

Abstract
Channelrhodopsins (ChRs) are light-gated ion channels widely used for activating selected cells in large cellular networks. ChR variants with a red-shifted absorption maximum, such as the modified Volvox carteri ChR1 red-activatable channelrhodopsin (“ReaChR,” λmax = 527 nm), are of particular interest because longer wavelengths allow optical excitation of cells in deeper layers of organic tissue. In all ChRs investigated so far, proton transfer reactions and hydrogen bond changes are crucial for the formation of the ion-conducting pore and the selectivity for protons versus cations, such as Na+, K+, and Ca2+ (1). By using a combination of electrophysiological measurements and UV-visible and FTIR spectroscopy, we characterized the proton transfer events in the photocycle of ReaChR and describe their relevance for its function. 1) The central gate residue Glu130 (Glu90 in Chlamydomonas reinhardtii (Cr) ChR2) (i) undergoes a hydrogen bond change in D → K transition and (ii) deprotonates in K → M transition. Its negative charge in the open state is decisive for proton selectivity. 2) The counter-ion Asp293 (Asp253 in CrChR2) receives the retinal Schiff base proton during M-state formation. Starting from M, a photocycle branching occurs involving (i) a direct M → D transition and (ii) formation of late photointermediates N and O. 3) The DC pair residue Asp196 (Asp156 in CrChR2) deprotonates in N → O transition. Interestingly, the D196N mutation increases 15-syn-retinal at the expense of 15-anti, which is the predominant isomer in the wild type, and abolishes the peak current in electrophysiological measurements. This suggests that the peak current is formed by 15-anti species, whereas 15-syn species contribute only to the stationary current.
Keywords: electrophysiology, Fourier transform IR (FTIR), gating, ion channel, photobiology, ultraviolet-visible spectroscopy (UV-vis spectroscopy), channelrhodopsin, photocycle, proton dynamics, proton transfer
Introduction
Channelrhodopsins (ChRs)6 are directly light-gated ion channels (2, 3) naturally located in the eyespot of motile green algae where they serve as sensory photoreceptors for phobic responses and phototaxis (4, 5). Light-induced cation influx leads to cell depolarization (6), a property that can be exploited for activation of ChR-expressing host cells (7, 8).
The red-activatable channelrhodopsin (ReaChR) (9) was designed as an optogenetic tool because its bathochromically shifted absorption maximum compared with CrChR2 allows activation at deeper tissue location (10). Under continuous illumination, the transient peak current in ReaChR decays to a steady-state level with smaller amplitude (9, 11). This phenomenon is called inactivation or desensitization and was interpreted previously in CrChR2 as an equilibration process between two conducting states with different conductivities, which become populated by two distinct dark states (D and D′) under continuous illumination (1, 12–14). It was suggested (11, 15, 16) that the dark states represent protein subspecies with a 15-anti- and 15-syn-retinal conformation, respectively, whereby their conducting states differ with respect to ion selectivity, conductivity, and channel kinetics.
The concept of parallel 15-anti and 15-syn photocycles also delivers an explanation for the observed isomeric light-dark adaptation of ChRs by assuming transitions between the different subspecies by C13=C14 and C15=N double isomerization: the initial dark state (IDA) of ChRs, which is formed after hours of dark adaptation, adopts a pure all-trans-retinal isomer according to recent NMR studies (16, 17). The apparent dark state (DAapp) instead is formed within minutes after recovery of the illuminated state and represents a mixture of 13-trans,15-anti- (D) and 13-cis,15-syn-retinal (D′) as shown by retinal extraction and subsequent HPLC analysis (15, 18, 19) and resonance Raman experiments (16, 18, 19). Although the parallel photocycle model is well based on experimental data, the actual determinants for the equilibrium between syn and anti cycle remain elusive.
The light-induced photocycles of ChRs involve a number of changes in hydrogen bonding and proton transfers that affect a variety of channel properties, such as ion selectivity, conductivity, and photocycle kinetics. The photocycle of the best studied ChR, ChR2 from Chlamydomonas reinhardtii (CrChR2), involves proton dynamics of the central gate residue Glu90 (Glu130 in ReaChR; Fig. 1), the counter-ion residue Asp253 (Asp293 in ReaChR), and the DC pair residue Asp156 (Asp196 in ReaChR). Glu90 plays a major role for both formation of the conducting pore and ion selectivity (20, 21). Asp253, in the following referred to as Ci2 (counter-ion 2), receives the RSBH+ proton that is released prior to channel opening (22). Alternatively, it was proposed that both Ci1 (Glu123) and Ci2 (Asp253) serve as proton acceptors (21). Furthermore, it was suggested that the DC pair residue Asp156 reprotonates the RSB in the P390 → P520 transition (M → N in ReaChR) (22) as mutations of this residue highly decelerate the photocycle kinetics (23). However, it is unclear whether the proton dynamics of CrChR2 also apply to red-shifted ChRs, such as ReaChR, as the bathochromically shifted absorption maximum points to molecular alterations near the retinal-binding pocket and the active site as compared with CrChR2; e.g. threonine 159, in the direct environment of Asp156, is exchanged for a cysteine in ReaChR (Cys199).
Figure 1.
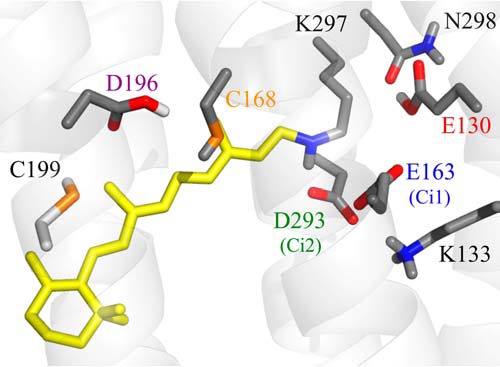
Schematic representation of the active site of ReaChR. Residues discussed in the text are highlighted. The homology model (SWISS-MODEL (50)) is based on the 3D structure of C1C2 (Protein Data Bank code 3ug9). Protonation states were estimated based on a pKa calculation (64).
The present study, based on a combination of FTIR difference spectroscopy, time-resolved UV-visible spectroscopy, electrophysiological measurements, and site-directed mutagenesis, showed that Glu130, Ci2, and the DC pair residue Asp196 (Fig. 1) also experience proton dynamics in ReaChR. Our data show that the Glu90-helix 2-tilt-model for CrChR2 (21) applies to ReaChR as well. Additionally, we now present a more detailed mechanism for ReaChR and show that deprotonation of Glu130 occurs in two distinct mechanistic steps. It undergoes a change in hydrogen bonding in the D → K transition and deprotonates before formation of the M-state. Consistent with CrChR2, the M-state (P390 in CrChR2) is formed by a proton transfer from the RSBH+ to Ci2, but after M formation a photocycle branching occurs that was not observed in CrChR2 WT. The main branch involves M → N → O transitions, and water is likely to serve as the proton donor for the reprotonation of the RSB during M → N transition because no amino acid residue could be identified as proton donor. In parallel, a direct M → D transition, involving reprotonation of the RSB by Ci2, takes place.
In either case, Asp196 does not serve as the proton donor for reprotonation of the RSB in ReaChR as its homologue Asp156 in CrChR2 does (22). Instead, Asp196 deprotonates in the N → O transition. Neutralization of Asp156 as in the D196N mutant increases D′ at the expense of D under conditions of continuous illumination and abolishes the peak current in electrophysiological measurements. This interesting correlation provides experimental evidence for the concept that the peak current is mainly formed by a conducting state with 13-cis,15-anti-retinal conformation, whereas the stationary current contains contributions of both 13-cis,15-anti- and 13-trans,15-syn-retinal isomers.
Results
Electrophysiological characterization of key residues
To characterize ion conductance, we expressed ReaChR WT and selected mutants in HEK cells. We mutated the two RSB counter-ions Ci1 (Glu163) and Ci2 (Asp293), the central gate residue Glu130, and the DC pair residue Asp196. A structural model of the protein region with these residues is given in Fig. 1. The amplitudes of both transient and stationary photocurrent (It and Is, respectively), of E163T resemble WT-like currents, whereas Is of E130Q and D293N at standard conditions decreased to 41 and 8%, respectively (Fig. 2, A and B). In E163T, the decay of the photocurrent after activation was accelerated (Fig. 2C; 34 ± 2 ms versus 132 ± 7 ms of WT) in line with findings for homologous mutations in CrChR2 (WT, τoff = 9.8 ms; E123T, τoff = 5.2 ms (24)) and the CrChR1/2 chimera (C1C2-E162A) (25). Off-kinetics were moderately accelerated in E130Q (89 ± 7 ms) similar to E90A in CrChR2 (26).
Figure 2.

Electrophysiological properties of wild type ReaChR and the mutants E130Q, E163T, D293N, and D196N. A, representative photocurrent traces recorded upon 500-ms illumination. B, average size of peak (left) and stationary (right) photocurrents; significance was tested with the Mann-Whitney U test. *, 0.05 > p > 0.01; **, 0.01 > p > 0.001; ns, not significant. C, time constants of apparent closing kinetics after light switched off; wild type, E130Q, and D196N were fitted monoexponentially; E163T was fitted biexponentially; and due to decreased currents there is no value for D293N. D, current–voltage relation of wild type (black) and E130Q (gray) under different extracellular ionic conditions (intracellular NaCl, pH 7.2) with corresponding I–E plot (normalized to standard conditions); holding potential (Ehold) = 0 mV in red. E, calculated reversal potentials (Erev) for wild type (filled bars) and E130Q (empty bars) (liquid junction potential-corrected; n = 5–8). F, reversal potential shifts (ΔErev) with respect to standard conditions (NaCl, pH 7.2) after substitution of extracellular Na+ for non-permeable NMG (NMG, pH 7.2; black), reduction of extracellular protons (NaCl, pH 9.0; dark gray), and reduction of both extracellular NaCl and protons (NMG, pH 9.0; light gray) (n = 5–8; liquid junction potential-corrected). Error bars represent standard errors (S. E.).
Most striking, however, was the lack of the transient current It during illumination in the DC pair mutant D196N, whereas the off-kinetics (202 ± 23 ms) of wild type and mutant are in the same order of magnitude. This is in contrast to the homologous mutations in CrChR2 where both the off-kinetics (WT, τoff = 10 ms; D156A, τoff > 1.5 × 105 ms (23)) and the photocurrents are significantly different (23, 27).
As expected from previous studies on CrChR2 (20, 28), the central gate mutant E130Q conducts less protons in favor of Na+, most clearly seen from the enlarged current and shifted reversal potential upon replacement of Na+ by N-methyl-d-glucamine (NMG) at extracellular pH 9 (Fig. 2, D–F). This suggests that Glu130, like in other ChRs, is part of the central selectivity filter. Based on this functional characterization, we asked whether there are internal alterations of the proton conducting network within the conducting pore and how these changes might be linked to changes of selectivity and to photocurrent inactivation and channel closure after light switched off.
Steady-state and time-resolved UV-visible spectroscopy of selected mutants
Next, light adaptation and photocycle intermediates of selected mutants were characterized by UV-visible spectroscopy and compared with the wild type (11). The IDA, i.e. the state before any light exposure of the protein, is slightly red-shifted for the counter-ion mutants, E163T (λmax = 530 nm) and D293N (λmax = 528 nm), with respect to the wild type (λmax = 527 nm; Fig. 3A). However, the bathochromic shift is minor compared with CrChR2, C1C2, and ChR2 from Platymonas subcordiformis (PsChR2), which show 10–25-nm shifts upon counter-ion neutralization (29, 30). In contrast, the IDA of E130Q (λmax = 513 nm) and D196N (λmax = 523 nm) are blue-shifted, which was not reported for analogous mutations in CrChR2 (23, 31). Upon extended illumination (60 s, 530 nm) and subsequent recovery in the dark (10 min), the DAapp is formed. Corresponding UV-visible spectra of all samples except E163T are shifted toward shorter wavelengths. A likely explanation for these hypsochromic shifts from IDA to DAapp is a relative increase of 15-syn-retinal (D′) compared with 15-anti-retinal (D) in DAapp assuming a blue-shifted absorption of the underlying 13-cis,15-syn chromophore as observed in bacteriorhodopsin (32–35). This effect is most pronounced in D196N (shift from 523 to 506 nm).
Figure 3.

UV-visible spectroscopy of ReaChR WT and the mutants E130Q, E163T, D293N, and D196N. IDA and DAapp spectra of recombinant wild type and mutants at pH 7.4 are shown in A. The wild type IDA spectrum is adapted from Ref. 11. DAapp is formed upon extended illumination (60 s, 530 nm) and subsequent recovery in the dark (10 min). The observed spectral shifts from IDA to DAapp indicate that the dark-state composition is altered upon first illumination. The significant blue shift of DAapp of D196N as compared with WT can be explained by the retinal fingerprint region in FTIR spectra of ReaChR wild type (gray filled curves) and the mutants E130Q, D196N, E163T, and D293N (B). D196N shows an additional negative band at 1183 cm−1 and a strong positive band at 1173 cm−1, hinting to a relative gain in 15-syn-retinal isomer at the expense of 15-anti-retinal in comparison with the wild type. C, the proposed ReaChR photocycle comprises two branches, a main branch starting from 13-trans,15-anti-retinal (D) and a side branch starting from 13-cis,15-syn-retinal (D′), which together form DAapp (11). In either branch, light induces a photoreaction involving photointermediates K, L, M, N, and O as well as K′, L′, M′, N′, and O′, respectively. Half-life times (t½) of the evolution-associated difference spectra are derived by global analysis of flash photolysis data (D--G) using a sequential model comprising four or five components. The values of t½ are summarized in the table (C, right); life-times of the wild type are taken from Ref. 11. Transient absorption changes (in mOD) induced by green laser flashes (10 ns, 530 nm, 5 mJ) were measured for E130Q (D), E163T (E), D196N (F), and D293N (G) for at least 15 cycles from 40 ns to 10 s. Single-wavelength kinetics of E163T and D293N are illustrated in H and I, respectively. Raw data (dots) and global fits (lines) are shown. Wild type data are taken from Ref. 11 (J). abs., absorbance; norm., normalized.
To further evaluate this interpretation, FTIR difference spectra of the wild type (gray filled curves), E130Q, E163T, D293N, and D196N were recorded (Fig. 3B). In the retinal fingerprint region, two negative bands at 1234 and 1199 cm−1 in the WT spectrum reflect depletion of the 13-trans,15-anti dark state (D) (18, 36), and the positive band at 1176 cm−1 is assigned to 13-trans-retinal (37–39). This band pattern is only slightly altered by mutations of Glu130, Ci1, and Ci2. However, in the D196N spectrum, an additional negative band at 1183 cm−1 is present, indicating a 13-cis,15-syn-retinal dark state (D′) (39–42). Additionally, a more pronounced positive band at 1173 cm−1 points to the 13-trans photoproduct of a photoconverted D′ state. This finding implies that 1) DAapp is composed of a mixture of 13-trans,15-anti-retinal (D) and 13-cis,15-syn-retinal (D′) isomers and 2) the contribution of the syn conformation (D′) to DAapp is relatively increased in the D196N mutant.
D and D′, which together form DAapp, undergo light-induced photocycles comprising the intermediates K, L, M, N, and O as well as K′, L′, M′, N′, and O′, respectively (Fig. 3C, left) (11). Due to similar absorption maxima, which are not more than 10 nm shifted (39), it is not a simple task to separately investigate both photocycles. Additionally, the contribution of the syn photocycle is small as compared with the anti photocycle because 1) ReaChR WT populates only around 20% of D′ in DAapp (11) and 2) a lower quantum efficiency of the photoactivation of the 13-cis,15-syn D′ is assumed (43). Therefore, the observed photocycle dynamics and half-life times are primarily assigned to the anti photocycle branch. The influence of key residues on the photocycle was studied by flash photolysis experiments on the mutants E130Q, E163T, D196N, and D293N (Fig. 3, D–G). Purified proteins were excited with green laser flashes (10 ns, 530 nm, 5 mJ), and UV-visible absorption changes were recorded. Half-life times (t½) were derived from global analysis (table in Fig. 3C; wild type values are taken from Ref. 11). In the photocycle of the WT at pH 7.4, an early red-shifted intermediate, termed K, evolves into the L-state succeeded by the deprotonated M-state. M decays biphasically (Fig. 3J and Ref. 11), indicating the presence of two different M-states, M1 and M2. Although M1 undergoes transition to N, M2 directly relaxes back to D. At pH 5 and 9, the red-shifted N intermediate (λmax = 530 nm) is observed (11), whereas it is superimposed by the strong-absorbing dark state at pH 7.4 (λmax = 527 nm). This general reaction sequence of the wild type is also found in the mutants at pH 7.4 but with significant variations. L is not observed in E130Q (Fig. 3D) and D196N (Fig. 3F), but all mutants show the N-state. The latter finding could again be explained by the hypsochromic shift of DAapp compared with the N-state for E130Q and D196N and by the reduced full width at half-maximum of the chromophore peaks of E163T and D293N. Neutralization of Ci1 or Ci2, either E163T (Fig. 3E) or D293N (Fig. 3G), reduces the life-time of K from 1.01 ± 0.01 μs to 455 ± 8 and 452 ± 5 ns, respectively, but in turn decelerates M-state formation from 10.8 ± 0.3 to 18.8 ± 0.2 μs in E163T and 245 ± 1 μs in D293N. The deprotonated M intermediate progresses either to the reprotonated N-state with 8.54 ± 0.27 and 12.7 ± 0.2 ms for E163T and D293N, respectively (M1), or directly to D within seconds (M2; Fig. 3, H and I). The strong impact of the D293N mutant on the M-state formation kinetics indicates that Ci2 receives the RSBH+ proton, similar to CrChR2 (22).
The central gate residue substitution, E130Q, accelerates M-formation (Fig. 3D), whereas D196N results in a slight life-time extension of K and M (1.87 ± 0.03 μs and 263 ± 4 ms, respectively; Fig. 3F). Furthermore, it is noticeable that D196N shows stronger accumulation of N as compared with the other mutants.
Monitoring proton transfer processes and changes in hydrogen bonding by FTIR difference spectroscopy
The formation of the photocycle intermediates is accompanied by changes of hydrogen bonding and/or proton transfer processes involving acidic and alkaline amino acid side chains. To address the question in which stage of the photocycle these events occur and to elucidate the role of specific amino acids, FTIR difference spectra of WT and selected mutants were recorded at cryogenic temperatures to stabilize photocycle intermediates. The steady-state spectrum obtained at 80 K mainly represents the transition from the dark state to the early K intermediate (11). Fig. 4A shows the FTIR difference spectra of the D → K transition in H2O (gray filled curves) and in D2O buffer (black) of ReaChR WT and the mutants E130Q, E163T, D293N, and D196N.
Figure 4.
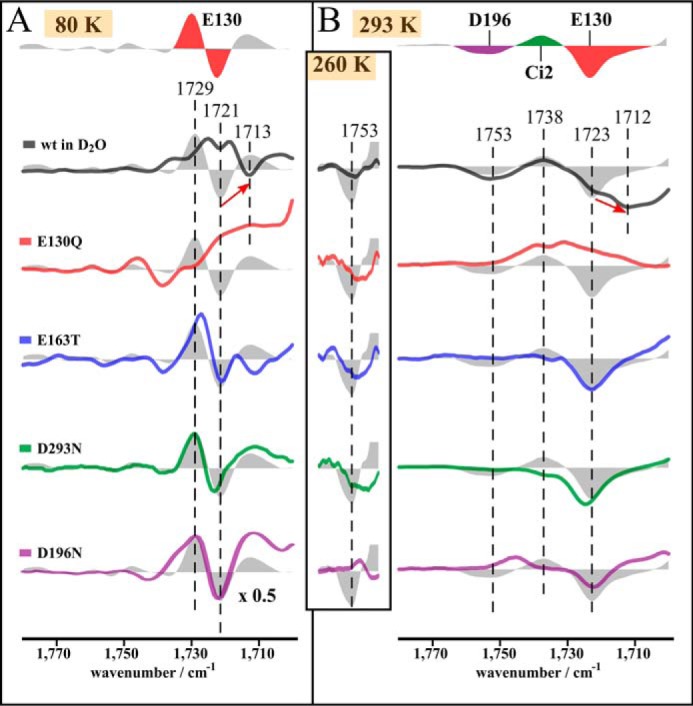
Proton transfer processes in ReaChR. Shown is the C=O stretching region of the steady state of the wild type at pH 7.4 in H2O (gray filled curves) and D2O (black) and the mutants E130Q (red), E163T (blue), D293N (green), and D196N (purple) at 80 (A) and 293 K (B), respectively. Additionally, the region around the 1753 cm−1 band is shown for measurements at 260 K. Spectra at 80 K represent the D → K transition. At 293 K, the steady states represent a mixture of the succeeding intermediates with a large contribution of O. Band assignment is indicated by colored bands in the upper line: mutational analysis at 80 K indicates an early hydrogen bond change of Glu130. Spectra at 293 K show that Glu130 deprotonates, the counter-ion complex protonates, and Asp196 also deprotonates during the photocycle, the latter being supported by spectra at 260 K.
Early proton dynamics in Glu130
Bands in the spectral region between 1800 and 1700 cm−1 are predominantly caused by carbonyl C=O stretching modes of protonated acidic amino acid side chains, and therefore corresponding difference bands reflect changes in hydrogen bonding strength and/or proton transfer processes. In the WT spectrum (Fig. 4A), a prominent difference band at 1729(+)/1721(−) cm−1 and a positive band at 1713 cm−1 are observed. In D2O, the negative band at 1721 cm−1 is downshifted to 1713 cm−1, whereas its positive counterpart at 1729 cm−1 is shifted to 1721 cm−1. However, this band maximum is superimposed by the negative band at 1721 cm−1 due to incomplete deuteration so that two apparent maxima at 1725 and 1719 cm−1 are seen. The band intensities and positions of the 1729(+)/1721(−) cm−1 difference band in the wild type are only slightly affected by mutations of the counter-ions, E163T and D293N, or by the DC pair mutation D196N. As this difference band is completely absent in E130Q, it is assigned to a vibration of Glu130. The characteristic pattern with one positive and one negative band suggests that Glu130 undergoes a hydrogen bond change in the D → K transition rather than a deprotonation, which would be indicated by only one single negative band.
The impact of the D196N mutation on this difference band might be explained by its influence on the ratio of 15-anti and 15-syn retinal (see Fig. 3B). The slight effect of E163T and D293N on the Glu130 difference band is explained by an altered interaction of these residues with Glu130. In the E163T spectrum, a negative band at 1712 cm−1 instead of the positive WT band at 1713 cm−1is observed. The origin of the positive wild type band is unclear; however, protonation of Ci1 is unlikely because in that case the positive band would be absent in the spectrum of the E163T mutant rather than being replaced by a negative band.
Proton transfer reactions in later photocycle intermediates
In the FTIR difference spectrum of the wild type at 293 K representing the O-state with minor contributions of M, N, and L (Fig. 4B, gray filled curve), we observe a band pattern at 1753(−)/1738(+)/1723(−) cm−1. The spectra of the mutants (Fig. 4B) support the assignment of the 1723 cm−1 band to Glu130 because it is absent only in E130Q. In D2O, this negative band is downshifted by 11 cm−1 to 1712 cm−1. The residual band at 1723 cm−1 indicates only partial sample deuteration. The existence of only one band assigned to Glu130 instead of a difference band as observed in the spectrum at 80 K (Fig. 4A) now shows deprotonation of Glu130. Deprotonation of the homologous residue Glu90 was observed by various authors for the photocycle of CrChR2, although it is still under debate in which intermediate this deprotonation occurs (20–22).
The positive band at 1738 cm−1 is affected by all mutants analyzed. Its intensity is significantly reduced in both the E163T and D293N mutants and thus reflects protonation of Ci2 during M-state formation. The influence of both mutations on the 1738 cm−1 band can be explained by an accelerated proton release from the unmutated counter-ion residue due to a lowered pKa value if the other respective residue is mutated. In E130Q, instead of one positive band at 1738 cm−1, two positive bands arise at 1739 and 1731 cm−1. This observation is explained by an overlap of the positive counter-ion band with a negative band that arises in E130Q only, possibly due to deprotonation of a so far unidentified residue.
In the spectrum of the D196N mutant, the band at 1738 cm−1 is upshifted to 1748 cm−1. The high upshift of this band can be explained by the influence of this mutation on the ratio of the 15-anti and 15-syn photocycle branches (see Fig. 3B), leading to a weaker chromophore–counter-ion interaction in line with the accumulation of the red-shifted N-intermediate in the steady state (Fig. 3F).
At 293 K, a negative band arises at 1753 cm−1 in the WT spectrum that is not observed in the spectrum of any mutant. An assignment of this band is nevertheless possible based on the spectra at 260 K. In these spectra, this vibration of the WT is slightly altered in E130Q, E163T, and D293N, but D196N is the only mutant in which it is not observed (Fig. 4B, inset column). Thus, we assign this band to a vibration of Asp196, reflecting deprotonation of this residue. Interestingly, this band is not D2O-sensitive, indicating that Asp196 is buried within the receptor and not accessible to bulk water, similar to observations made for C1C2 (36). The absence of this band in any mutant at 293 K indicates that Asp196 is already reprotonated in the steady state of the mutants at this temperature, which is in agreement with faster photocycle progression of the mutants compared with WT (Fig. 3C).
M-state-related proton transfer
Although proton transfer reactions involving Glu130, the counter-ion complex, and Asp196 were shown to occur in the photocycle of ReaChR, it is still unclear in which photocycle intermediate(s) these transfers actually occur. Accordingly, we investigated proton transfers related to M-state formation and decay. The slow-cycling DC pair mutant C168S (C128S in CrChR2) is suitable to investigate proton transfer reactions in the M-state because it highly accumulates M upon illumination (60 s, 530 nm) in the steady state at 293 K (Fig. 5A, inset). In the FTIR difference spectra of C168S in H2O (orange) and D2O (magenta) reflecting D → M transition (Fig. 5A), the positive band at 1738 cm−1, which was assigned to the protonation of Ci2 (Fig. 4B), is split into two positive bands arising at 1742 and 1729 cm−1, similar to the E130Q mutant (Fig. 4B). This observation can again be explained by an overlap of a single positive counter-ion band with an additional negative band arising in the M-state. The negative band at 1721 cm−1 assigned to deprotonation of Glu130 is downshifted to 1711 cm−1 in D2O, similar to WT. This indicates that Glu130 deprotonates at the latest during M-formation. An early deprotonation of the homologous residue Glu90 was observed during the photocycle of CrChR2 (21). Asp196 is not deprotonated in M because the spectra lack the negative band at 1753 cm−1.
Figure 5.

M-state-related proton transfer. A, C=O stretching region of the slow-cycling C168S mutant at pH 7.4 in H2O (orange) and D2O (magenta), respectively. Under continuous illumination, C168S accumulates M at 293 K (inset, UV-visible difference spectrum (light-dark) after illumination (60 s, 530 nm)). The presence of the positive bands at 1742 and 1729 cm−1 and the negative band at 1721 cm−1 indicates that proton transfer processes of Glu130 and the counter-ions occur before M-state decay. B, application of an illumination protocol with alternating green and UV light to ReaChR WT at 293 K. Spectral and kinetic components obtained by SVD and rotational analysis are shown. The first spectral component represents the formation of the steady state, mainly the O-state. The second spectral component strictly follows the light protocol as reflected by the corresponding kinetics. Thus, it represents molecular rearrangements occurring with transition from the steady state to the M-state. Presence of the 1766(+)/1756(−) cm−1 difference band implies that Asp196 undergoes a hydrogen bond change with formation and decay of M. C, C=O stretching region of the steady state of the wild type at 293 K and its two decay components revealed by SVD and a rotational analysis and fitted by a global fit using a biexponential function (R2 > 0.99). Time constants are indicated below the respective components. Dark-state recovery is accomplished by a fast and a slow transition that occur in parallel: the fast decay component mainly represents M → D transition, which is inferred from its similarity to the second component in B, whereas the slow decay component represents O → D transition.
Ion conductance of channelrhodopsins can be rapidly switched on and off with alternating illumination of suitable wavelengths as shown in electrical studies (44, 45). We applied a similar illumination protocol with alternating green (520 nm) and UV (390 nm) light to the wild type. To estimate the independent spectral components contributing to the signal change, we evaluated the results using a combination of singular value decomposition (SVD) and a rotation procedure (46, 47) as performed for CrChR2-C128T (15) (Fig. 5B and see “Experimental procedures” for details). The first component represents formation of the late intermediates, mainly O, which can be derived from similarity of this spectral component to the steady-state spectrum at 293 K (Fig. 4B, gray filled curves). This component displays a negative band at 1753 cm−1, indicating deprotonation of Asp196. It arises with green illumination, remains throughout the whole illumination period, and slowly decays after the end of the illumination. A further component strictly follows the illumination protocol. Green illumination induces a steady state including M, which can be depopulated by UV light, so that formation and decay of M, which contains contributions of both M1 and M2 (see Fig. 3, H–J), can be directly observed. This component involves a difference band at 1766(+)/1756(−) cm−1, which demonstrates a change of hydrogen bonding of still protonated Asp196 coincident with formation and decay of M, whereas the negative band in the first component confirms later deprotonation of Asp196, i.e. after formation of the N-state. Thus, it can be excluded that Asp196 serves as proton donor for the RSB during M decay in contrast to the homologue Asp156 in CrChR2 (22). Another likely candidate for reprotonation of the RSB is Ci2, which receives the RSBH+ proton during M formation. Indeed, the component following the illumination protocol comprises a band pattern with two maxima at 1744 and 1731 cm−1 that was already assigned to Ci2 protonation (see Fig. 5A). This shows that Ci2 receives a proton during M formation and releases it during light-induced decay. In contrast, the band at 1737 cm−1 in the first component shows that Ci2 remains partially protonated in the O state. This finding shows that a photocycle branching takes place after M formation, involving a light-induced shortcut from M to D, as observed earlier in ReaChR (11).
To elucidate the role of the observed proton transfers during the thermal M → D and O → D transitions, respectively, we investigated the decay process starting from the steady state at 293 K (Fig. 5C). As revealed by SVD and rotation procedure, two components, which were fitted by a global analysis procedure using a biexponential function (31), contributed to the decay process. The first decay component almost mirrors the component following alternating green and UV illumination (Fig. 5B) and thus reflects the direct thermal M → D transition involving Ci2 deprotonation. The second component, which represents the O → D transition as can be inferred from its slow kinetics (τ ≈ 71 s; see Fig. 3C), comprises a band at 1738 cm−1 that implies late deprotonation of Ci2. These findings support the above mentioned concept that the photocycle branches after the M-state, involving 1) a direct M → D transition where the RSB is presumably reprotonated by Ci2 and 2) a late O → D transition. In the latter case, both Asp196 and Ci2 are excluded as proton donors to the RSB.
Discussion
In this study, the electrical properties and the photocycle dynamics of ReaChR WT were compared with properties of selected mutants by a combined spectroscopic and electrophysiological approach. In the following, the key proton transfers are characterized with respect to their functional relevance (Fig. 6).
Figure 6.
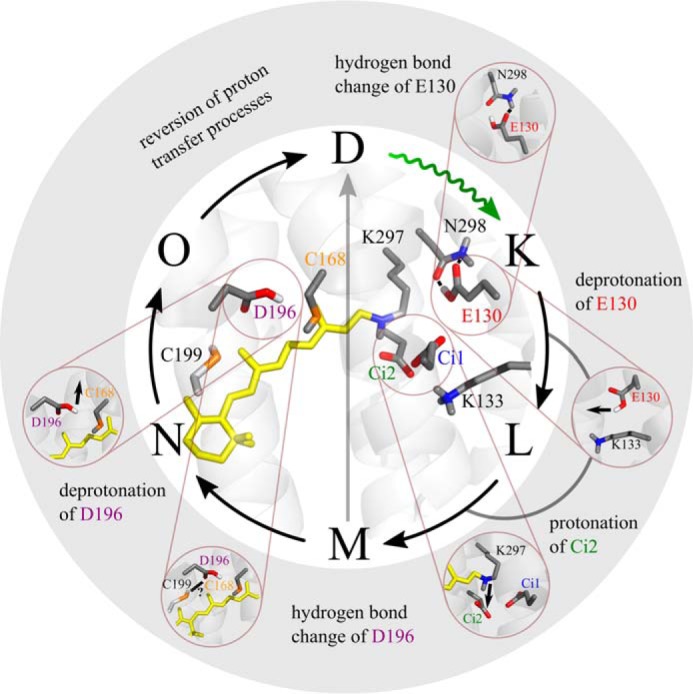
Summary of proton transfer processes in ReaChR. 1) During D → K transition, Glu130 experiences a hydrogen bond change and then 2) deprotonates at the latest in the M state. 3) Asp293 receives a proton from the RSBH+ during M formation. 4) M decays via two pathways: (i) direct transition to D and (ii) transition to N. 5) Asp196 undergoes a hydrogen bond change during M → N transition, and 6) Asp196 deprotonates after M decay, presumably in the N → O transition. The proton transfer reactions are reversed in the O → D and M → D transitions, respectively. The structure was obtained by homology modeling with SWISS-MODEL using the crystal structure of C1C2 (Protein Data Bank code 3ug9) (25) as a template. Residues discussed in this work are highlighted.
Glu130 is a determinant for ion selectivity
Our observation that replacement of Glu130 by Gln reduces H+ conductance in favor of Na+ (Fig. 2D) raised questions about the dynamics of this residue during the photocycle, which were then addressed by spectroscopic measurements. We show that Glu130 undergoes a hydrogen bond change in the D → K transition and deprotonates in the K → M transition (Figs. 4B and 5A). This finding agrees well with the Glu90-helix 2-tilt (EHT) model, that we postulated for the homologue Glu90 of CrChR2 based on quantum mechanics/molecular mechanics calculations (21). The low-temperature measurements on ReaChR allowed us now for the first time to observe hydrogen bond change and deprotonation as separate events and thus provide experimental evidence for validity of the EHT model to ChRs other than CrChR2.
The EHT model suggests that in CrChR2 one of the two hydrogen bonds between Glu90 and Asn258 is disrupted after retinal isomerization. Subsequently, Glu90 moves outward and deprotonates, eventually leading to the preformation of the conducting pore. Based on the presented experiments and because of the far reaching sequence homologies of the Glu130 environment in ReaChR as compared with the environment of Glu90 in CrChR2, we propose a similar scenario for ReaChR. Accordingly, the observed upshift of the Glu130 band by 8 cm−1 in the K spectrum (Fig. 4A) reflects weakening of hydrogen bonding of Glu130 and is in agreement with a reduction of the number of hydrogen bonds between Glu130 and Asn298 from two (weak hydrogen bonds) to one (48). Subsequently, Glu130 deprotonates in the K → M transition, moves outward, and presumably forms a hydrogen bond to Lys133 (Lys93 in CrChR2) as it does in CrChR2 (21).
Now we focus on how the change of the protonation state of Glu130 is linked to ion selectivity. We showed that, upon light activation of ReaChR, Glu130 deprotonates, and therefore a negative charge in the central gate is created that persists in the conducting M2- and N-states (11). The E130Q mutation has two consequences. First, in the dark state, no hydrogen bond to Asn298 is formed, and Gln130 is already oriented outward as proposed for the chloride-conducting ChRs “ChloC” and derivatives (27, 49). This would explain the about 10-fold accelerated M formation in the E130Q mutant (Fig. 3D). Outward orientation of Gln130 and thus a preactive conformation might be stabilized by a hydrogen bond of its γ-NH2 group to Ci1 as estimated from a structural model (50) and molecular dynamics calculations on CrChR2-E90Q (20). The second consequence of the ReaChR-E130Q mutation is neutralization of the negative charge normally present in the conducting state(s). It was shown previously that mutation of the homologous Glu90 in CrChR2 alters Na+ selectivity in a positive or negative sense depending on the charge of the introduced residue (20, 28). Exchange for aspartate hardly affects Na+ selectivity as the charge remains unchanged. Neutralization of Glu90 by glutamine or alanine increases Na+ selectivity, whereas lysine or arginine eliminates the Na+ conductance in favor of Cl− conductance (27). Accordingly, increased Na+ selectivity in the E130Q mutant of ReaChR as in E90Q appears to be predominantly achieved by altered electrostatics within the ion pore rather than changes of pore geometry near the restriction site.
Asp196 is a determinant of dark adaptation
The two-photocycle model, comprising two closed (D and D′) and two conducting states, was proposed to explain the photocurrent kinetics and amplitudes of the transient and photostationary photocurrents (1, 12–14, 51). This model was supported by spectroscopic data (52) and chromophore isomer analysis and extended with the finding that during one photocycle the retinal remains in 15-anti conformation and during the other the retinal remains in 15-syn conformation (11, 15, 16). Similarly, in bacteriorhodopsin, two different dark states exist, BR548 with 13-cis,15-syn-retinal and BR568 with 13-trans,15-anti-retinal (32, 53). Based on the observed differences between IDA and DAapp (Fig. 3A), we assume that this applies to ReaChR as well. The photocycle of ReaChR is anticipated to start from IDA with 100% 13-trans,15-anti-retinal (D) by both trans/cis and, with lower efficiency, C13=C14 and C15=N double isomerization of the retinal (16). Incomplete thermal back-reaction leads to an altered syn/anti ratio in DAapp as compared with IDA (see Fig. 3A). This effect is most prominent in D196N (see Fig. 3, A and B). The observation that the D196N mutation abolishes the transient current It (Fig. 2A) can thus be explained by the assumption that the 15-anti branch, which is reduced in D196N, causes the peak current, whereas both the 15-anti and 15-syn branches contribute to the stationary current. Accordingly, we provide further evidence that the two open states observed in electrophysiological experiments represent the conducting states of the respective photocycle branches as described earlier (15).
For the question how Asp196 affects the retinal syn/anti ratio, especially because it is located at more than 9-Å distance from the RSB (Fig. 6), the protonation state of Asp196 is decisive: Asp196 deprotonates during N → O transition, rendering a negative charge close to the chromophore. The electrostatic interaction of deprotonated Asp196 with the retinal polyene chain facilitates the back-reaction from 15-syn- to 15-anti-retinal during dark adaptation. Due to the D196N mutation, this negative charge at the chromophore is neutralized, and the syn → anti reaction is impaired. Moreover, the DC pair residues or other amino acid pairs (DT in bacteriorhodopsin) at this position in other microbial rhodopsins form a hydrogen bond between helix 3 and helix 4 that is essential for the protein stability and the retinal-binding pocket in particular. Destruction of this bond in many cases destabilizes the retinal-binding pocket and the protein stability in general (54), resulting in a slowdown of both M formation and decay, and might also alter the syn/anti ratio accordingly.
It remains unclear whether the influence of Asp196 on the syn/anti equilibrium is applicable to CrChR2 as well: although mutations of Asp156 abolish the peak current of CrChR2 wild type similarly to D196N in ReaChR (23, 27), the retinal fingerprint region in FTIR spectra does not imply a significant alteration of the retinal isomer composition (22). The environment of Asp196 is different from its homologues in CrChR2 or C1C2, which is reflected by the higher frequency of the C=O stretch vibration (1753 cm−1) as compared with the respective vibrations at 1738–1737 cm−1 (22, 55). This could, among other things, be due to the adjacent Cys199, which is exchanged for a threonine in these blue-absorbing ChRs.
Proton transfers in the RSBH+ counter-ion complex coincide with photocycle branching
The counter-ion complex of the RSBH+ is formed by Ci1 (Glu163), Ci2 (Asp293), and presumably water molecules (56–58). Ci2 receives the RSB proton during M-state formation (Fig. 3G), which is in agreement with the findings for Ci2 in CrChR2 (22) but different to CaChR1 where Ci1 was proposed as proton acceptor (59). Although in both CrChR2 and ReaChR the counter-ion complex interacts with a lysine (Lys133 in ReaChR) that is exchanged for a phenylalanine in CaChR1, the frequency of the Ci2 stretch vibration (1738 cm−1) is more similar to that of CaChR1 (1753–1740 cm−1) (59) as compared with CrChR2 (1695 cm−1) (22).
Reprotonation of the RSB coincides to M-state decay. As shown by UV/visible and FTIR spectroscopy, the M-state decays thermally by two distinct parallel paths: a major fraction undergoes an M → N → O transition (main path), eventually leading back to D, and a minor fraction directly reacts back to the dark state in a shortcut. A similar bifurcation of the late photocycle was stated for CrChR2 with the P520 intermediate (homologue to N) as the branching point (39) or for its slow-cycling mutants (P390 → D470) (23). Similarly to the proposed P520 → D470 shortcut in CrChR2, the actual molecular determinant of this branching process as well as its functional relevance remains elusive in ReaChR. Nevertheless, the branching process is correlated with the protonation state of Ci2: in the side path, Ci2 deprotonates during dark-state recovery and might thus serve as proton donor for reprotonation of the RSB. In the main path, Ci2 deprotonates in the O → D transition (Fig. 5C), which excludes Ci2 as proton donor for reprotonation of the RSB. The DC pair residue Asp196 is excluded as well as proton donor for the RSB (see Figs. 3F and 5B) because RSB reprotonation occurs prior to deprotonation of Asp196. This is in contrast to its homologue Asp156 that was proposed to be the proton donor in CrChR2 (22). As no further carboxylic amino acids could be identified as proton donors to the RSB, the most likely proton source is bulk water, which is abundant during the conducting state.
Conclusion
The proton dynamics observed in the early stage of the ReaChR photocycle, i.e. until formation of the M intermediate, are similar to the blue-absorbing CrChR2, involving (i) hydrogen bond change and subsequent deprotonation of Glu130 and (ii) proton transfer from the RSBH+ to Ci2 during M formation. Neutralization of Glu130 enhances Na+ conductivity by altered electrostatics of the central gate.
After formation of the M-state, important mechanistic differences between ReaChR and CrChR2 become evident: (i) the photocycle branches with M-state decay, (ii) Asp196 is excluded as proton donor for RSB reprotonation, and (iii) deprotonation of Asp196 facilitates thermal syn → anti isomerization of the chromophore during dark-state recovery. These differences are explained by distinct structural differences between ReaChR and blue-absorbing ChRs at the active site and Asp196 environment, respectively. Asp196 represents an interesting target for elucidating the actual functional relevance of 15-syn-retinal species in ChRs that is poorly understood so far.
Experimental procedures
Molecular biology
For spectroscopic experiments, DNA encoding ReaChR (GenBankTM accession number KF448069.1; amino acids 1–345) with a C-terminal 1D4 epitope (TETSQVAPA) was inserted into the pMT4 expression vector (60) between EcoRI and NotI (Thermo Fischer Scientific, Waltham, MA), whereas for patch clamp recordings ReaChR was C-terminally fused to the mCerulean3 fluorophore (61) and cloned into the pEGFP-N1 vector between HindIII and XbaI (Thermo Fischer Scientific). The mutations E130Q, E163T, C168S, D196N, and D293N were created via site-directed mutagenesis (QuikChange, Agilent Technologies, Santa Clara, CA).
Electrical measurements
HEK293 cells were seeded (0.75 × 105 cells/ml) in Petri dishes on poly-d-lysine-coated glass coverslips and supplemented with 1 μm all-trans-retinal. One day later, cells were transiently transfected with DNA encoding the respective construct using FuGENE HD (Promega, Madison, WI). Recordings took place 48 h after transfection. Signals were amplified and digitized using an AxoPatch 200B and a DigiData 1440 (Molecular Devices, Sunnyvale, CA). Light for activation was provided by a Polychrome V (TILL Photonics, Planegg, Germany) coupled to the optical path of an inverted IX-70 microscope (Olympus, Shinjuku, Japan) and controlled with a programmable shutter (Vincent Associates, Rochester, NY). Standard buffer conditions were as follows: internal 140 mm NaCl, 2 mm MgCl2, 2 mm CaCl2, 1 mm KCl, 1 mm CsCl, 10 mm HEPES, 10 mm EGTA, pH 7.2, and external 140 mm NaCl, 2 mm MgCl2, 2 mm CaCl2, 1 mm KCl, 1 mm CsCl, 10 mm HEPES. If not indicated otherwise, data were recorded under that conditions in the whole-cell configuration at a holding potential of −60 mV. Raw traces were baseline-corrected and filtered with a low-pass Gaussian filter (1000-Hz cutoff). Data are displayed as mean ± S.E.
For selectivity measurements, the patch was established at standard conditions, and then extracellular solution was exchanged (five times) for 140 mm NMGCl, 2 mm MgCl2, 2 mm CaCl2, 1 mm KCl, 1 mm CsCl, 10 mm HEPES, pH 7.2 (NMG, pH 7.2); then 140 mm NMGCl, 2 mm MgCl2, 2 mm CaCl2, 1 mm KCl, 1 mm CsCl, 10 mm Tris, pH 9.0 (NMG, pH 9.0); and finally 140 mm NaCl, 2 mm MgCl2, 2 mm CaCl2, 1 mm KCl, 1 mm CsCl, 10 mm Tris, pH 9.0 (NaCl, pH 9.0). Reversal potentials (Erev) were calculated from the respective stationary currents via linear interpolation between the two nearest data points and corrected for the respective liquid junction potential (23 °C) except for NMG, pH 9.0, where the values were obtained through linear extrapolation from the currents recorded at −60 and −40 mV.
Expression and purification of ReaChR
Expression in HEK293T and purification via immunoaffinity chromatography (1D4) were performed as described before (11).
UV-visible spectroscopy
Absorption spectra were recorded by a Cary 50 Bio spectrophotometer (Varian Inc., Palo Alto, CA) at 22 °C. IDA spectra represent spectral properties of protein purified under safe light (>600 nm), but DAapp spectra were measured after preillumination (530 nm, 60 s, 1 × 1020 photons m−2 s−1; Luxeon light-emitting diode, Phillips, Amsterdam, Netherlands) and subsequent recovery phase in the dark (10 min). The difference spectrum of ReaChR-C168S was achieved upon prolonged illumination (60 s, 530 nm). Flash photolysis experiments and data processing were performed as described elsewhere (49). In short, a tunable optical parametric oscillator (Rainbow, MagicPrismTM, Opotek Inc., Carlsbad, CA) was pumped by the third harmonic (355 nm) of a neodymium-doped yttrium aluminium garnet laser (Rainbow, BrilliantB, Quantel, Les Ulis, France). Excitation wavelength was adjusted manually by a micrometer drive and calibrated with an Andor iStar intensified charge-coupled device camera (Andor Technology Ltd., Belfast, Ireland). Absorption changes were probed by a 150-watt xenon short-arc XBO lamp (Osram, München, Germany) and detected by the intensified charge-coupled device camera. Samples with an optical density of A280 = 1.0 were excited with green flashes (10 ns, 530 nm, 5 mJ/flash). Global analysis is based on a sequential model with four or five spectral components and was performed with Glotaran (62, 63).
FTIR measurements
FTIR samples in Dulbecco's PBS, pH 7.4, and 0.03% (w/v) n-dodecyl β-d-maltopyranoside were prepared on a BaF2 window by repeated drying under a nitrogen stream and subsequent rehydration. After preparation, the sample was sealed with a second BaF2 window. Until use, the samples were stored at −40 °C for cryostatic samples and at 4 °C for room temperature samples. For deuteration experiments, pD was adjusted to 7.8. Samples were deuterated by repeated buffer exchange using Centricon centrifugal filter units (GE Healthcare) and subsequent equilibration for 3 days minimum.
Samples were illuminated with light-emitting diodes (maximum emission wavelengths of ∼530 nm and ∼390 nm for alternating illumination experiments). For cryostatic measurements, the cryostat DN (Oxford Instruments, Abingdon, UK) was used. Samples were equilibrated at the respective temperature for at least 45 min. After measurement, the sample was heated up again to a minimum 20 °C to allow relaxation.
FTIR measurements were performed using an ifs66v/s FTIR spectrometer (Bruker Optics, Karlsruhe, Germany) with an LN2-cooled mercury cadmium telluride detector (Kolmar Technologies, Newburyport, MA). A 1850 cm−1 optical cutoff filter was used. Spectra were recorded with a 200-kHz sampling rate and a spectral resolution of 2 cm−1. For each data set, >2500 spectra of the samples were collected and averaged in the dark and after illumination. At cryotemperatures, this procedure was performed for every sample at least twice (n ≥ 2), and measurements at 293 K were conducted at least 18 times (n ≥ 18) to exclude instabilities of pH and temperature that might affect the reproducibility of the data set.
The difference spectra were corrected for baseline drifts using a spline algorithm and the baseline correction mode implemented in the OPUS 6.5 software package (Bruker Optics). The “steady state” was defined by the steady state of the kinetics of the strongest absorption band in the amide I region (∼1660 cm−1). Results obtained from alternating illumination were evaluated by a combination of SVD and a rotation procedure to allow for the estimation of independent spectral components (15). Briefly, the first set of independent spectral components (b-spectra; Si) and corresponding kinetics components (Vi) were estimated by SVD. These data were then subjected to a rotation procedure based on the autocorrelation function of Vi to increase the signal content in a smaller number of vectors as described previously (47). Recovery kinetics were obtained by application of a method combining SVD with a rotation procedure and a global fitting approach using a multiexponential function, the latter supplying the time constants of the decay of the steady state (46).
Author contributions
J. C. D. K. performed FTIR spectroscopic measurements. B. S. K. expressed and purified protein and performed UV-visible spectroscopic measurements. C. G. conducted electrophysiological experiments. E. R. provided evaluation software and experimental expertise. E. R., P. H., and F. J. B. designed and interpreted experiments. J. C. D. K. and F. J. B. wrote the manuscript with further contributions from all authors. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Christina Schnick, Altina Klein, and Maila Reh for excellent technical assistance and Thomas P. Sakmar for providing 1D4 antibody.
This work was supported in part by the Deutsche Forschungsgemeinschaft via Sonderforschungsbereich 1078 Projects B2 (to P. H.) and B5 (to F. J. B.) and Cluster of Excellence 314 “Unifying Concepts in Catalysis” Project E4/D4 (to P. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- ChR
- channelrhodopsin
- C1C2
- chimera of CrChR1 and CrChR2 from C. reinhardtii
- CaChR1
- Chlamydomonas augustae channelrhodopsin-1
- Ci1
- counter-ion 1 (Glu163 in ReaChR)
- Ci2
- counter-ion 2 (Asp293 in ReaChR)
- CrChR1
- C. reinhardtii channelrhodopsin-1
- CrChR2
- C. reinhardtii channelrhodopsin-2
- D
- 13-trans,15-anti-retinal
- D′
- 13-cis,15-syn-retinal
- DAapp
- apparent dark state
- IDA
- initial dark state
- NMG
- N-methyl-d-glucamine
- PsChR2
- P. subcordiformis channelrhodopsin-2
- ReaChR
- red-activatable channelrhodopsin
- RSB
- retinal Schiff base
- RSBH+
- protonated retinal Schiff base
- SVD
- singular value decomposition
- It
- transient photocurrent
- Is
- stationary photocurrent.
References
- 1. Schneider F., Gradmann D., and Hegemann P. (2013) Ion selectivity and competition in channelrhodopsins. Biophys. J. 105, 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nagel G., Ollig D., Fuhrmann M., Kateriya S., Musti A. M., Bamberg E., and Hegemann P. (2002) Channelrhodopsin-1: a light-gated proton channel in green algae. Science 296, 2395–2398 [DOI] [PubMed] [Google Scholar]
- 3. Nagel G., Szellas T., Huhn W., Kateriya S., Adeishvili N., Berthold P., Ollig D., Hegemann P., and Bamberg E. (2003) Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. U.S.A. 100, 13940–13945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sineshchekov O. A., Jung K.-H., and Spudich J. L. (2002) Two rhodopsins mediate phototaxis to low- and high-intensity light in Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. U.S.A. 99, 8689–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berthold P., Tsunoda S. P., Ernst O. P., Mages W., Gradmann D., and Hegemann P. (2008) Channelrhodopsin-1 initiates phototaxis and photophobic responses in Chlamydomonas by immediate light-induced depolarization. Plant Cell 20, 1665–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harz H., and Hegemann P. (1991) Rhodopsin-regulated calcium currents in Chlamydomonas. Nature 351, 489–491 [Google Scholar]
- 7. Boyden E. S., Zhang F., Bamberg E., Nagel G., and Deisseroth K. (2005) Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 8, 1263–1268 [DOI] [PubMed] [Google Scholar]
- 8. Li X., Gutierrez D. V., Hanson M. G., Han J., Mark M. D., Chiel H., Hegemann P., Landmesser L. T., and Herlitze S. (2005) Fast noninvasive activation and inhibition of neural and network activity by vertebrate rhodopsin and green algae channelrhodopsin. Proc. Natl. Acad. Sci. U.S.A. 102, 17816–17821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin J. Y., Knutsen P. M., Muller A., Kleinfeld D., and Tsien R. Y. (2013) ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat. Neurosci. 16, 1499–150823995068 [Google Scholar]
- 10. Tromberg B. J., Shah N., Lanning R., Cerussi A., Espinoza J., Pham T., Svaasand L., and Butler J. (2000) Non-invasive in vivo characterization of breast tumors using photon migration spectroscopy. Neoplasia 2, 26–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krause B. S., Grimm C., Kaufmann J. C., Schneider F., Sakmar T. P., Bartl F. J., and Hegemann P. (2017) Complex photochemistry within the green-absorbing channelrhodopsin ReaChR. Biophys. J. 112, 1166–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berndt A., Prigge M., Gradmann D., and Hegemann P. (2010) Two open states with progressive proton selectivities in the branched channelrhodopsin-2 photocycle. Biophys. J. 98, 753–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hegemann P., Ehlenbeck S., and Gradmann D. (2005) Multiple photocycles of channelrhodopsin. Biophys. J. 89, 3911–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nikolic K., Grossman N., Grubb M. S., Burrone J., Toumazou C., and Degenaar P. (2009) Photocycles of channelrhodopsin-2. Photochem. Photobiol. 85, 400–411 [DOI] [PubMed] [Google Scholar]
- 15. Ritter E., Piwowarski P., Hegemann P., and Bartl F. J. (2013) Light-dark adaptation of channelrhodopsin C128T mutant. J. Biol. Chem. 288, 10451–10458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bruun S., Stoeppler D., Keidel A., Kuhlmann U., Luck M., Diehl A., Geiger M.-A., Woodmansee D., Trauner D., Hegemann P., Oschkinat H., Hildebrandt P., and Stehfest K. (2015) Light-dark adaptation of channelrhodopsin involves photoconversion between the all-trans and 13-cis retinal isomers. Biochemistry 54, 5389–5400 [DOI] [PubMed] [Google Scholar]
- 17. Becker-Baldus J., Bamann C., Saxena K., Gustmann H., Brown L. J., Brown R. C., Reiter C., Bamberg E., Wachtveitl J., Schwalbe H., and Glaubitz C. (2015) Enlightening the photoactive site of channelrhodopsin-2 by DNP-enhanced solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 112, 9896–9901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nack M., Radu I., Bamann C., Bamberg E., and Heberle J. (2009) The retinal structure of channelrhodopsin-2 assessed by resonance Raman spectroscopy. FEBS Lett. 583, 3676–3680 [DOI] [PubMed] [Google Scholar]
- 19. Muders V., Kerruth S., Lórenz-Fonfría V. A., Bamann C., Heberle J., and Schlesinger R. (2014) Resonance Raman and FTIR spectroscopic characterization of the closed and open states of channelrhodopsin-1. FEBS Lett. 588, 2301–2306 [DOI] [PubMed] [Google Scholar]
- 20. Eisenhauer K., Kuhne J., Ritter E., Berndt A., Wolf S., Freier E., Bartl F., Hegemann P., and Gerwert K. (2012) In Channelrhodopsin-2 Glu-90 is crucial for ion selectivity and is deprotonated during the photocycle. J. Biol. Chem. 287, 6904–6911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuhne J., Eisenhauer K., Ritter E., Hegemann P., Gerwert K., and Bartl F. (2015) Early formation of the ion-conducting pore in channelrhodopsin-2. Angew. Chem. Int. Ed. Engl. 54, 4953–4957 [DOI] [PubMed] [Google Scholar]
- 22. Lórenz-Fonfría V. A., Resler T., Krause N., Nack M., Gossing M., Fischer von Mollard G., Bamann C., Bamberg E., Schlesinger R., and Heberle J. (2013) Transient protonation changes in channelrhodopsin-2 and their relevance to channel gating. Proc. Natl. Acad. Sci. U.S.A. 110, E1273–E1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bamann C., Gueta R., Kleinlogel S., Nagel G., and Bamberg E. (2010) Structural guidance of the photocycle of channelrhodopsin-2 by an interhelical hydrogen bond. Biochemistry 49, 267–278 [DOI] [PubMed] [Google Scholar]
- 24. Gunaydin L. A., Yizhar O., Berndt A., Sohal V. S., Deisseroth K., and Hegemann P. (2010) Ultrafast optogenetic control. Nat. Neurosci. 13, 387–392 [DOI] [PubMed] [Google Scholar]
- 25. Kato H. E., Zhang F., Yizhar O., Ramakrishnan C., Nishizawa T., Hirata K., Ito J., Aita Y., Tsukazaki T., Hayashi S., Hegemann P., Maturana A. D., Ishitani R., Deisseroth K., and Nureki O. (2012) Crystal structure of the channelrhodopsin light-gated cation channel. Nature 482, 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sugiyama Y., Wang H., Hikima T., Sato M., Kuroda J., Takahashi T., Ishizuka T., and Yawo H. (2009) Photocurrent attenuation by a single polar-to-nonpolar point mutation of channelrhodopsin-2. Photochem. Photobiol. Sci. 8, 328–336 [DOI] [PubMed] [Google Scholar]
- 27. Wietek J., Wiegert J. S., Adeishvili N., Schneider F., Watanabe H., Tsunoda S. P., Vogt A., Elstner M., Oertner T. G., and Hegemann P. (2014) Conversion of channelrhodopsin into a light-gated chloride channel. Science 344, 409–412 [DOI] [PubMed] [Google Scholar]
- 28. Ruffert K., Himmel B., Lall D., Bamann C., Bamberg E., Betz H., and Eulenburg V. (2011) Glutamate residue 90 in the predicted transmembrane domain 2 is crucial for cation flux through channelrhodopsin 2. Biochem. Biophys. Res. Commun. 410, 737–743 [DOI] [PubMed] [Google Scholar]
- 29. Govorunova E. G., Sineshchekov O. A., Li H., Janz R., and Spudich J. L. (2013) Characterization of a highly efficient blue-shifted channelrhodopsin from the marine alga Platymonas subcordiformis. J. Biol. Chem. 288, 29911–29922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li H., Govorunova E. G., Sineshchekov O. A., and Spudich J. L. (2014) Role of a helix B lysine residue in the photoactive site in channelrhodopsins. Biophys. J. 106, 1607–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ritter E., Stehfest K., Berndt A., Hegemann P., and Bartl F. J. (2008) Monitoring light-induced structural changes of channelrhodopsin-2 by UV-visible and Fourier transform infrared spectroscopy. J. Biol. Chem. 283, 35033–35041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harbison G. S., Smith S. O., Pardoen J. A., Winkel C., Lugtenburg J., Herzfeld J., Mathies R., and Griffin R. G. (1984) Dark-adapted bacteriorhodopsin contains 13-cis,15-syn and all-trans,15-anti retinal Schiff bases. Proc. Natl. Acad. Sci. U.S.A. 81, 1706–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Farrar M. R., Lakshmi K. V., Smith S. O., Brown R. S., Raap J., Lugtenburg J., Griffin R. G., and Herzfeld J. (1993) Solid state NMR study of [ϵ-13C]Lys-bacteriorhodopsin: Schiff base photoisomerization. Biophys. J. 65, 310–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oshima K., Shigeta A., Makino Y., Kawamura I., Okitsu T., Wada A., Tuzi S., Iwasa T., and Naito A. (2015) Characterization of photo-intermediates in the photo-reaction pathways of a bacteriorhodopsin Y185F mutant using in situ photo-irradiation solid-state NMR spectroscopy. Photochem. Photobiol. Sci. 14, 1694–1702 [DOI] [PubMed] [Google Scholar]
- 35. Aton B., Doukas A. G., Callender R. H., Becher B., and Ebrey T. G. (1977) Resonance Raman studies of the purple membrane. Biochemistry 16, 2995–2999 [DOI] [PubMed] [Google Scholar]
- 36. Ito S., Kato H. E., Taniguchi R., Iwata T., Nureki O., and Kandori H. (2014) Water-containing hydrogen-bonding network in the active center of channelrhodopsin. J. Am. Chem. Soc. 136, 3475–3482 [DOI] [PubMed] [Google Scholar]
- 37. Smith S. O., Lugtenburg J., and Mathies R. A. (1985) Determination of retinal chromophore structure in bacteriorhodopsin with resonance Raman spectroscopy. J. Membr. Biol. 85, 95–109 [DOI] [PubMed] [Google Scholar]
- 38. Miranda M. R., Choi A. R., Shi L., Bezerra A. G. Jr., Jung K.-H., and Brown L. S. (2009) The photocycle and proton translocation pathway in a cyanobacterial ion-pumping rhodopsin. Biophys. J. 96, 1471–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lórenz-Fonfría V. A., Schultz B.-J., Resler T., Schlesinger R., Bamann C., Bamberg E., and Heberle J. (2015) Pre-gating conformational changes in the ChETA variant of channelrhodopsin-2 monitored by nanosecond IR spectroscopy. J. Am. Chem. Soc. 137, 1850–1861 [DOI] [PubMed] [Google Scholar]
- 40. Smith S. O., Pardoen J. A., Lugtenburg J., and Mathies R. A. (1987) Vibrational analysis of the 13-cis-retinal chromophore in dark-adapted bacteriorhodopsin. J. Phys. Chem. 91, 804–819 [Google Scholar]
- 41. Ogren J. I., Mamaev S., Russano D., Li H., Spudich J. L., and Rothschild K. J. (2014) Retinal chromophore structure and Schiff base interactions in red-shifted channelrhodopsin-1 from Chlamydomonas augustae. Biochemistry 53, 3961–3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kawanabe A., and Kandori H. (2009) Photoreactions and structural changes of Anabaena sensory rhodopsin. Sensors 9, 9741–9804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wand A., Gdor I., Zhu J., Sheves M., and Ruhman S. (2013) Shedding new light on retinal protein photochemistry. Annu. Rev. Phys. Chem. 64, 437–458 [DOI] [PubMed] [Google Scholar]
- 44. Bamann C., Kirsch T., Nagel G., and Bamberg E. (2008) Spectral characteristics of the photocycle of channelrhodopsin-2 and its implication for channel function. J. Mol. Biol. 375, 686–694 [DOI] [PubMed] [Google Scholar]
- 45. Berndt A., Yizhar O., Gunaydin L. A., Hegemann P., and Deisseroth K. (2009) Bi-stable neural state switches. Nat. Neurosci. 12, 229–234 [DOI] [PubMed] [Google Scholar]
- 46. Elgeti M., Ritter E., and Bartl F. J. (2008) New insights into light-induced deactivation of active rhodopsin by SVD and global analysis of time-resolved UV/Vis- and FTIR-data. Z. Phys. Chem. 222, 1117–1129 [Google Scholar]
- 47. Henry E. R., and Hofrichter J. (1992) Singular value decomposition: application to analysis of experimental data. Methods Enzymol. 210, 129–192 [Google Scholar]
- 48. Nie B., Stutzman J., and Xie A. (2005) A vibrational spectral maker for probing the hydrogen-bonding status of protonated Asp and Glu residues. Biophys. J. 88, 2833–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wietek J., Broser M., Krause B. S., and Hegemann P. (2016) Identification of a natural green light absorbing chloride conducting channelrhodopsin from Proteomonas sulcata. J. Biol. Chem. 291, 4121–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Szundi I., Bogomolni R., and Kliger D. S. (2015) Platymonas subcordiformis channelrhodopsin-2 (PsChR2) function. II. Relationship of the photochemical reaction cycle to channel currents. J. Biol. Chem. 290, 16585–16594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Szundi I., Li H., Chen E., Bogomolni R., Spudich J. L., and Kliger D. S. (2015) Platymonas subcordiformis channelrhodopsin-2 function. I. The photochemical reaction cycle. J. Biol. Chem. 290, 16573–16584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smith S. O., Myers A. B., Pardoen J. A., Winkel C., Mulder P. P., Lugtenburg J., and Mathies R. (1984) Determination of retinal Schiff base configuration in bacteriorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 81, 2055–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Joh N. H., Min A., Faham S., Whitelegge J. P., Yang D., Woods V. L., and Bowie J. U. (2008) Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature 453, 1266–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Inaguma A., Tsukamoto H., Kato H. E., Kimura T., Ishizuka T., Oishi S., Yawo H., Nureki O., and Furutani Y. (2015) Chimeras of channelrhodopsin-1 and-2 from Chlamydomonas reinhardtii exhibit distinctive light-induced structural changes from channelrhodopsin-2. J. Biol. Chem. 290, 11623–11634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Watanabe H. C., Welke K., Schneider F., Tsunoda S., Zhang F., Deisseroth K., Hegemann P., and Elstner M. (2012) Structural model of channelrhodopsin. J. Biol. Chem. 287, 7456–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Watanabe H. C., Welke K., Sindhikara D. J., Hegemann P., and Elstner M. (2013) Towards an understanding of channelrhodopsin function: simulations lead to novel insights of the channel mechanism. J. Mol. Biol. 425, 1795–1814 [DOI] [PubMed] [Google Scholar]
- 58. Guo Y., Beyle F. E., Bold B. M., Watanabe H. C., Koslowski A., Thiel W., Hegemann P., Marazzi M., and Elstner M. (2016) Active site structure and absorption spectrum of channelrhodopsin-2 wild-type and C128T mutant. Chem. Sci. 7, 3879–3891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ogren J. I., Yi A., Mamaev S., Li H., Spudich J. L., and Rothschild K. J. (2015) Proton transfers in a channelrhodopsin-1 studied by Fourier transform infrared (FTIR) difference spectroscopy and site-directed mutagenesis. J. Biol. Chem. 290, 12719–12730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Karnik S. S., Sakmar T. P., Chen H.-B., and Khorana H. G. (1988) Cysteine residues 110 and 187 are essential for the formation of correct structure in bovine rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 85, 8459–8463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Markwardt M. L., Kremers G.-J., Kraft C. A., Ray K., Cranfill P. J., Wilson K. A., Day R. N., Wachter R. M., Davidson M. W., and Rizzo M. A. (2011) An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS One 6, e17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Snellenburg J. J., Laptenok S. P., Seger R., Mullen K. M., and van Stokkum I. H. M. (2012) Glotaran: a Java-based graphical user interface for the R package TIMP. J. Stat. Softw. 49, 1–22 [Google Scholar]
- 63. Mullen K. M., and van Stokkum I. H. M. (2007) TIMP: an R package for modeling multi-way spectroscopic measurements. J. Stat. Softw. 18, 1–46 [Google Scholar]
- 64. Olsson M. H., Søndergaard C. R., Rostkowski M., and Jensen J. H. (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 [DOI] [PubMed] [Google Scholar]