as an anti-HCV target, little is known about how the level of claudin-1 at the cell surface is regulated by host vesicular transport. Here, we identified an interaction between claudin-1 and Sec24C, a cargo-sorting component of the coat protein complex II (COPII) vesicular transport system. By interacting with Sec24C through its C-terminal YV, claudin-1 is transported from the endoplasmic reticulum (ER) and is eventually targeted to the cell surface. Blocking COPII transport inhibits HCV entry by reducing the level of claudin-1 at the cell surface. These findings provide mechanistic insight into the role of COPII vesicular transport in HCV entry.
IMPORTANCE Tight junction protein claudin-1 is one of the cellular receptors for hepatitis C virus, which infects 185 million people globally. Its cellular distribution plays important role in HCV entry; however, it is unclear how the localization of claudin-1 to the cell surface is controlled by host transport pathways. In this paper, we not only identified Sec24C as a key host factor for HCV entry but also uncovered a novel mechanism by which the COPII machinery transports claudin-1 to the cell surface. This mechanism might be extended to other claudins that contain a C-terminal YV or V motif.
KEYWORDS: HCV, virus entry, claudin-1, COPII, Sec24C
INTRODUCTION
Hepatitis C virus (HCV) is a positive-strand RNA virus that belongs to the genus Hepacivirus within the Flaviviridae family. HCV causes chronic liver diseases, and it is estimated that 185 million people are infected globally (1). No approved vaccine for HCV is available due to the high variability of the virus. The development of novel direct-acting antivirals (DAAs) against HCV has greatly improved the efficacy of anti-HCV therapy, and the majority of patients receiving DAA treatment achieve a sustained virological response (SVR) (2). Current treatment strategies would greatly benefit from alternative strategies to control HCV, such as by targeting host factors involved in the life cycle of HCV (3, 4). This approach would not only raise a high barrier to viral resistance but also provide therapeutic options of suppressing HCV at multiple complementary steps.
The first step of the HCV life cycle is viral entry, which requires several host receptors and coreceptors, including CD81, SRB1 (scavenger receptor class B type I), occludin, and claudin-1 (CLDN1) (5). Claudin-1 interacts with CD81 to facilitate virus internalization during the HCV postbinding steps. It is a structural component of the hepatocyte tight junction and is highly expressed in liver tissue (6). By constituting the backbone of tight junction strands, claudins mediate cell adhesion and determine the permeability of epithelia. Proteins belonging to the CLDN family contain four transmembrane domains and two extracellular loops, with both the N and C termini located in the cytoplasm (7). The C-terminal PSD95–DlgA–ZO-1 homology (PDZ) binding motif of CLDNs binds to scaffolding proteins at cell junctions, such as ZO-1, ZO-2, and ZO-3. ZO-1 and ZO-2 further cross-link CLDNs to the actin cytoskeleton, and these junctional complexes are necessary to maintain the proper cellular permeability. The tight junction level of CLDNs at a given time is determined by elaborately regulated trafficking processes, including the transport of newly synthesized receptors from the endoplasmic reticulum (ER) to the tight junction, the internalization and recycling of the receptors between the tight junction and the endosomal compartment, and the transport of receptors to lysosomes for degradation. Over the past few decades, most studies of CLDN trafficking have focused on the events involved in the internalization, recycling, and degradation of CLDNs; these studies have greatly advanced our understanding of the intracellular trafficking of CLDNs (8–10). However, the molecular mechanisms by which CLDNs exit from the ER are largely unexplored.
Conservative estimates suggest that the coat protein complex II (COPII) machinery supports the ER export of a third of the translated proteins in eukaryotic cells (11). The exit of nascent protein from the ER is mediated by COPII-derived transport vesicles. The core components of COPII include five conserved proteins: Sar1, Sec23/Sec24, and Sec13/Sec31 (12). Sar1 initiates the coat assembly on the ER membrane. Sec23/Sec24 form the inner layer of the COPII coat, and Sec13/Sec31 form the outer layer that promotes the budding of the nascent COPII vesicle from the ER (13). Mammalian cells express four isoforms of Sec24, including Sec24A, Sec24B, Sec24C, and Sec24D (13), which are responsible for the recruitment of protein cargo molecules into nascent COPII vesicles.
Numerous studies have demonstrated that protein export from the ER is a selective process, and the recruitment of cargo molecules to COPII vesicles is mediated by the ER export motifs of the recruited cargo (12, 14). Several ER export motifs, such as diacidic and dihydrophobic motifs, have been identified and well characterized (15, 16). Diacidic motifs, such as DXE, have been found in the cytoplasmic C termini of several membrane proteins, including the vesicular stomatitis virus glycoprotein (VSV-G), cystic fibrosis transmembrane conductance regulator, ion channels, and the yeast (Saccharomyces cerevisiae) membrane proteins Sys1p and Gap1p. The dihydrophobic motifs are required for the efficient transport from the ER to Golgi intermediate compartment 53 (ERGIC-53), the p24 family of proteins, and the complex Erv41 to Erv46.
In the present study, we performed an unbiased interactome screen and identified key COPII components as claudin-1 binding partners. We identified the C-terminal YV motif of claudin-1 as the ER export motif required for the interaction between claudin-1 and Sec24C, a cargo-sorting component of the COPII transport machinery. Claudin-1 was confirmed to be a COPII cargo. Importantly, we demonstrated that the COPII transport machinery controls HCV entry by regulating claudin-1 localization at cell surface.
RESULTS
Identification of the interaction between claudin-1 and COPII components.
To elucidate the role of intracellular transport of claudin-1 during HCV entry, we performed proteomic interaction studies to identify novel host proteins in complex with the claudin-1 tail, which represented the C-terminal cytoplasmic tail of claudin-1 (Fig. 1A). Lysates derived from Huh7.5.1 cells were subjected to pulldown using a glutathione S-transferase (GST)–claudin-1 tail or GST, followed by liquid chromatography tandem mass spectrometry (LC-MS/MS). The resulting mass spectrometry data were processed using compPASS to identify high-confidence candidate interacting proteins (HCIPs). To identify the specific binding partners for the claudin-1 tail, we subtracted the total hits with GST pulldown proteins as background. As shown in Fig. 1A and Table 1, the claudin-1 tail was found to associate with its known partner, ZO-2 (17). Interestingly, COPII coat proteins Sec24C, Sec23A, and Sec23B were identified as HCIPs with sequence coverage of 12%, 16%, and 5%, respectively.
FIG 1.

Identification of the interactions between claudin-1 and COPII components. (A) A schematic drawing showing the primary sequence (aa 183 to 211) of claudin-1 fused to GST. Cell lysates from Huh7.5.1 cells were incubated with purified GST–claudin-1 tail or GST, and proteins were pulled down with glutathione-Sepharose beads. Bound proteins were detected by LC-MS/MS-based interactome analysis. Candidate claudin-1 tail-interacting proteins were the GST–claudin-1-tail interactome. (B) Validation of the interaction between the claudin-1 tail and COPII components. Cell lysates from Huh7.5.1 cells were pulled down by GST–claudin-1 tail or GST followed by Western blotting. (C) Cell lysates from Huh7.5.1 cells were pulled down by GST-CD81-N-tail, GST-CD81-C-tail, GST-occludin-tail, GST-SRB1-tail, GST-claudin-1 tail or GST with two extra washes. Bound proteins were subjected to Western blotting for analysis. (D and E) Immunoblots showing the association of Sec24C with claudin-1 in transfected HEK293T cells by a coimmunoprecipitation assay. HEK293T cells were transfected with Flag–claudin-1, Flag-SRB1, or PCMV-Tag2 empty vector for 48 h. Cell lysates were incubated with anti-Flag antibody-coated beads, and co-IP proteins were subjected to Western blotting for analysis. (F) Coimmunoprecipitation of endogenous claudin-1 with Sec24C in Huh7.5.1 cells. The lysates were immunoprecipitated with anti-claudin-1 antibody followed by Western blotting. (G) Colocalization of claudin-1 with Sec24C in Huh7.5.1 cells. Huh7.5.1 cells were fixed by PFA and immunofluorescently labeled for claudin-1 (green) and Sec24C (red). DAPI staining indicates the nucleus (blue). Scale bar, 15 μm.
TABLE 1.
Candidate proteins associated with claudin-1 tail from a GST pulldown screen in Huh7.5.1 cells
| GenBank accession no. | Protein name | Description | Confidence scorea | No. of unique peptides |
|---|---|---|---|---|
| Q9UDY2 | ZO-2 | Tight junction protein ZO-2 | 619 | 32 |
| P12956 | XRCC6 | X-ray repair cross-complementing protein 6 | 476 | 16 |
| Q15436 | Sec23A | Protein transport protein Sec23A | 279 | 9 |
| Q13509 | TUBB3 | Tubulin beta-3 chain | 259 | 7 |
| Q08211 | DHX9 | ATP-dependent RNA helicase A | 238 | 11 |
| Q15084 | PDIA6 | Protein disulfide-isomerase A6 | 209 | 6 |
| P53992 | Sec24C | Protein transport protein Sec24C | 185 | 10 |
| P19338 | NCL | Nucleolin | 166 | 3 |
| Q71U36 | TUBA1A | Tubulin alpha-1A chain | 155 | 8 |
| P54652 | HSPA2 | Heat shock-related 70-kDa protein 2 | 150 | 4 |
| P52272 | HNRNPM | Heterogeneous nuclear ribonucleoprotein M | 110 | 6 |
| P51659 | HSD17B4 | Peroxisomal multifunctional enzyme type 2 | 108 | 5 |
| Q8N448 | LNX2 | Ligand of Numb protein X 2 | 101 | 6 |
| P09874 | PARP1 | Poly(ADP-ribose) polymerase 1 | 99 | 4 |
| Q7Z3Y8 | KRT27 | Keratin, type I cytoskeletal 27 | 99 | 3 |
| Q9H4B7 | TUBB1 | Tubulin beta-1 chain | 94 | 3 |
| P31040 | SDHA | Succinate dehydrogenase | 84 | 2 |
| P28482 | MAPK1 | Mitogen-activated protein kinase 1 | 79 | 2 |
| P14618 | PKM | Pyruvate kinase PKM | 58 | 3 |
| A5A3E0 | POTEF | POTE ankyrin domain family member F | 53 | 2 |
| Q8WZ42 | TTN | Titin | 50 | 15 |
| Q92841 | DDX17 | Probable ATP-dependent RNA helicase DDX17 | 44 | 3 |
| P0C0S8 | HIST1H2AG | Histone H2A type 1 | 42 | 2 |
| Q15437 | Sec23B | Protein transport protein Sec23B | 39 | 3 |
| P01857 | IGHG1 | Ig gamma-1 chain C region | 36 | 2 |
| Q9BYE9 | CDHR2 | Cadherin-related family member 2 | 36 | 3 |
| P13639 | EEF2 | Elongation factor 2 | 33 | 3 |
| Q5NUL3 | FFAR4 | Free fatty acid receptor 4 | 32 | 2 |
| Q14146 | URB2 | Unhealthy ribosome biogenesis protein 2 homolog | 32 | 2 |
| P12271 | RLBP1 | Retinaldehyde-binding protein 1 | 28 | 1 |
| P53804 | TTC3 | E3 ubiquitin-protein ligase TTC3 | 27 | 2 |
| P01861 | IGHG4 | Ig gamma-4 chain C region | 26 | 2 |
| Q05397 | PTK2 | Focal adhesion kinase 1 | 25 | 2 |
| Q00839 | HNRNPU | Heterogeneous nuclear ribonucleoprotein U | 25 | 1 |
| Q4G0N4 | NADK2 | NAD kinase 2, mitochondrial | 24 | 1 |
| Q86YC3 | NRROS | Negative regulator of reactive oxygen species | 24 | 1 |
| P23246 | SFPQ | Splicing factor, proline and glutamine rich | 24 | 2 |
| P49792 | RANBP2 | E3 SUMO-protein ligase RanBP2 | 24 | 2 |
| Q9Y3T9 | NOC2L | Nucleolar complex protein 2 homolog | 23 | 1 |
| Q6P1X5 | TAF2 | Transcription initiation factor TFIID subunit 2 | 22 | 2 |
Individual protein scores are indicated in the form of a Mascot-derived confidence score [−10 log(PEP)]. The default significance threshold is a P value of <0.05.
To validate the interaction between the claudin-1 tail and Sec24C, Sec23A, and Sec23B, we performed GST pulldown experiments in Huh7.5.1 cells using the recombinant GST–claudin-1 tail and confirmed each interaction by performing a Western blot analysis (Fig. 1B). The interaction between claudin-1 tail and Sec24C was highly specific since it did not associate with other Sec24 isoforms, such as Sec24A, Sec24B, and Sec24D (Fig. 1C). Next, to further confirm the interaction between full-length claudin-1 and Sec24C, Flag–claudin-1 or the empty Flag-Tag2 vector was transfected into 293T cells, and the cell lysates were immunoprecipitated with an antibody against Flag. As shown in Fig. 1D, claudin-1 associated with Sec24C. To prove the specificity of the interaction between Sec24C and claudin-1, Flag-SRB1 was used as a control. We found that Sec24C interacted with claudin-1 but not SRB1 (Fig. 1E). To determine whether the claudin-1–Sec24C interaction occurs in Huh7.5.1 cells, we performed endogenous immunoprecipitation (IP) using a claudin-1 antibody, which confirmed the interaction (Fig. 1F). Taken together, these findings indicated that the claudin-1 tail associated specifically with the COPII component Sec24C.
To assess the physical interaction of claudin-1 with COPII components in vivo, Huh7.5.1 cells were stained with antibodies against claudin-1 and Sec24C. Consistent with the interaction results described above, claudin-1 partially colocalized with Sec24C (Fig. 1G).
Sec24C is required to maintain the claudin-1 level at the cell surface.
Since claudin-1 is a transmembrane protein that associates with Sec24C, we wondered whether claudin-1 is a COPII cargo molecule. To investigate whether claudin-1 is sorted into COPII-coated vesicles, we performed a well-established in vitro COPII vesicle reconstitution assay (Fig. 2A) (14). To reconstitute the COPII vesicles, semi-intact cells (SICs) were incubated with rat liver cytosol, GTP, and an ATP regeneration system. Vesicle fractions were obtained by differential centrifugation and probed for the presence of various proteins by Western blotting. The ER-resident protein ribophorin 1, which is not sorted into COPII vesicles, was used as a control to assess the specificity of the vesicle reconstitution assay. The cycling cargo adaptor protein ERGIC-53 served as a reference for the quantity of vesicles generated. As shown in Fig. 2A, claudin-1 was detected in membrane vesicles budding from the ER, whereas another HCV receptor, SRB1, was not. These results demonstrated that claudin-1 was packed into COPII vesicles.
FIG 2.

Knocking down Sec24C reduced the cell surface level of claudin-1. (A) Schematic representation of the in vitro budding assay. Claudin-1 was transported from the ER in an in vitro budding assay. Huh7.5 cells were treated with digitonin to prepare the semi-intact cells (SICs). The vesicle formation reaction was performed in the presence or absence of an ATP regenerating system. Adapted from Molecular Biology of the Cell (44) with permission of the publisher. (B) Huh7.5 cells treated with Sec24C siRNA or a control siRNA for 72 h were stained with antibodies against claudin-1 (green) and the tight junction marker ZO-1 (red). Nuclei were stained with DAPI (blue). Scale bar, 15 μm. Sec24C knockdown was confirmed by quantitative PCR and immunoblotting. ***, P < 0.001. (C) Cell surface claudin-1 was quantified by FACS assay in nonpermeabilized cells. Huh7.5 cells treated with Sec24C siRNA or a control siRNA for 72 h were harvested and stained with an anti-claudin-1 antibody (MAB4618; R&D Systems) which specifically recognizes surface claudin-1 without permeabilization. A total of 104 cells were analyzed, and isotype control antibody was used to delineate the gate. FITC-A−, fluorescein isothiocyanate negative area. FITC-A+, fluorescein isothiocyanate positive area. (D) Intercellular claudin-1 was quantified by FACS assay in permeabilized cells. Huh7.5 cells were treated with Sec24C siRNA or a control siRNA for 72 h. Then cells were permeabilized by saponin and stained with an anti-claudin-1 antibody (MAB4618; R&D Systems). A total of 104 cells were analyzed, and isotype control antibody was used to delineate the gate.
Next, we wondered how silencing Sec24C affected the claudin-1 level at the cell surface. To address this question, we silenced Sec24C by a small interfering RNA (siRNA) and examined the localization of claudin-1 and ZO-1. Exposure to an siRNA duplex against Sec24C reduced the claudin-1 level at the cell surface but not the total protein levels of claudin-1 (Fig. 2B). Consistent with the fact that claudin-1 recruits ZO-1 to the cell surface (17), the localization of ZO-1 at the cell surface was also reduced by an Sec24C siRNA (Fig. 2B).
To clearly distinguish intracellular localization of claudin-1 from cell surface staining of claudin-1, we use a fluorescence-activated cell sorting (FACS) technique to analyze the expression of claudin-1 in permeabilized and nonpermeabilized cells. Sec24C siRNA reduced the surface level of claudin-1 (nonpermeabilization condition) and increased the intracellular level of claudin-1 (permeabilization condition) (Fig. 2C and D).
To confirm that COPII activity is required for the cell surface localization of claudin-1, we employed xanthohumol (XN), a COPII inhibitor that blocks COPII coat assembly by inhibiting Sec24C (18). We applied two fixation methods in our immunofluorescence experiments. The methanol fixation method provided clear cell surface staining of claudin-1, whereas paraformaldehyde (PFA) fixation stained more intracellular claudin-1. We found that XN decreased the amount of claudin-1 at the cell surface and increased the intracellular level of claudin-1 in a concentration-dependent manner (Fig. 3A and B). Next, we use FACS to analyze the expression of claudin-1 in the presence of XN in permeabilized and nonpermeabilized cells. XN reduced the surface level of claudin-1 (nonpermeabilization condition) and increased the intracellular level of claudin-1 (permeabilization condition) (Fig. 3C and D). Taking these results together, we concluded that the COPII inhibitor (XN) reduced the cell surface level of claudin-1.
FIG 3.

Blocking COPII transport by XN decreased the cell surface level of claudin-1. (A) Huh7.5 cells treated with different concentrations of XN for 12 h were fixed with methanol and stained with antibodies against claudin-1 (red) and the tight junction marker ZO-1 (green). Nuclei were stained with DAPI (blue). Scale bar, 15 μm. (B) Huh7.5 cells treated with different doses of XN for 12 h were fixed with PFA and stained with antibodies against claudin-1 (red) and the tight junction marker ZO-1 (green). Nuclei were stained with DAPI (blue). Scale bar, 15 μm. (C) Cells surface claudin-1 was quantified by FACS assay in nonpermeabilized cells. Huh7.5 cells treated with 1.562 μg/ml XN or dimethyl sulfoxide (mock) for 12 h were harvested and stained with an anti-claudin-1 antibody (MAB4618; R&D Systems) without permeabilization. A total of 104 cells were analyzed, and isotype control antibody was used to delineate the gate. (D) Intercellular claudin-1 was quantified by FACS assay in permeabilized cells. Huh7.5 cells were treated with 1.562 μg/ml XN or dimethyl sulfoxide for 12 h. Then cells were permeabilized by saponin and stained with an anti-claudin-1 antibody (MAB4618; R&D Systems). A total of 104 cells were analyzed and isotype control antibody was used to delineate the gate.
Sec24C is critical for HCV entry.
Having established a key role for Sec24C in regulating the level of claudin-1 at the cell surface, we hypothesized that Sec24C is important for HCV entry. To determine if endogenous Sec24C is required for HCV infection, we silenced Sec24C by siRNA and examined its effect on HCV entry. Knocking down Sec24C by siRNA inhibited the entry of HCV pseudoparticles (HCVpp) but not VSV pseudoparticles (VSVpp) without detectable cytotoxicity, as determined by the cellular ATP content (Fig. 4A and B). Sec24C depletion also inhibited HCV entry without detectable cytotoxicity in the Jc1 infectious HCV model (Fig. 4C).
FIG 4.

Blocking COPII transport reduced HCV entry. (A and B) Huh7.5.1 cells treated with an siRNA targeting Sec24C or claudin-1 5′ UTR or with a control siRNA for 72 h were infected with genotype 1b HCVpp (Con1) or VSVpp for another 72 h, followed by measurement of the luciferase activity and cellular ATP levels. The relative luciferase activity was normalized to the cellular ATP level. (C) Huh7.5.1 cells treated with Sec24C siRNA or a control siRNA for 72 h were infected with Jc1 for another 24 h, followed by measurement of the luciferase activity and cellular ATP levels. The relative luciferase activity was normalized to the cellular ATP level. (D and E) Huh7.5.1 cells treated with different doses of XN for 12 h were infected with genotype 1b HCVpp (Con1) or VSVpp for 3 days, followed by measurement of the luciferase activity. For all panels, data are presented as the means ± SD. **, P < 0.01; ***, P < 0.001.
To confirm the role of Sec24C in HCV entry, we assessed whether the Sec24C inhibitor XN also inhibited HCVpp entry. As shown in Fig. 4D and E, HCVpp entry was inhibited by XN in a dose-dependent fashion without detectable cytotoxicity, demonstrating that Sec24C is required for HCV entry. Treatment with XN slightly decreased VSVpp entry (Fig. 4E). We reasoned that the modest inhibition of VSVpp entry by XN was independent of Sec24C and claudin-1.
Characterization of the interaction between claudin-1 and Sec24C.
An inspection of the protein sequences of the cytoplasmic tails of claudin-1 from different species revealed that the C-terminal cytoplasmic domain (C183 to V211) of claudin-1 is highly conserved (Fig. 5A). To determine which residues in the cytoplasmic tail of claudin-1 are important for the association of claudin-1 with Sec24C, we first divided the tail of human claudin-1 into two parts: amino acids (aa) 183 to 197 and aa 198 to 211 (claudin-1 tail183–197 and claudin-1 tail198–211, respectively). As shown in Fig. 5B, GST–claudin-1 tail198–211, but not GST–claudin-1 tail183–197, bound Sec24C. Next, we generated a series of GST-conjugated mutants of the claudin-1 tail by alanine scanning and tested their ability to bind Sec24C. We also tested the binding affinities of claudin-1 tail mutants with ZO-2, a known claudin-1 binding partner, which was also detected by mass spectrometry, as shown in Fig. 1A. Previous study identified that 210Y and 211V in claudin-1 are critical for association with ZO-2 (17). We found that 209D, 210Y, and 211V are critical for the association of claudin-1 with ZO-2, while 207G, 208K, 210Y, and 211V were important for the interaction between claudin-1 and Sec24C (Fig. 5C to G). The most critical residues in claudin-1 for claudin-1–COPII interaction are the very C-terminal two residues of the claudin-1 tail (Fig. 5H).
FIG 5.

Characterization of the interaction between claudin-1 and Sec24C. (A) Multiple alignments of the sequences of the claudin-1 tails (aa 183 to 211) from different species. Asterisks, identical; one or two dots, similarity degree. (B to F) Mapping the regions of the claudin-1 tail associated with Sec24C. Lysates from Huh7.5.1 cells were pulled down by GST fusion proteins as indicated or by GST. The bound proteins were analyzed by Western blotting. (G) Summary of the expression levels of GST fusion proteins and the interaction affinities between Sec24C and GST fusion proteins. (H) The results of a mutation analysis of the regions in the claudin-1 tail required for the claudin-1–Sec24C interaction, as determined by a GST pulldown assay. Huh7.5.1 cells were subjected to pulldown of the wild-type or mutant GST-claudin-1 tail. G. gallus, Gallus gallus; O. hoazin, Opisthocomus hoazin; M. musculus, Mus musculus; S. scrofa, Sus scrofa; H. sapiens, Homo sapiens; B. taurus, Bos taurus; F. damarensis, Fukomys damarensis; L. crocea, Larimichthys crocea.
To further establish that the claudin-1 YV motif is responsible for the interaction between claudin-1 and Sec24C, coimmunoprecipitation experiments were performed on HEK293T cells transfected with a Flag–claudin-1 construct or Flag–claudin-1 with the YV/AA substitutions [Flag–claudin-1(YV/AA)]. A clear band corresponding to Sec24C was detected in the cells transfected with Flag–claudin-1 but not in Flag–claudin-1(YV/AA) transfected cells. These results indicated that the YV motif in claudin-1 is required for binding Sec24C (Fig. 6A). To test whether the YV motif is sufficient for the association between claudin-1 and Sec24C, we constructed GST-YV and GST-GKDYV. As shown in Fig. 6B, neither GST-YV nor GST-GKDYV interacted with Sec24C, suggesting that the entire length of the cytoplasmic tail is important for the association of claudin-1 with Sec24C.
FIG 6.

The carboxy-terminal YV motif in claudin-1 is required for the interaction between claudin-1 and Sec24C. (A) Immunoblots showing the coimmunoprecipitation of Sec24C with full-length wild-type (WT) claudin-1 or claudin-1 mutants in transfected HEK293T cells. Lysates from HEK293T cells expressing Flag–claudin-1 (WT), Flag–claudin-1 (Y/A), Flag–claudin-1 (V/A), Flag–claudin-1 (YV/AA), or PCMV-Tag2 empty vector were incubated with anti-Flag antibody-coated beads, and co-IP proteins were subjected to Western blotting for analysis. (B) Huh7.5.1 cells were subjected to pulldown of the GST-claudin-1 tail or truncation mutants. GST-GKDYV or GST-YV was directly added with KGDYV or YV amino acids at GST C terminus. (C) Huh7.5.1 cell lysates were incubated with different peptides for 30 min, followed by GST–claudin-1 tail pulldown assays. The bound proteins were subjected to Western blotting for analysis. (D) HEK293T cells were transfected with wild-type Sec24C or an Sec24C cargo-deficient mutant (895LIL897/895AAA897) and subjected to GST-claudin-1 tail pulldown.
Next, we performed a peptide competition experiment. Peptide 1 mimicking the claudin-1 tail, but not peptide 2 with C-terminal YV/AA mutation, blocked the interaction of Sec24C with the GST–claudin-1 tail, which further demonstrated the requirement of YV for this interaction (Fig. 6C).
Previous studies have suggested that 895LIL897 are the key residues in Sec24C required for binding IXM, the known ER export signal motif (19). To investigate whether these residues are critical for claudin-1 binding, we generated the Sec24C-AAA mutant in which the residues 895LIL897 were mutated to 895AAA897. As shown in Fig. 6D, Sec24C-AAA no longer bound to the GST–claudin-1 tail.
We wondered whether the defects of HCV entry by Sec24C siRNA can be restored by expression of an siRNA-resistant Sec24C cDNA. First, we tested the expression of siRNA-resistant wild-type (WT) Sec24C and the Sec24C-AAA mutant, which could not interact with claudin-1 in the Sec24C silencing cells. Both the siRNA-resistant wild-type Sec24C and Sec24C-AAA mutant cDNA were expressed efficiently (Fig. 7A). These cells were then infected with HCVpp or Jc1. As shown in Fig. 7B and C, the inhibitory effect of HCV entry, which knocked down endogenous Sec24C, could be rescued by expression of wild-type Sec24C. However, the cargo binding-deficient mutant Sec24C-AAA could not rescue HCV entry. These results indicated that the effect of Sec24C on HCV entry was through its cargo binding function.
FIG 7.

HCV entry could be rescued by expression of wild-type Sec24C but not the cargo binding-deficient mutant. (A) Protein expression levels of wild-type or mutant (895LIL897 to 895AAA897) siRNA-resistant Sec24C were measured by Western blotting. Huh7.5.1 cells were treated with siRNA targeting the Sec24C or a control siRNA for 24 h and transfected with a construct expressing wild-type or mutant siRNA-resistant Sec24C for 48 h. (B and C) Then the cells were infected with genotype 1b HCVpp (Con1) for another 72 h (B) or infected with Jc1 for another 24 h (C). The infection activity and ATP levels were measured. The relative luciferase activity was normalized to the cellular ATP levels. Data are presented as the means ± SD. *, P < 0.1; ***, P < 0.001.
We also wanted to investigate whether a similar motif is present in other members of the CLDN family. Sequence analysis of the C-terminal cytoplasmic tails from 27 members of CLDN family proteins revealed that there is a conserved YV motif within the carboxy terminus of several claudins (Fig. 8A). This aromatic/hydrophobic motif was previously suggested to be an ER export signal consensus sequence recognized by Sec24C (20, 21). We fused GST to a series of CLDN tails and performed GST pulldown experiments. As shown in Fig. 8B, Sec24C interacted with claudin-1, claudin-4, claudin-5, claudin-11, claudin-16, and claudin-19, all of which contained YV or V in the C terminus. However, Sec24C did not bind to claudin-22 or claudin-23, which lacked the C-terminal YV or V. Claudin-6 and claudin-9 are reported to be the cellular receptors for some HCV strains (22, 23). Claudin-6 and claudin-9 also have a predicted Sec24C interaction site (Fig. 8C). We tested these interactions by GST pulldown. Claudin-6 and claudin-9 could interact with Sec24C (Fig. 8C). YV/AA mutants of claudin-6 and -9 significantly lost the ability to interact with Sec24C (Fig. 8C). From these results, we concluded that CLDNs with a C-terminal YV or V motif can associate with the COPII component Sec24C.
FIG 8.

Claudins with V or YV in the C terminus interact with Sec24C. (A) Sequence alignments of the C termini of human claudins. (B) The results of a GST pulldown assay performed to test the interaction between Sec24C and claudin family members. The C-terminal tails were predicted by the TMHMM server, version 2.0. (C) Claudin-6 or -9 interacted with Sec24C. Huh7.5.1 cells were subjected to pulldown of the GST–claudin-1 tail, GST–claudin-6 tail, GST–claudin-9 tail, GST–claudin-1 tail(YV/AA), GST–claudin-6 tail(YV/AA), or GST–claudin-9 tail(YV/AA).
The YV motif of claudin-1 is critical for determining the claudin-1 level at the cell surface and for HCV entry.
Since the YV motif in claudin-1 is critical for its association with Sec24C, we wondered whether this motif is also critical for the cell surface level of claudin-1. Indeed, as shown in Fig. 9A, the level of claudin-1(YV/AA) at the cell surface was reduced compared with that of claudin-1(WT). To investigate the role of the C-terminal YV motif of claudin-1 in HCV entry, we generated an siRNA-resistant, full-length claudin-1 expression construct and a construct expressing claudin-1(YV/AA) (in which the C-terminal YV motif was mutated to AA). The expression of siRNA-resistant claudin-1 rescued HCVpp and Jc1 entry even in the presence of claudin-1 siRNA (Fig. 9B to D). However, the effect of silencing endogenous claudin-1 on HCV entry could not be rescued by the exogenous expression of claudin-1(YV/AA) (Fig. 9B to D), demonstrating that the dependence of HCV entry on claudin-1 is YV specific.
FIG 9.

The YV motif of claudin-1 is critical for determining the cell surface level of claudin-1 in and HCV entry. (A) Huh7.5 cells were transfected with wild-type Flag–claudin-1 or Flag–claudin-1 with a mutation in the C terminus (210YV211/210AA211) and fixed by PFA, followed by staining with Flag antibody. DAPI staining indicates the nucleus (blue). Scale bar, 15 μm. (B) Immunoblots showing the protein expression of wild-type or mutant siRNA-resistant claudin-1. Endo, endogenous. (C and D) Huh7.5.1 cells were treated with siRNA targeting the claudin-1 5′ UTR or a control siRNA for 24 h and transfected with a construct expressing wild-type or mutant siRNA-resistant claudin-1 for 48 h. Then the cells were infected with genotype 1b HCVpp (Con1) for another 72 h (C) or infected with Jc1 for another 24 h (D). The infection activity and ATP levels were measured. The relative luciferase activity was normalized to the cellular ATP levels. Data are presented as the means ± SD. ***, P < 0.001; NS, not significant.
DISCUSSION
In this paper, we provided several lines of evidence that Sec24C, a cargo-sorting component of COPII, regulates claudin-1 trafficking and HCV entry (Fig. 10). We showed that Sec24C associates with the cytoplasmic tail of claudin-1, a coreceptor for HCV entry. Claudin-1 was packed into COPII vesicles in our in vitro budding assay. Moreover, we demonstrated that Sec24C is crucial for HCV entry. Perturbation of Sec24C by siRNA or xanthohumol treatment as well as the use of a claudin-1 binding-deficient Sec24C mutant reduced the cell surface localization of claudin-1.
FIG 10.
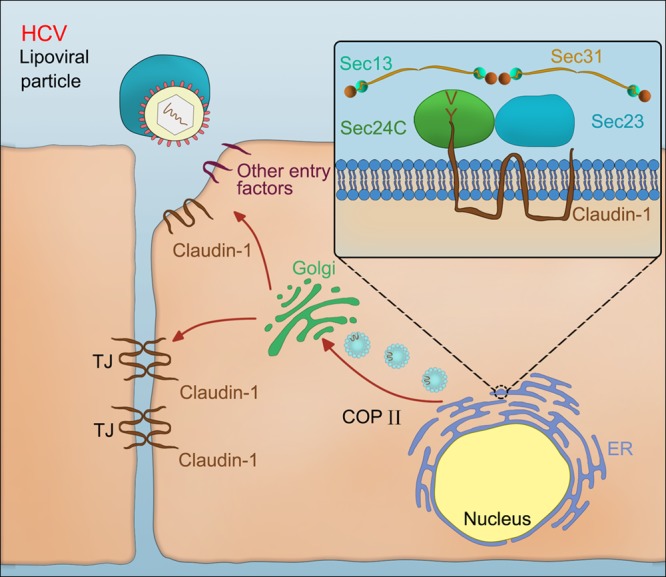
Schematic representation of the key role for Sec24C in regulating HCV entry through claudin-1 trafficking. Claudin-1 utilizes the YV motif-Sec24C interaction to exit from the ER to the Golgi compartment and eventually reach its final destination, the cell surface, where claudin-1 functions as an HCV coreceptor for viral entry.
We also showed that claudin-1 is a COPII cargo molecule by investigating cargo sorting and vesicle formation. First, we demonstrated that claudin-1 contains a COPII cargo-sorting motif associated with Sec24C. Through GST–claudin-1 tail pulldown experiments, we identified the interactome of the cytoplasmic tail of claudin-1, which included three key components of the COPII machinery: Sec24C, Sec23A, and Sec23B. Based on sequence alignment, GST pulldown mapping, and immunoprecipitation experiments, we demonstrated that the C-terminal YV motif in claudin-1 is critical for the interaction between claudin-1 and Sec24C. Second, we showed that claudin-1 is incorporated into COPII vesicles. Our confocal microscopy studies indicated that endogenous claudin-1 markedly colocalizes with Sec24C (Fig. 1G). Finally, our in vitro budding assays showed that claudin-1 buds off from the ER (Fig. 2A). Taken together, these findings indicate that claudin-1 is a cargo molecule for COPII-mediated transport.
There are four Sec24 isoforms in humans. We revealed that Sec24C, but not Sec24A, Sec24B, or Sec24D, associates with claudin-1. Sec24C interacts with different motifs of COPII cargo molecules through multiple sites. Sec24C-AAA, in which the binding site for another ER export signal motif (IXM) was mutated, did not interact with claudin-1. Further studies, including structural studies, are needed to explore the exact position of the motif in Sec24C that binds to claudin-1.
The inhibition of COPII-mediated transport has been shown to impair the formation of tight junctions (24, 25). However, the mechanism underlying how COPII-mediated transport contributes to functional tight junctions has been unclear. Our present study provides an explanation: COPII-mediated transport is required for the proper tight junction localization of claudin-1 as blocking COPII-mediated transport by silencing Sec24C or by treatment with XN reduces the localization of claudin-1 to the cell surface (Fig. 2B to D and 3A to D). The level of the claudin-1(YV/AA) mutant at the cell surface was lower than that of claudin-1(WT) (Fig. 9A); this finding supports the key role of Sec24C in the cargo sorting of claudin-1. The export of claudin-1 from the ER represents a critical checkpoint for the tight junction localization of claudin-1, which in turn forms tight junction strands.
As a coreceptor for HCV, claudin-1 plays a key role in HCV entry (26). Consistent with the fact that claudin-1 is a COPII cargo protein, perturbation of the COPII transport machinery reduced cell culture-adapted HCV (HCVcc) and HCVpp entry (Fig. 4). We predict that COPII also plays an important role in dengue virus entry because claudin-1 is a receptor for dengue virus (27).
Previous studies have shown that several pathogens hijack the COPII machinery (28–31). Non-LEE-encoded effector A (NleA, where LEE is locus for enterocyte effacement) is a type III secretion system (T3SS) effector common to enterohemorrhagic Escherichia coli, enteropathogenic E. coli, and Citrobacter rodentium. The NleA disrupts tight junctions by interacting with Sec24C (28). It was hypothesized that NleA inhibits the trafficking of tight junction proteins to the sites of tight junctions by inhibiting the COPII machinery (25). However, the tight junction protein(s) involved in that process is currently unknown. Based on the results of our study, we propose that the inhibition of Sec24C by NleA will block the exit of claudin-1 from the ER and will eventually reduce the tight junction localization of claudin-1, which is required for establishing tight junctions.
Xanthohumol, a natural product derived from hops, is an inhibitor of HCV replication (32). Here, we extended the knowledge of the role of xanthohumol against HCV by providing evidence that this compound reduces the cell surface levels of claudin-1 and HCV entry. Thus, xanthohumol not only inhibits HCV replication but also reduces HCV entry.
We found that many CLDNs containing a C-terminal YV or V motif associate with Sec24C (Fig. 8B and C), suggesting that the COPII machinery plays key roles in the proper localization and functions of CLDNs. For instance, the expression of claudin-2 increases the paracellular permeability of the intestinal lumen and is associated with the transepithelial electrical resistance (TER) (33, 34). We predict that xanthohumol would reduce the cell surface expression of claudin-2, thereby relieving inflammatory diseases caused by the increased expression of claudin-2, such as inflammatory bowel disease (IBD) and necrotizing enterocolitis (NEC) (33, 35). Although there are differential CLDN usages by HCV genotypes (36, 37), another two HCV coreceptors, claudin-6 and claudin-9, also contain a C-terminal YV motif for association with Sec24C (Fig. 8C). We anticipated that the cell surface levels of claudin-6 and claudin-9 were also dependent on Sec24C. Thus, Sec24C will play an important role in the entry of pan-HCV genotypes.
In conclusion, our study provides clear evidence that the C-terminal YV sequence in claudin-1 serves as an ER export signal by selectively binding to Sec24C. Claudin-1 utilizes the YV motif-Sec24C interaction to exit from the ER and reach its final destination, the cell surface, where claudin-1 functions as an HCV coreceptor for viral entry. We also identified a natural product derived from hops, xanthohumol, that can function as a novel inhibitor of HCV entry by reducing the cell surface levels of claudin-1. Our study provides insights that will have important implications for the development of therapeutic strategies against the hepatitis C virus.
MATERIALS AND METHODS
Cells and virus.
Huh7.5.1 and Huh7.5 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Thermo Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS; Thermo Scientific, Waltham, MA). Jc1Flag (p7-nsGluc2A) was obtained from Charles Rice. HCV pseudovirus particles (HCVpp) or VSV pseudovirus particles (VSVpp) were produced in HEK293T cells by transfection with expression plasmids encoding HIV Gag/Pol (pLP1), HIV Rev (pLP2), pLenti6 encoding luciferase, and HCV E1E2 from genotype 1b strain Con1 or the VSV-G expression plasmid (provided by Ping Zhao) (38, 39).
Plasmids.
The constructs encoding the GST-CD81-N-tail (aa 1 to 22), GST-CD81-C-tail (aa 227 to 236), GST-occludin-tail (aa 266 to 522), GST-SRB1-C-tail (aa 462 to 509), GST-claudin-1-C-tail, GST-claudin-4 tail, GST-claudin-5 tail, GST-claudin-6 tail, GST-claudin-6 tail(YV/AA), GST-claudin-9 tail, GST-claudin-9 tail(YV/AA), GST-claudin-11 tail, GST-claudin-16 tail, GST-claudin-19 tail, GST-claudin-22 tail, and GST-claudin-23 tail were synthesized and inserted into the pGEX-4T-1 vector. The GST-claudin-1-C-tail mutants were generated using a QuikChange site-directed mutagenesis kit. The GST-claudin-1-C-tail truncations were generated by PCR and inserted into the pGEX-4T-1 vector. Constructs encoding full-length claudin-1 or SRB1 fused to Flag were inserted into the PCMV-Tag2 vector. The Flag–claudin-1(Y/A), Flag–claudin-1(V/A), and Flag–claudin-1(YV/AA) constructs were generated using mutant primers. Constructs encoding wild-type Sec24C and the Sec24C mutant fused to Flag were synthesized and inserted into the PCMV-Tag2 vector. The siRNA-resistant wild-type Sec24C and the Sec24C mutant were generated using a QuikChange site-directed mutagenesis kit. The plasmid constructs used in this study are listed in Table 2.
TABLE 2.
Plasmids used in this study
| Vector and plasmid |
|---|
| pGEX-4T-1 plasmids |
| pGEX-4T-1-claudin-1-tail-WT |
| pGEX-4T-1-claudin-1-tail-183C/A |
| pGEX-4T-1-claudin-1-tail-184C/A |
| pGEX-4T-1-claudin-1-tail-185S/A |
| pGEX-4T-1-claudin-1-tail-186C/A |
| pGEX-4T-1-claudin-1-tail-187P/A |
| pGEX-4T-1-claudin-1-tail-188R/A |
| pGEX-4T-1-claudin-1-tail-189K/A |
| pGEX-4T-1-claudin-1-tail-190T/A |
| pGEX-4T-1-claudin-1-tail-191T/A |
| pGEX-4T-1-claudin-1-tail-192S/A |
| pGEX-4T-1-claudin-1-tail-193Y/A |
| pGEX-4T-1-claudin-1-tail-194P/A |
| pGEX-4T-1-claudin-1-tail-195T/A |
| pGEX-4T-1-claudin-1-tail-196P/A |
| pGEX-4T-1-claudin-1-tail-197R/A |
| pGEX-4T-1-claudin-1-tail-198P/A |
| pGEX-4T-1-claudin-1-tail-199Y/A |
| pGEX-4T-1-claudin-1-tail-200P/A |
| pGEX-4T-1-claudin-1-tail-201K/A |
| pGEX-4T-1-claudin-1-tail-202P/A |
| pGEX-4T-1-claudin-1-tail-204P/A |
| pGEX-4T-1-claudin-1-tail-205S/A |
| pGEX-4T-1-claudin-1-tail-206S/A |
| pGEX-4T-1-claudin-1-tail-207G/A |
| pGEX-4T-1-claudin-1-tail-208K/A |
| pGEX-4T-1-claudin-1-tail-209D/A |
| pGEX-4T-1-claudin-1-tail-210Y/A |
| pGEX-4T-1-claudin-1-tail-211V/A |
| pGEX-4T-1-claudin1-tail-YV/AA |
| pGEX-4T-1-claudin-1-tail-1–15 |
| pGEX-4T-1-claudin-1-tail-16–29 |
| pGEX-4T-1-YV |
| pGEX-4T-1-GKDYV |
| pGEX-4T-1-CD81-N-tail |
| pGEX-4T-1-CD81-C-tail |
| pGEX-4T-1-occludin-tail |
| pGEX-4T-1-SRB1-tail |
| pGEX-4T-1-claudin-4-tail |
| pGEX-4T-1-claudin-5-tail |
| pGEX-4T-1-claudin-11-tail |
| pGEX-4T-1-claudin-16-tail |
| pGEX-4T-1-claudin-19-tail |
| pGEX-4T-1-claudin-22-tail |
| pGEX-4T-1-claudin-23-tail |
| pGEX-4T-1-claudin-6-tail |
| pGEX-4T-1-claudin-9-tail |
| pGEX-4T-1-claudin-6-tail-YV/AA |
| pGEX-4T-1-claudin-9-tail-YV/AA |
| PCMV-Tag2b plasmids |
| PCMV-Tag2b-claudin-1-WT |
| PCMV-Tag2b-SRB1 |
| PCMV-Tag2b-claudin-1-YA |
| PCMV-Tag2b-claudin-1-VA |
| PCMV-Tag2b-claudin-1-YV/AA |
| PCMV-Tag2b-Sec24C |
| PCMV-Tag2b-Sec24C-AAA |
| PCMV-Tag2b-Sec24C-siRNA-resistant |
| PCMV-Tag2b-Sec24C-AAA-siRNA-resistant |
Antibodies.
The following primary mouse antibodies were used: anti-actin (for Western blotting [WB] at 1:3,000) (catalog no. A2228; Sigma-Aldrich, St. Louis, MO), anti-Flag (WB, 1:3,000) (A2220; Sigma-Aldrich, St. Louis, MO), IgG control (M075-3; MBL, Nagoya, Japan), anti-SRB1 (610883; BD Biosciences), anti-ZO-1 (immunofluorescence [IF], 1:100) (33-9100; Thermo Scientific, Waltham, MA), and anti-claudin-1 (IF, 1:100) (sc-166338; Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
The following primary rabbit antibodies were used: anti-claudin-1 (for WB, 1:1,500; IF, 1:200) (catalog no. ab15098; Abcam, Cambridge, MA), anti-ribophorin 1 (WB, 1:1,000) (ab198508; Abcam, Cambridge, MA), anti-ERGIC-53 (WB, 1:1,000) (125006; Abcam, Cambridge, MA), anti-Sec24A (WB, 1:1,000) (9678; Cell Signaling Technology, Inc., Boston, MA), anti-Sec24B (WB, 1:1,000) (12042; Cell Signaling Technology, Inc., Boston, MA), anti-Sec24C (WB, 1:1,000; IF, 1:100) (14676; Cell Signaling Technology, Inc., Boston, MA), anti-ZO-2 (WB, 1:1,000) (2847; Cell Signaling Technology, Inc., Boston, MA), anti-Sec24D (WB, 1:1,000) (191566; Abcam, Cambridge, MA), anti-Sec23A (WB, 1:1,000; IF, 1:100) (8162; Cell Signaling Technology, Inc., Boston, MA), and anti-Sec23B (WB, 1:1,000) (151258; Abcam, Cambridge, MA).
The following primary rat antibodies were used for FACS analysis (at 1:50): anti-claudin-1 (MAB4618; R&D Systems, Inc., Minneapolis, MN) and claudin-1 isotype control antibody (MAB006; R&D Systems, Inc., Minneapolis, MN).
The secondary antibodies included horseradish peroxidase (HRP)-conjugated enhanced chemiluminescent (ECL) goat anti-rabbit IgG (A6154; Sigma-Aldrich, St. Louis, MO), HRP-conjugated ECL goat anti-mouse IgG (A4416; Sigma-Aldrich, St. Louis, MO), and donkey anti-mouse–Alexa Fluor 488, donkey anti-rabbit–Alexa Fluor 555, donkey anti-rabbit–Alexa Fluor 488, donkey anti-mouse–Alexa Fluor 555, and donkey anti-rat–Alexa Fluor 488 (all, Invitrogen, Carlsbad, CA).
Immunofluorescence microscopy.
Immunofluorescence microscopy has been described previously (40). Briefly, cells seeded onto glass coverslips were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (PFA) in PBS buffer (for intracellular staining) or in −20°C methanol (for cell surface staining) for 5 min at room temperature. Fixed cells were incubated with blocking solution (PBS containing 10% normal goat serum) for 5 min at room temperature. Next, the coverslips were incubated with primary antibodies in permeabilizing buffer (0.1% Triton X-100 in PBS containing 10% normal goat serum) for 1 h. The coverslips were then washed three times with blocking solution, followed by incubation with Alexa Fluor 488- and Alexa Fluor 555-conjugated secondary antibodies for 1 h at room temperature (RT). After being washed three times with blocking solution, the coverslips were mounted with mounting medium containing 4′,6′-diamidino-2-phenylindole (DAPI). The cells were imaged using a Leica TCS SP5 microscope (Germany) equipped with a 40× oil immersion lens.
Immunoprecipitation.
The immunoprecipitation (IP) experiment has been described previously (41). Briefly, cells were lysed with lysis buffer (1% Triton X-100, 50 mM Tris, pH 7.4, 90 mM KCl, 2.5 mM MgCl2, and protease inhibitor cocktail) and precleared by the addition of protein A/G beads for 30 min at 4°C. The lysates were then incubated with protein A/G beads prebound with 1 μg of antibody for 1 h at 4°C. The samples were washed three times with PBS, eluted in SDS sample buffer, and analyzed by Western blotting.
GST pulldown.
The GST pulldown assay been described previously (42). Briefly, the expression of a GST control protein or the GST fusion proteins was induced in E. coli Rosetta(DE3) by treatment with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 37°C for 5 h. The cells were then lysed with lysis buffer (1% Triton X-100, 50 mM Tris, pH 7.4, 90 mM KCl, 2.5 mM MgCl2, and protease inhibitor cocktail), and the GST or GST fusion proteins were purified using glutathione-Sepharose beads. For pulldown assays, the cell lysates were mixed with GST or GST fusion proteins. After incubation at 4°C for 1 h, the glutathione beads were pelleted and washed three times with PBS buffer. The protein samples were then boiled in SDS-containing loading buffer for gel electrophoresis, followed by Western blotting.
In vitro COPII budding assay.
Preparation of semi-intact Huh7.5 cells and the COPII budding assay were carried out as described previously (43, 44). Briefly, Huh7.5 cells were cultured in 100-mm plates to 90 to 100% confluence, washed in PBS, removed from the plates with trypsin, and washed in B88-0 buffer (20 mM HEPES, pH 7.2, 250 mM sorbitol, 150 mM potassium acetate [KOAc], and 5 mM MgOAc) containing protease inhibitor cocktail. In brief, the cells were permeabilized with 40 μg/ml digitonin for 5 min in ice-cold B88-0 buffer, washed, and resuspended in 100 μl of B88-0 buffer. To form vesicles, donor membranes were mixed with 4 mg/ml mouse liver cytosol, an ATP regeneration system (40 mM creatine phosphate, 0.2 mg/ml creatine phosphokinase, and 1 mM ATP), and 0.2 mM GTP, followed by incubation at 30°C for 1 h. Newly formed vesicles were separated from the donor membranes by centrifugation at 15,000 rpm for 15 min. The collected vesicles were analyzed by SDS-PAGE.
Knockdown by siRNA.
The siRNAs were transfected into cells using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA). The siRNAs used for Sec24C and claudin-1 knockdown were obtained from GenePharma (China) and had the following sequences: sense (5′–3′), GGCUGCUGUGUAGAUCUCUTT and antisense (5′–3′), AGAGAUCUACACAGCAGCCTT for Sec24C; and sense (5′–3′), GCUUCUAGUAUCCAGACUCTT and antisense (5′–3′), GAGUCUGGAUACUAGAAGCTT for the claudin-1 5′ untranslated region (UTR).
Fluorescence-activated cell sorting (FACS).
Huh7.5 cells were harvested and washed with PBS twice. For permeabilization, the cells were permeabilized by 0.1% saponin in PBS containing 10% normal donkey serum for 10 min. For the nonpermeabilization condition, the cells were blocked in PBS containing 10% normal donkey serum. Then the cells were washed twice and stained with the anti-claudin-1 antibody (1:50) (MAB4618; R&D Systems) or the isotype control antibody (1:50) (MAB4618; R&D Systems) for 40 min at 4°C. The cells were washed twice and stained with donkey anti-rat–Alexa Fluor 488 antibody for 40 min at 4°C. The cells were washed twice, and 1 × 104 cells were analyzed for each sample. Isotype control antibody was used to delineate the gate.
Reporter assay.
Jc1Flag2(p7-nsGluc2A) or HCVpp-infected Huh 7.5.1 cells were assessed by monitoring the luciferase activity (Promega, Madison, WI). Cell viability was determined by measuring the cellular ATP level using a CellTiter-Glo luminescent cell viability assay kit (Promega, Madison, WI) according to the manufacturer's protocol. The normalized luciferase activity was determined by dividing the luciferase activity by the ATP level.
Mass spectrometry.
To identify claudin-1 tail-interacting proteins, lysates from Huh7.5.1 cells were pulled down by GST–claudin-1 tail or GST. The interacting proteins were analyzed by electrospray ionization tandem mass spectrometry (MS) on a Thermo LTQ Orbitrap instrument. Proteins identified from the GST–claudin-1 tail sample but not from the GST control sample were detected. Individual ion scores are indicated in the form of a Mascot-derived confidence score [calculated from the posterior error probability (PEP) as −10 log(PEP)]. The default significance threshold is a P value of <0.05.
Statistics.
Statistically significant differences were assessed using a paired Student t test in GraphPad Prism, version 5 (GraphPad Software, Inc., La Jolla, CA). Unless otherwise stated, the data represent the means from at least three independent experiments ± the standard deviations (SD).
ACKNOWLEDGMENTS
This work was supported by grants from National Natural Science Foundation of China (81672035, 81471955), CAMS Innovation Fund for Medical Sciences (2016-I2M-3-020), National Key Plan for Research and Development of China (2016YFD0500300), the Fundamental Research Funds for the Central Universities (3332013118 and 2016ZX310047), and Program for Changjiang Scholars and Innovative Research Team in University (IRT13007).
We thank Charles Rice, Francis Chisari, Xiaowei Chen, Sidney Yu, and Ping Zhao for reagents.
We declare that we have no competing interests.
REFERENCES
- 1.Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. 2014. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 61:S45–57. doi: 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 2.Gogela NA, Lin MV, Wisocky JL, Chung RT. 2015. Enhancing our understanding of current therapies for hepatitis C virus (HCV). Curr HIV/AIDS Rep 12:68–78. doi: 10.1007/s11904-014-0243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou LY, Zhang LL. 2016. Host restriction factors for hepatitis C virus. World J Gastroenterol 22:1477–1486. doi: 10.3748/wjg.v22.i4.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colpitts CC, Baumert TF. 2016. Hepatitis C virus cell entry: a target for novel antiviral strategies to address limitations of direct acting antivirals. Hepatol Int 10:741–748. doi: 10.1007/s12072-016-9724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding Q, von Schaewen M, Ploss A. 2014. The impact of hepatitis C virus entry on viral tropism. Cell Host Microbe 16:562–568. doi: 10.1016/j.chom.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colpitts CC, Baumert TF. 2017. Claudins in viral infection: from entry to spread. Pflugers Arch 469:27–34. doi: 10.1007/s00424-016-1908-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Itallie CM, Anderson JM. 2014. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol 36:157–165. doi: 10.1016/j.semcdb.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dukes JD, Fish L, Richardson JD, Blaikley E, Burns S, Caunt CJ, Chalmers AD, Whitley P. 2011. Functional ESCRT machinery is required for constitutive recycling of claudin-1 and maintenance of polarity in vertebrate epithelial cells. Mol Biol Cell 22:3192–3205. doi: 10.1091/mbc.E11-04-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farquhar MJ, Hu K, Harris HJ, Davis C, Brimacombe CL, Fletcher SJ, Baumert TF, Rappoport JZ, Balfe P, McKeating JA. 2012. Hepatitis C virus induces CD81 and claudin-1 endocytosis. J Virol 86:4305–4316. doi: 10.1128/JVI.06996-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin P, Zhang L. 2016. Aspirin inhibits hepatitis C virus entry by downregulating claudin-1. J Viral Hepat 23:62–64. doi: 10.1111/jvh.12446. [DOI] [PubMed] [Google Scholar]
- 11.Dancourt J, Barlowe C. 2010. Protein sorting receptors in the early secretory pathway. Annu Rev Biochem 79:777–802. doi: 10.1146/annurev-biochem-061608-091319. [DOI] [PubMed] [Google Scholar]
- 12.Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 13.Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. 2011. COPII and the regulation of protein sorting in mammals. Nat Cell Biol 14:20–28. doi: 10.1038/ncb2390. [DOI] [PubMed] [Google Scholar]
- 14.Campbell JL, Schekman R. 1997. Selective packaging of cargo molecules into endoplasmic reticulum-derived COPII vesicles. Proc Natl Acad Sci U S A 94:837–842. doi: 10.1073/pnas.94.3.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller EA, Barlowe C. 2010. Regulation of coat assembly–sorting things out at the ER. Curr Opin Cell Biol 22:447–453. doi: 10.1016/j.ceb.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lord C, Ferro-Novick S, Miller EA. 2013. The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the Golgi. Cold Spring Harb Perspect Biol 5:a013367. doi: 10.1101/cshperspect.a013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. 1999. Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol 147:1351–1363. doi: 10.1083/jcb.147.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyata S, Inoue J, Shimizu M, Sato R. 2015. Xanthohumol improves diet-induced obesity and fatty liver by suppressing sterol regulatory element-binding protein (SREBP) activation. J Biol Chem 290:20565–20579. doi: 10.1074/jbc.M115.656975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mancias JD, Goldberg J. 2008. Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J 27:2918–2928. doi: 10.1038/emboj.2008.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nufer O, Guldbrandsen S, Degen M, Kappeler F, Paccaud JP, Tani K, Hauri HP. 2002. Role of cytoplasmic C-terminal amino acids of membrane proteins in ER export. J Cell Sci 115:619–628. [DOI] [PubMed] [Google Scholar]
- 21.Wendeler MW, Paccaud JP, Hauri HP. 2007. Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep 8:258–264. doi: 10.1038/sj.embor.7400893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meertens L, Bertaux C, Cukierman L, Cormier E, Lavillette D, Cosset FL, Dragic T. 2008. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J Virol 82:3555–3560. doi: 10.1128/JVI.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng A, Yuan F, Li Y, Zhu F, Hou P, Li J, Song X, Ding M, Deng H. 2007. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J Virol 81:12465–12471. doi: 10.1128/JVI.01457-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thanabalasuriar A, Kim J, Gruenheid S. 2013. The inhibition of COPII trafficking is important for intestinal epithelial tight junction disruption during enteropathogenic Escherichia coli and Citrobacter rodentium infection. Microbes Infect 15:738–744. doi: 10.1016/j.micinf.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Thanabalasuriar A, Koutsouris A, Weflen A, Mimee M, Hecht G, Gruenheid S. 2010. The bacterial virulence factor NleA is required for the disruption of intestinal tight junctions by enteropathogenic Escherichia coli. Cell Microbiol 12:31–41. doi: 10.1111/j.1462-5822.2009.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 27.Torres-Flores JM, Arias CF. 2015. Tight junctions go viral! Viruses 7:5145–5154. doi: 10.3390/v7092865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Thanabalasuriar A, Chaworth-Musters T, Fromme JC, Frey EA, Lario PI, Metalnikov P, Rizg K, Thomas NA, Lee SF, Hartland EL, Hardwidge PR, Pawson T, Strynadka NC, Finlay BB, Schekman R, Gruenheid S. 2007. The bacterial virulence factor NleA inhibits cellular protein secretion by disrupting mammalian COPII function. Cell Host Microbe 2:160–171. doi: 10.1016/j.chom.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 29.Yamayoshi S, Noda T, Ebihara H, Goto H, Morikawa Y, Lukashevich IS, Neumann G, Feldmann H, Kawaoka Y. 2008. Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. Cell Host Microbe 3:168–177. doi: 10.1016/j.chom.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang J, Patarroyo C, Garcia Cabanillas D, Zheng H, Laliberte JF. 2015. The vesicle-forming 6K2 protein of turnip mosaic virus interacts with the COPII coatomer Sec24a for viral systemic infection. J Virol 89:6695–6710. doi: 10.1128/JVI.00503-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Fuchs S, Zhang J, Wellford S, Schuldiner M, Wang X. 2016. An unrecognized function for COPII components in recruiting the viral replication protein BMV 1a to the perinuclear ER. J Cell Sci 129:3597–3608. doi: 10.1242/jcs.190082. [DOI] [PubMed] [Google Scholar]
- 32.Yang M, Li N, Li F, Zhu Q, Liu X, Han Q, Wang Y, Chen Y, Zeng X, Lv Y, Zhang P, Yang C, Liu Z. 2013. Xanthohumol, a main prenylated chalcone from hops, reduces liver damage and modulates oxidative reaction and apoptosis in hepatitis C virus infected Tupaia belangeri. Int Immunopharmacol 16:466–474. doi: 10.1016/j.intimp.2013.04.029. [DOI] [PubMed] [Google Scholar]
- 33.Krug SM, Schulzke JD, Fromm M. 2014. Tight junction, selective permeability, and related diseases. Semin Cell Dev Biol 36:166–176. doi: 10.1016/j.semcdb.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Gong Y, Hou J. 2017. Claudins in barrier and transport function-the kidney. Pflugers Arch 469:105–113. doi: 10.1007/s00424-016-1906-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mankertz J, Schulzke JD. 2007. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol 23:379–383. doi: 10.1097/MOG.0b013e32816aa392. [DOI] [PubMed] [Google Scholar]
- 36.Haid S, Grethe C, Dill MT, Heim M, Kaderali L, Pietschmann T. 2014. Isolate-dependent use of claudins for cell entry by hepatitis C virus. Hepatology 59:24–34. doi: 10.1002/hep.26567. [DOI] [PubMed] [Google Scholar]
- 37.Hopcraft SE, Evans MJ. 2015. Selection of a hepatitis C virus with altered entry factor requirements reveals a genetic interaction between the E1 glycoprotein and claudins. Hepatology 62:1059–1069. doi: 10.1002/hep.27815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao J, Chen Z, Ren Y, Luo Y, Cao M, Lu W, Zhao P, Qi Z. 2011. Oral immunization with attenuated Salmonella carrying a co-expression plasmid encoding the core and E2 proteins of hepatitis C virus capable of inducing cellular immune responses and neutralizing antibodies in mice. Vaccine 29:3714–3723. doi: 10.1016/j.vaccine.2011.02.083. [DOI] [PubMed] [Google Scholar]
- 39.Guan M, Wang W, Liu X, Tong Y, Liu Y, Ren H, Zhu S, Dubuisson J, Baumert TF, Zhu Y, Peng H, Aurelian L, Zhao P, Qi Z. 2012. Three different functional microdomains in the hepatitis C virus hypervariable region 1 (HVR1) mediate entry and immune evasion. J Biol Chem 287:35631–35645. doi: 10.1074/jbc.M112.382341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin P, Hong Z, Yang X, Chung RT, Zhang L. 2016. A role for retromer in hepatitis C virus replication. Cell Mol Life Sci 73:869–881. doi: 10.1007/s00018-015-2027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Yang X, Yang G, Hong Z, Zhou L, Yin P, Xiao Y, Chen L, Chung RT, Zhang L. 2014. Hepatitis C virus NS5A hijacks ARFGAP1 to maintain a phosphatidylinositol 4-phosphate-enriched microenvironment. J Virol 88:5956–5966. doi: 10.1128/JVI.03738-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang N, Yin P, Zhou L, Li H, Zhang L. 2016. ARF1 activation dissociates ADRP from lipid droplets to promote HCV assembly. Biochem Biophys Res Commun 475:31–36. doi: 10.1016/j.bbrc.2016.05.024. [DOI] [PubMed] [Google Scholar]
- 43.Lee H, Noh H, Mun J, Gu C, Sever S, Park S. 2016. Anks1a regulates COPII-mediated anterograde transport of receptor tyrosine kinases critical for tumorigenesis. Nat Commun 7:12799. doi: 10.1038/ncomms12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adolf F, Rhiel M, Reckmann I, Wieland FT. 2016. Sec24C/D-isoform-specific sorting of the preassembled ER-Golgi Q-SNARE complex. Mol Biol Cell 27:2697–2707. doi: 10.1091/mbc.E16-04-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]