

Abstract
Recently newer therapies have been designed to more specifically target rejection in an effort to improve efficacy and limit unwanted toxicity. Belatacept, a CD28-CD80/86 specific reagent is associated with superior patient survival and graft function than traditional therapy but its adoption as a mainstay immunosuppressive therapy has been tempered by increased rejection rates. It is essential that the underlying mechanisms associated with this rejection be elucidated before belatacept is more widely employed. To that end we designed a study in a non-human primate kidney transplant model where animals were treated with either a belatacept- or a tacrolimus-based immunosuppressive regimen. Interestingly we found that elevated pre-transplant frequencies of CD28+CD8+TEMRA cells are associated with rejection on belatacept but not tacrolimus treatment. Further analysis showed that the CD28+CD8+TEMRA cells rapidly lose CD28 expression after transplant in those animals that go on to reject with the allograft infiltrate being predominantly CD28−. These data suggest that CD28+ memory T cells may be resistant to belatacept, capable of further differentiation including loss of CD28 expression while maintaining effector function. The unique signaling requirements of CD28+ memory T cells provide opportunities for the development of targeted therapies, which may synergize with belatacept to prevent costimulation independent rejection.
Introduction
Solid organ transplantation has become the primary treatment for end-stage organ failure. Success over the last 30 years has largely been driven by the advent of increasingly potent immunosuppressants. In particular calcineurin inhibitors (CNIs) such as cyclosporine and tacrolimus were instrumental in reducing the incidence of early graft failure due to acute rejection. Despite these advances long-term transplant outcomes have remained largely unchanged over the past twenty years (1). Although this is likely multi-factorial, the non-immune side effects of CNIs, including nephrotoxicity, diabetes, hyperlipidemia and increased overall cardiovascular risk, play an important role in diminishing long-term outcomes (2–4). In 2011 the first non-CNI alternative, belatacept, was approved for use in kidney transplantation (5). Belatacept is a high affinity variant of the fusion protein CTLA4-Ig which specifically blocks CD28 mediated T cell costimulation, providing a more targeted and less toxic form of immunosuppression than CNIs (6). Kidney transplant patients treated with belatacept live longer and enjoy better renal function than those patients treated with cyclosporine (43% reduction in the risk of death or graft loss at 7 year follow-up) (7–10). Despite these improvements use of belatacept has to date been tepid mostly due to its association with more frequent and severe rejection episodes in a subset of patients (11). Costimulation blockade based strategies hold immense promise for improved long-term outcomes, but resistance to belatacept-based immunosuppression in a subset of patients demands further investigation into the underlying mechanisms of costimulation independence. Indeed, wider utilization of this more targeted, less toxic approach to transplant immunosuppression may hinge on the ability to phenotypically identify costimulation independent cell subsets and understand their unique signaling requirements (12). This knowledge will enhance both the clinical utility of belatacept as well as inform future strategies to optimize belatacept-based therapy.
Belatacept binds to CD80 and/or CD86, preventing the ligation of the CD28 costimulatory molecule expressed on the majority of T cells. The ability of cells to dispense with CD28 costimulation, potentially marked by CD28 loss, is one mechanism by which T cells may become belatacept resistant. Increases in CD28− cells are associated with advanced age and chronic inflammation (13 −14). More specifically, inflammatory cytokines such as TNF (15), IL-2 (16), and IL-15 (17) drive the loss of CD28. Antigen exposure and the development of terminally differentiated T cell memory is also marked by the down-regulation of CD28 (14, 18–19). Contextually, loss of CD28 or CD28 independence is part of a program of differentiation and cellular maturation marked by changes in receptor expression and cell functionality. Classic models of memory classification define CD4 and CD8 T cells by CCR7 and CD45RA or by CD28 and CD95 (20–22). Multi-parametric flow cytometry has revealed the phenotypic and functional heterogeneity of T cell subsets (23–27). These studies reveal functional differences between subsets defined by four or more phenotypic markers at a time, with a potentially critical transition marked by CD28 loss (28). Previous studies demonstrate CD28+ cells retain enhanced anti-viral capacity (22) and proliferative potential (29), whereas loss of CD28 is associated with increased cytotoxicity, reduced responsiveness to T cell activation via the TCR (29), and the development of a dependence on homeostatic cytokine signaling for survival and effector function (30–32). One study suggests that CD28−CD57+CD4+ T cells may be associated with increased risk of belatacept resistant rejection (33). How and when CD28 is lost following transplantation, and whether the loss of CD28 potentiates belatacept resistance remains formally untested.
We investigated the mechanisms that underlie belatacept resistance in a pre-clinical non-human primate model of kidney transplantation. Therapy was withdrawn day 140 post-transplantation, giving rise to two distinct study populations: belatacept resistant animals which experienced early acute rejection during belatacept therapy, and belatacept susceptible animals which demonstrated excellent graft function for the duration of belatacept treatment. Together with data from a separate clinical study of patients treated with belatacept (34), we observe that a critical threshold frequency of CD28+, not CD28−, memory T cells is associated with belatacept resistance. These CD28+ memory cells retain proliferative capacity and may eventually lose CD28 expression as they fully differentiate into cytotoxic effector T cells. These findings suggest that pre-transplant immunophenotyping using the frequency of CD8+CD28+TEMRA T cells may provide a strategy to identify individuals who are susceptible to belatacept therapy thereby reducing the risk of rejection.
Materials and Methods
Donor-recipient pair selection and kidney transplantation
All experiments described herein were performed in compliance with the principles set forth in The Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, DHHS). Outbred rhesus monkeys (Macaca mulatta) ranging between 3 and 5 years old were obtained from AlphaGenesis, Inc. (Yemassee, SC) and Yerkes National Primate Research Center (Lawrenceville, GA). Donor–recipient pairs were chosen to maximize genetic disparity at both MHC class I and class II alleles based on 454 deep sequencing analysis (University of Wisconsin, Madison, WI). Kidney transplantation was performed using standard microvascular techniques (35). Animals were heparinized (100 Units/kg) during organ procurement and implantation. Left native nephrectomy was performed at least 3 weeks prior to transplantation, and a completion right native nephrectomy was performed at the time of transplantation. All transplanted animals were monitored with daily clinical assessment and serial laboratory evaluations, including complete blood count and serum chemistry.
NHP experimental groups and immunomodulation
All animals received methylprednisolone (subcutaneous injection according to the following schedule, d0: 20 mg, d1: 16 mg, d2: 12 mg, d3: 8 mg, d4: 4 mg, d5–14: 3mg, d15–140: 1mg) and mycophenalate mofetil (30mg/kg bid, d0-d140) to recapitulate clinically relevant immunosuppression strategy. A subset (n=8/16) of belatacept treated animals received basiliximab induction therapy (0.3mg/kg d0, d4). All Tacrolimus treated animals received basiliximab induction (Figure 1). Belatacept therapy was discontinued at day 140 (d0: 10 mg/kg, d4: 15 mg/kg, d14-d56: 20 mg/kg bi-weekly, d56-d140: 20 mg/kg every 4 weeks). Tacrolimus levels were monitored weekly (8–12ng/ml d0-d56, 5–8ng/ml d57-d168, Supplemental Figure 4). Expanded analysis of pre-transplant immunophenotype of included animals treated with CD28 domain antibody (36) or CD154 domain antibody (37).
Figure 1. Kidney Transplant Treatment Schema.

All animals received methylprednisolone (subcutaneous injection according to the following schedule, d0: 20 mg, d1: 16 mg, d2: 12 mg, d3: 8 mg, d4: 4 mg, d5–14: 3mg, d15–140: 1mg) and mycophenalate mofetil (30mg/kg bid, d0-d140) to recapitulate clinically relevant immunosuppression strategy. A subset (n=8/16) of belatacept treated animals received basiliximab induction therapy (0.3mg/kg d0, d4). All Tacrolimus treated animals received basiliximab induction. Belatacept therapy was discontinued at day 140 (d0: 10 mg/kg, d4: 15 mg/kg, d14-d56: 20 mg/kg bi-weekly, d56-d140: 20 mg/kg every 4 weeks). Tacrolimus trough levels were monitored weekly (8–12ng/ml d0–56, 5–8ng/ml d57–168, Supplemental Figure 4).
Mixed Lymphocyte Reaction and Cell Sorting
PBMCs were isolated from unmanipulated rhesus monkeys and fluorescently sorted based on CD28, CD45RA and CCR7 expression. Responder cells were labeled with CTV (Invitrogen, C34557), and stimulators were labeled with CFSE (Invitrogen, C34554). 1×105 Responder cells were plated in 96 well-flat-bottom plates with 1×105 irradiated MHC-mismatched stimulators, and cultured for 5 days. Cells were cultured in RPMI 1640 (Corning cellgro, Manassas, VA) supplemented with 10% fetal bovine serum +/− 100ug/ml belatacept (Bristol Myers-Squibb, Princeton NJ).
Isolation of graft infiltrating lymphocytes
To isolate graft-infiltrating lymphocytes, rejected allografts were mechanically disrupted, filtered through 70 µM cell strainers, washed in PBS and then filtered again through 40 µM cell strainers. Cell suspensions were separated by Ficoll-Paque density gradient centrifugation (GE Healthcare Life Sciences, Pittsburgh, PA) to isolate mononuclear cells. Mononuclear cells were washed twice and counted prior to antibody staining.
Antibodies and flow cytometric analysis
Flow cytometric analysis was performed up to 3 times pre-transplant and serially post-transplant to characterize peripheral blood immune cell phenotypes. Total T cells and T cell subsets were quantified by complete blood cell count and flow cytometry. Fresh PBMCs were isolated by Ficoll density gradient centrifugation (BD Biosciences, Franklin Lakes, NJ). PBMCs were stained with the following mAbs: CD3 PacBlue, CD95 V450, CD3 Alexa 700, CD4 PerCP-Cy5.5, CD8 V500, CD28 PE-Cy7, CD25 PE-Cy7, IFNy PE-Cy7, CD28 APC, TNF APC, VLA-4 APC, CD11a PE, CD45RA FITC, CD40 FITC, CCR7 APC, CD20 APC (all BD Biosciences). PBMCs (1.5×106) were incubated with appropriately titered antibodies for 15min at 20°C and washed twice. Samples were acquired immediately on a BD LSR II multicolor flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (Tree Star, San Carlos, CA). For the stimulation assay, 1.5×106 PBMCs were cultured in RPMI 1640 (Corning cellgro, Manassas, VA) supplemented with 10% fetal bovine serum and stimulated with 10µM phorbol 12-myristate 13-acetate (PMA) and 200nM ionomycin (Sigma-Aldrich, St. Louis, MO), with 1ul/ml GolgiPlug protein transport inhibitor for 5 h, +/− IL-15 (10ng/mL). PBMCs were were processed with BD Cytofix/Cytoperm Plus kit (BD 555028) per the manufactures recommendation prior to data acquisition.
Statistics
Survival statistics were calculated using a log-rank test. T cell frequencies were compared using a parametric unpaired t-test. Decision tree analysis was performed with the programming language and statistical software R (v. 3.2.3), and utilized the “rpart” package. Data were analyzed using Prism 6 (GraphPad Software, La Jolla, CA). A two-tailed p-value of <0.05 was considered statistically significant.
Transcriptome Analysis
RNA was prepared from biopsies of transplanted kidneys (time of sacrifice), as well as from isolated graft infiltrating cells. The quality of the total RNA from samples were monitored by the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) and RNA quantity was measured with NanoDrop (NanoDrop Technologies, Inc. Wilmington, DE) following the manufacturer's instructions. 200ng of total RNA were amplified and labeled with 3' IVT Express Kit (Affymetrix, Santa Clara, CA). Labeled cRNA were hybridized on Affymetrix GeneChip Rhesus Macaque Genome Array (Cat.900657, Affymetrix, Santa Clara, CA). Scanned images were subjected to visual inspection and a chip quality report was generated by Expression console (Affymetrix). The image data was processed using the RMA to determine the specific hybridizing signal for each gene. All Chip data were loaded into BMS database (Bristol Myers Squibb, Princeton NJ) for further analysis. Gene expression data were also analyzed utilizing DAVID Bioinformatics Database as described (35–36).
Results
Transplant recipients segregate based on response to belatacept therapy
In order to investigate the mechanisms of rejection during belatacept treatment 24 rhesus macaques were randomized to receive either belatacept- or tacrolimus-based immunosuppression following life-sustaining kidney transplant (Figure 1). Animals continued to receive immunosuppressive therapy until day 140 at which time therapy was discontinued. We observed two distinct populations within each treatment group: belatacept “resistant” animals which experienced rejection and graft failure during treatment, and belatacept “susceptible” animals which maintained excellent graft function during administration of therapy (Figure 2a). Tacrolimus therapy similarly gave rise to resistant (n=2) and susceptible (n=6) animals (Figure 2b). Once all treatments were stopped the remaining “susceptible” animals eventually succumbed to immune mediated graft destruction in the absence of immunosuppression.
Figure 2. Survival and Therapeutic Resistance.

Belatacept and tacrolimus based immunosuppression gave rise to two distinct study populations: those animals which were “susceptible” to therapy (grey lines) and those which were “resistant” to treatment (black lines). Resistant animals experienced rejection and graft loss while treatment was ongoing (between day 0 and day 140). Susceptible animals experienced prolonged allograft survival for the entire duration of the therapy and only experienced rejection after withdrawal of therapy (after day 140). (a) Survival with belatacept therapy (n=6 “resistant’ animals and n=10 “susceptible” animals). (b) Survival with tacrolimus treatment (n=2 “resistant” animals and n=6 “susceptible” animals).
IL-2Rα blockade is utilized clinically as induction therapy and has demonstrated efficacy in combination with primary immunosuppressive agents such as calcineurin inhibitors or belatacept (5,37). There has been some concern that the addition of an antibody targeting CD25 may impact beneficial cell populations such as regulatory T cells (Tregs) and may negatively impact outcomes, particularly with newer strategies such as costimulatory blockade (38). The addition of basiliximab to a subset of the belatacept treated animals (n=8) did not have a significant impact on the rate of rejection (rejectors on therapy n=3/group) when compared to belatacept treated animals that did not receive basiliximab induction (n=3/group). Similarly there was no significant difference in survival between belatacept (MST=167 days), belatacept with basiliximab induction (MST=189.5 days), or tacrolimus (MST=183 days) based immunosuppression (Supplemental Figure 1). All animals demonstrated excellent renal function as measured by serum creatinine until just prior to allograft rejection, defined as two consecutive serum Cr values >5 (Supplemental Figure 1).
Elevated frequency of CD28+CD8+TEMRA is associated with costimulation-resistant rejection
An important question in the field of transplantation is the role of pre-transplant immune memory status in transplant recipients, particularly in the context of costimulatory blockade based immunosuppression. We and others have demonstrated elevated pre-existing immune memory, and more broadly, increasing prior pathogen exposure constitute a potent barrier to transplant tolerance in murine and non-human primate models (38–40). Other groups have found unique regulatory memory subsets in alternative models of transplantation (41). To assess whether pre-transplant memory immune phenotype could predict whether animals would respond to belatacept-based immunosuppression we examined peripheral blood samples from all recipients prior to kidney transplantation using multi-parameter flow cytometry. We then examined the association of the frequency of memory cell subsets and rejection. We found that belatacept-resistant animals, which rejected while on therapy, had higher baseline frequencies of CD28+CD95+CD8+ T cells prior to transplantation, compared to non-rejectors (Figure 3c, P=0.0402)., A similar trend was not observed in the tacrolimus treated animals (Figure 3d, P=0.2005). Interestingly, the pre-transplant frequency of CD28− memory T cells (CD28−CD95+ CD8+ T cells), which have been implicated in costimulation-resistant rejection (42), was not significantly different between therapy resistant or susceptible animals in any treatment group (Figure 3e–f).
Figure 3. Pre-Transplant Immune Phenotyping with CD28 and CD95.

Prior to transplantation, CD8 T cells were analyzed by CD28 and CD95 expression (top row: belatacept treated animals, bottom row: tacrolimus treated animals). Subsets are expressed as a frequency of total CD8 T cells as follows: (a & b) CD28+CD95 (c & d) CD28+CD95+ and (e & f) CD28CD95+. Mean values of pre-transplant samples from resistant animals (black circles) were compared to those from susceptible animals (grey circles). Only the difference in the frequency of CD28+CD95+CD8+ T cells between resistant and susceptible animals treated with belatacept were significantly different (Figure 3c, P=0.0402).
The use of CD28 and CD95 is a standard approach for identifying naïve (CD28+CD95−), central memory (CD28+CD95+) and effector/effector memory (CD28−CD95+) T cells when phenotyping subsets in primates (21–22, 25). For a more granular analysis of the heterogeneous and functionally distinct memory and effector populations within CD3+ T cell compartment, we included additional conventional memory markers CD45RA and CCR7 (19, 25–27). Utilizing a gating strategy involving these four memory markers (Supplemental Figure 6) revealed belatacept resistant rejection was highly associated with elevated pre-transplant frequencies of CD28+CD95+CD45RA+CCR7− CD8 T cells, so-called CD28+ CD8+ TEMRA (Figure 4c–d, P<0.0001). The pre-transplant frequencies of CD28+ CD8+ TEMRA were significantly elevated in belatacept resistant animals irrespective of whether they received basiliximab induction (Supplemental Figure 2c–f). In contrast tacrolimus resistant animals did not exhibit similarly elevated pre-transplant frequencies of CD28+ CD8+ TEMRA (Figure 4a–b, P=0.2060).
Figure 4. Pre-transplant Multi-parametric Immune Phenotyping of CD8 T cells.

Pre-transplant immunophenotyping of therapy resistant (black circles) versus therapy susceptible animals (grey circles), with corresponding survival curves. (a) Tacrolimus treated animals demonstrate no significant difference in pre-transplant CD28+CD95+CD45RA+CCR7 cells as a frequency of total CD8+ T cells between therapy susceptible (4a grey circles) and therapy resistant animals (4a black circles, 4b corresponding survival curve). (c) Elevated CD28+CD95+CD45RA+CCR7 (CD28+TEMRA) as a frequency of total CD8+ T cells in pre-transplant peripheral blood samples discriminates belatacept resistant animals (black circles) from those susceptible to treatment (grey circles) with (d) corresponding survival curve (P<0.0001). (e) In an expanded cohort of animals treated with costimulatory blockade reagents (either CD28 directed or CD154 directed blockade) we observed a similar pre-transplant immunophenotype between resistant (black circles) and susceptible animals (grey circles), (f) corresponding aggregate survival curve. (g) Decision tree analysis determined that a cut-off value of 3.065% CD28+ CD8+ TEMRA segregated animals who would go on to experience costimulation resistant rejection from those animals susceptible to costimulation blockade therapy (87.5% sensitivity, 95.23% specificity, 93.33% PPV and 90.09% NPV).
In order to assess the generalizability of this biomarker to predict costimulation blockade resistant rejection beyond belatacept treatment we examined additional animals that had been treated with various costimulation blockade reagents that target either the CD28-CD80/86 or CD40-CD154 pathways. Similar to the belatacept-treated animals we saw a clear distinction between “susceptible” and “resistant” animals using the pre-transplant level of CD28+CD8+TEMRA (Figure 4e–f, n=37 P<0.0001). We next performed a decision tree analysis on this same larger cohort utilizing all CD4 and CD8 T cell subsets, including memory subsets such as CD4+ and CD8+, TEM or TEMRA, accounting for CD28 expression. We found that a frequency of greater than 3% CD28+CD95+CD45RA+CCR7− (CD28+TEMRA) of total CD8+ T cells provided the strongest predictor of costimulation blockade resistant rejection in our model with a 87.5% sensitivitd a 95.2% specificity (Figure 4g). For this model the positive predictive value was 93.3% and the negative predictive value was 90.1% (Figure 4g). Tacrolimus treated animals did not similarly segregate based on baseline frequencies of memory T cell subsets (Figure 4a–b), and thus we limited our decision tree analysis to the belatacept treated cohort.
CD28+ CD8+TEMRA cells lose CD28 expression and have superior proliferative capacity
We investigated the kinetics of peripheral blood CD28+ CD8+ TEMRA T cells over time and interestingly observed that while the overall frequency of CD45RA+CCR7− CD8+ T cells (CD8+ TEMRA) remained unchanged following transplantation, the percentage of CD28+ cells within the CD45RA+CCR7− CD8+ T cell (CD8+ TEMRA) compartment precipitously declined in the peripheral blood in belatacept “resistant” animals (Figure 5a–b). The rapid decline of the CD28+ fraction of CD8+ TEMRA was observed in both belatacept resistant animals irrespective of basiliximab induction (Supplemental Figure 3). When we examined the frequency of CD28+ cells within the CD8+ TEMRA subset 7 days prior to rejection we found a significant decrease compared to baseline values (−19.24%, P=0.0029, Figure 5c).
Figure 5. Kinetics of CD8+ TEMRA.

(a) overall percentage of CD45RA+CCR7 (CD8+ TEMRA, all CD95+) as a subset of CD8 T cells remained stable over time in both groups (b) while the fraction of CD8+ TEMRA which were CD28+ decreased rapidly post-transplantation and remained low in belatacept resistant animals (black lines), compared to animals susceptible to therapy (grey lines). (c) The frequency of CD28+ cells within the CD8 TEMRA subset is decreased by 19.24% (SEM= 3.543%, P=0.0029) 7 days prior to rejection compared to pre-transplant peripheral blood samples.
In an effort to measure the proliferative capacity and effector function of alloreactive NHP T cell subsets we sorted PMBCs from rhesus macaques into CD28+ and CD28− TEMRA and performed a standard mixed lymphocyte reaction using allogeneic stimulators. We found that CD28+CD8+ TEMRA had superior proliferative capacity compared to CD28−CD8+ TEMRA cells (77.5% relative reduction in proliferative capacity, Figure 6a, P<0.001). This difference in proliferative capacity was not affected by belatacept treatment. CD28+ CD8+ TEMRA exhibited a 71% proliferative advantage over conventional CD8+ TEM, CD8+CD45RA−CCR7−, as well (Figure 6b, P=0.0016). Of note, after 96 hours in co-culture, allo-stimulated CD8+ TEMRA failed to produce effector cytokines with classic re-stimulation reagents such as PMA and Ionomycin. Instead, exogenous administration of IL-15 to the culture was required to elicit effector function for either CD28+ or CD28− TEMRA. IL-15 provoked a potent response in both CD28+ and CD28− TEMRA (Figure 6c, increased frequency of IFNγ+TNF+ cells no cytokine vs cytokine CD28+ TEMRA 0.11% to 11.51%, P=0.0013, and CD28−TEMRA, 0.21% to 15.77%, P<0.0001). While there is a clear distinction between CD28+ and CD28− TEMRA in their proliferative capacity, both subsets seem to be reliant on exogenous IL-15 for effector cytokine production.
Figure 6. In-Vitro Functional Assessment of Alloreactive CD28+ CD8+ TEMRA.

(a) Depletion of CD28+ cells from sorted CD8+ TEMRA results in a 77.5% relative reduction in proliferative capacity (P=0.0001, black bars). Proliferation of CD28+ TEMRA is unaffected by belatacept treatment (100ug/ml, grey bars). (b) CD28+ CD8+ TEMRA exhibited a 71% relative proliferative advantage over CD8+CD45RACCR7, conventional CD8+ TEM (P=0.0016). (c) PMA and Ionomycin (P+I) was insufficient to elicit effector function from CD8+ TEMRA, but addition of IL-15 results in IFNγ+TNF+ CD8+ TEMRA in both CD28+ (P=0.0013) and CD28 TEMRA (P<0.0001).
Increased adhesion molecule expression is temporally associated with rejection
Mounting evidence has demonstrated a critical role for adhesion molecules such as CD49d (VLA-4) and CD11a (LFA-1) in mediating costimulation independent allograft rejection (43–46). Analysis of adhesion marker expression on T cell subsets prior to transplantation revealed no significant difference in the frequency or intensity of VLA-4 or LFA-1 expression on CD8+ or CD4+ T cells between belatacept “resistant” or “susceptible” animals. After transplantation, however, circulating CD8+ and CD4+ T cells increased expression of VLA-4 and CD11a early in those animals resistant to belatacept therapy, particularly in the weeks preceding allograft rejection (Figure 7a–b). Graft infiltrating cells analyzed at the time of rejection in all treatment groups expressed uniformly high levels of VLA-4 and CD11a compared to baseline peripheral blood samples (Figure 7c–d). Taken together these data suggest that CD28+TEMRA may drive a costimulation independent response where donor reactive cells rapidly expand and acquire effector function and then lose CD28 expression while upregulating adhesion cell molecules such as LFA-1 and VLA-4 allowing for access to the donor compartment. At the time of rejection, graft infiltrate of all animals demonstrated uniformly elevated levels of VLA-4 and CD11a expression, suggesting that expression of these molecules is part of “final common pathway” of allograft infiltration regardless of treatment.
Figure 7. Increased Adhesion Molecule Expression in Rejection.

Belatacept resistance demonstrates a trend towards increased expression of (a) LFA-1 and (b) VLA-4 early post-transplantation, particularly in the weeks preceding allograft rejection (resistant animals black lines, susceptible animals grey lines). Graft Infiltrating Cells isolated from rejecting allografts (Sac) uniformly express high levels of both (c) LFA-1 and (d) VLA-4 irrespective of response to therapy, compared to peripheral blood at baseline, representative histogram.
Belatacept resistant graft infiltrate is characterized by CD28− CD8+ TEMRA
As described above treatment with belatacept gave rise to two distinct groups: those animals that rejected on therapy (<140 days)- “resistant” and those which enjoyed excellent graft function with minimal graft infiltrate on biopsy until therapy was withdrawn at which time rejection ensued- “susceptible.” In other words “resistant” animals rejected on belatacept treatment while “susceptible” animals did not reject until therapy had been discontinued. We were interested to see if the character of the rejection response differed between these two groups. Accordingly, we more closely examined the rejection response between belatacept “resistant” and “susceptible” groups at the time of graft failure. In general allograft infiltrate was principally CD8+ T cells (60–70%), with a smaller CD4+ component (20–30%), in both groups (data not shown). These frequencies in graft infiltrate were reciprocal to the frequencies found in the peripheral blood where CD4+ T cells typically outnumbered CD8+ T cells 3 to 1. Belatacept “resistant” animals exhibited a significant increase in CD8+ TEMRA graft infiltrating cells extracted from rejected kidneys, whereas belatacept susceptible animals exhibited a larger frequency of less fully differentiated CD8+ TEM, even though no difference in the frequency of these subsets existed in the peripheral blood before transplant (Figure 8a and 8b). Further analysis including CD28 expression status revealed a significant increase CD28− CD8+ TEMRA in graft infiltrating cells in resistant animals (Figure 8e) whereas susceptible animals exhibited a higher frequency of CD28+ CD8+ TEM (Figure 8f). These differences were not observed in tacrolimus treated animals (Figure 8c–d, Figure 8g–h). In summary when examining the character of the infiltrate at the time of rejection in “resistant” animals the predominant cell subset was CD28− CD8+ TEMRA compared to CD28+ CD8+ TEM in “susceptible” animals suggesting a more terminally differentiated subset was responsible for rejection in belatacept “resistant” animals (representative flow plot Figure 8i).
Figure 8. Belatacept Resistance is Marked by Terminally Differentiated T cells in the Graft Infiltrate.

CD3+ CD8+ T cells extracted from rejected allografts were characterized by memory phenotype. (a) Belatacept resistant animals (black squares) demonstrate a unique, increased frequency of CD8 TEMRA infiltrate within the graft compared to belatacept susceptible animals (grey circles, P=0.009). (b) Belatacept susceptible animals (grey circles) who mount a rejection response in the absence of belatacept demonstrate a less differentiated CD8+ TEM infiltrate compared to belatacept resistant animals (black squares, P=0.0014). We next investigated the CD28 expression pattern on memory subsets. (e) Belatacept resistant animals (black squares) demonstrated increased CD28 CD8+ TEMRA compared to Belatacept susceptible animals (grey circles, (P=0.0166). (f) In contrast, belatacept susceptible animals (grey circles) had increased levels of less fully differentiated CD28+ CD8+ TEM compared to belatacept resistant animals (black squares, P=0.0245) (i) Example plots of graft infiltrating CD8 T cells in belatacept resistant rejection and belatacept susceptible animals who rejected after the withdrawal of therapy with the predominating phenotype distinctive of belatacept resistance (CD28CD8+ CD45RA+ TEMRA, lower right quadrant) vs belatacept susceptibility (CD28+ CD8+ CD45RA TEM, upper left quadrant). These differences were not observed in tacrolimus treated animals (c-d, g-h).
Graft infiltrating cells in belatacept “resistant” rejection exhibit a transcriptional signature consistent with exhaustion
In an effort to further characterize the graft infiltrate in belatacept resistant animals we analyzed the gene-expression profile of graft tissue from belatacept “resistant” vs. “susceptible” animals at the time of rejection. We identified unique modules of coordinated gene expression that were significantly divergent between the two groups. Unique chemokine and cytokines, increased trafficking and adhesion molecules, and increased expression of CD8+ T cell memory and exhaustion genes were associated with belatacept resistance (Figure 9a–c). Pro-inflammatory chemokines and cytokines such as CXCL12, IL-6, and IL-15 were coordinately up-regulated in belatacept resistant animals compared to those which were susceptible to therapy (Figure 9a). Cytokine and chemokine receptors IL-17RA, IL12RB2 and CCR6 were also up-regulated in belatacept resistant rejection, while CD25 (IL2RA), critical for regulatory T cell function, was down-regulated. Genes associated with adhesion and trafficking such as ITGA4 and ITGB2 were up-regulated in belatacept resistance (Figure 9b). Belatacept resistant animals demonstrated significant up-regulation of a number of co-inhibitory receptors and memory transcription factors characteristic of and critical for terminally differentiated CD8+ T cells, such as several of the Killer Cell Lectin-Like Receptors, EOMES, TBX21, BTLA, CTLA-4 and FAS (Figure 9c). Consistent with the flow cytometric analyses this gene expression data suggests that the infiltrate associated with belatacept resistance is of a more fully differentiated T cell phenotype.
Figure 9. Distinct Intragraft Transcriptome Modules Define Belatacept Resistant Rejection.

Transcriptome arrays from tissue acquired at the time of rejection revealed unique pathways augmented in belatacept resistance compared to belatacept susceptibility, as defined by KEGG and/or Biocarta pathway analysis. Analysis was restricted to differential gene expression between on-therapy rejection (belatacept resistant) compared off-therapy rejection (belatacept susceptible). Genes involved in (a) Cytokine and Chemokine systems, (b) Adhesion and Migration and (c) Exhaustion and Memory were up-regulated in belatacept resistant rejection. For example, CXCL12 was expressed 2.83 fold higher in belatacept resistant graft tissue compared to belatacept susceptible graft tissue, at the time of rejection.
Discussion
Current immunosuppressive strategies most commonly employ a calcineurin inhibitor, such as tacrolimus or cyclosporine, as the primary agent to prevent rejection following organ transplant. While these reagents provide for excellent initial outcomes, including one year patient and graft survival, long-term outcomes remain less than desirable (1–2, 4). There has been a concerted effort to develop new strategies to avoid unwanted side effects and improve late outcomes. While belatacept has established the promise of costimulation blockade to improve long-term outcomes, wide-spread adoption of this therapy has been limited by, among other things, increased rates of rejection and a perceived lack of efficacy. In an effort to better understand the underlying mechanisms leading to the resistance of costimulation blockade therapy we designed a study in a pre-clinical model of non-human primate kidney transplantation comparing animals treated with tacrolimus and belatacept. We identified a pre-transplant immunophenotype in the peripheral blood of elevated CD28+ CD8+ TEMRA cells that discriminates animals that go on to experience costimulation blockade resistant rejection from those that are susceptible to therapy. These CD28+ TEMRA retain proliferative capacity in addition to other effector functions unlike their CD28− counterparts. Further we show that these cells likely down-regulate CD28 or give rise to a population of CD28− effector/effector memory T cells, which constitute the major component of the infiltrate in rejected kidneys in resistant animals. Along with data presented in our companion study in human patients we suggest that this relatively straight-forward test could potentially be used as a pre-transplant screen to determine eligibility for belatacept therapy (34).
There are several differences between our findings in the non-human primate model and what was observed in patients. First there tended to be a tighter link between increased frequencies of CD28+CD4+ TEM/EMRA and a higher likelihood of rejection in human patients, whereas in our non-human primate study we found the pre-transplant level of CD28+CD8+ TEMRA was more highly associated with costimulation independent rejection. There are many differences between non-human primates and humans including age and immune experience that are likely contributors. Animals used in our studies are captive-bred juveniles and may not have the same degree of immune exposures as older adult humans. Moreover human transplant recipients, unlike the healthy primates, all have end stage renal disease with its accompanying uremia and almost universal dialysis requirement, factors which can contribute to an inflammatory environment causing immune dysregulation perhaps differentially affecting some cell subsets more than others.
We initially hypothesized that higher baseline levels of CD28− memory T cells may be predictive of costimulation independent rejection (15–16, 48). On a cursory level this subset would appear to be poised to avoid the effects of belatacept given the lack of the receptor for the targeted pathway. However, a more comprehensive view of this subset suggests that the lack of CD28 expression accompanies cells that have a more differentiated phenotype, usually following antigen exposure and activation (47). These cells often demonstrate diminished proliferative capacity despite their immediate effector capabilities. While not completely clear we suggest from our studies that CD28− cells, although potentially potent effector cells, may not possess the capacity to sustain a response that would lead to rejection of the transplanted organ. Rather we believe that CD28 expression on TEM/TEMRA designates a cells subset that has a combination of proliferative reserve and cytolytic capacity. In fact we showed that CD28+ CD8+ TEMRA proliferate readily in mixed lymphocyte reaction despite belatacept treatment, and that CD28− CD8+ TEMRA lack proliferative potential. Interestingly we observed that while CD8+ TEMRA cell frequencies remain stable during transplantation, there is a rapid loss of the CD28+ fraction. We further demonstrate CD28−CD8+ TEMRA uniquely compose a predominate fraction of the allograft infiltrate at the time of rejection in belatacept resistant rejection.
The stability of this phenotype is unknown, ie higher frequency of CD28+CD8+ TEMRA. Multiple factors may drive increased CD28 expression on TEMRA and TEM. Viral specific memory T cells from prior pathogen exposure exhibit distinct memory phenotypes based on CD45RA, CCR7, CD28 and CD95 expression. T cell memory phenotype is subject to change based on chronicity of antigen exposure, acuity of infection, or heterologous challenge (19). Consistent with our work, previous studies have identified a highly proliferative and potently cytolytic CD28+ CD8+ TEMRA cell subset in patients vaccinated for yellow fever (50). Additional investigation into the factors contributing to rejection associated with a higher level of CD28+ TEMRA is required. Work by our group and others suggests that differential expression of co-inhibitory molecules such as CTLA-4 may be responsible for preferential activation of some T cell subsets when belatacept is used. Further studies are needed to examine the longitudinal stability of this phenotype and whether interventions such as cytokine deprivation via antibody administration or periods of lower overall inflammation/immune stimulation may allow for a decrease in this subset. More importantly if the level of CD28+ TEMRA changes over time and drops below the 3% level are animals that were once “resistant” to costimulation blockade now “susceptible” to therapy? These important questions require additional investigation.
Transcriptomic analysis of the allograft infiltrate suggests an augmented pro-inflammatory milieu leads to a more fully differentiated T cell infiltrate consistent with the phenotype obtained from flow cytometry. Taken together these data suggest one mechanism of belatacept resistance is mediated by CD28 bearing memory cells which leverage a proliferative advantage in order to sustain alloreactivity, while further differentiating and increasingly relying on alternative signals (i.e. KIRs, γ-chain cytokines, inflammatory chemokines, adhesion molecules) to prosecute costimulation independent rejection. Studies aimed at high-resolution spatial and temporal tracking of memory T cells will increase our understanding of the ontology, kinetics and plasticity of T cell memory subsets. These studies may inform how and when to leverage dynamic T cell signaling sensitivities to promote transplant tolerance.
Supplementary Material
Animals were assessed weekly for renal function utilizing serum creatinine. Depressed renal function pursuant to allograft rejection was determined by two consecutive Cr > 5. Resistant animals (black lines) and susceptible animals (grey lines). Weekly serum creatinine and corresponding survival curves of belatacept treated animals (a-b), belatacept treated animals who received basiliximab induction (c-d) and tacrolimus treated animals (e-f). Survival analysis revealed no statistically significant differences between treatment groups, belatacept vs. basiliximab+belatacept (b, P1 =0 .1122) or belatacept vs. basiliximab+tacrolimus (b, P2 = 0.1175), or basiliximab+belatacept vs. basiliximab+tacrolimus (P = 0.6678). Belatacept and basiliximab+belatacept treated animals demonstrated an identical rate of therapeutic resistance (n=3/grp).
Pre-transplant immunophenotyping of therapy resistant (black circles) versus therapy susceptible animals (grey circles), with corresponding survival curves. Belatacept and basiliximab+belatacept treated animals demonstrate similarly elevated pre-transplant CD28+CD95+CD45RA+CCR7 cells as a frequency of total CD8+ T cells (c and e), as well as identical rates of therapy resistance (d and f). These differences were not observed in tacrolimus resistance (a and b)
Kinetics of CD8+ TEMRA in Belatacept Therapy overall percentage of CD45RA+CCR7 (CD8+ TEMRA, all CD95+) as a subset of CD8 T cells remained stable over time in (a) belatacept and (c) belatacept+basiliximab treated animals while the fraction of CD8+ TEMRA which were CD28+ decreased rapidly post-transplantation and remained low in belatacept resistant animals (black lines), compared to animals susceptible to therapy (grey lines), in both (b) belatacept and (d) basiliximab+belatacept treated animals. Belatacept and basiliximab+belatacept treated animals demonstrate similar kinetics of CD28+CD8+ TEMRA.
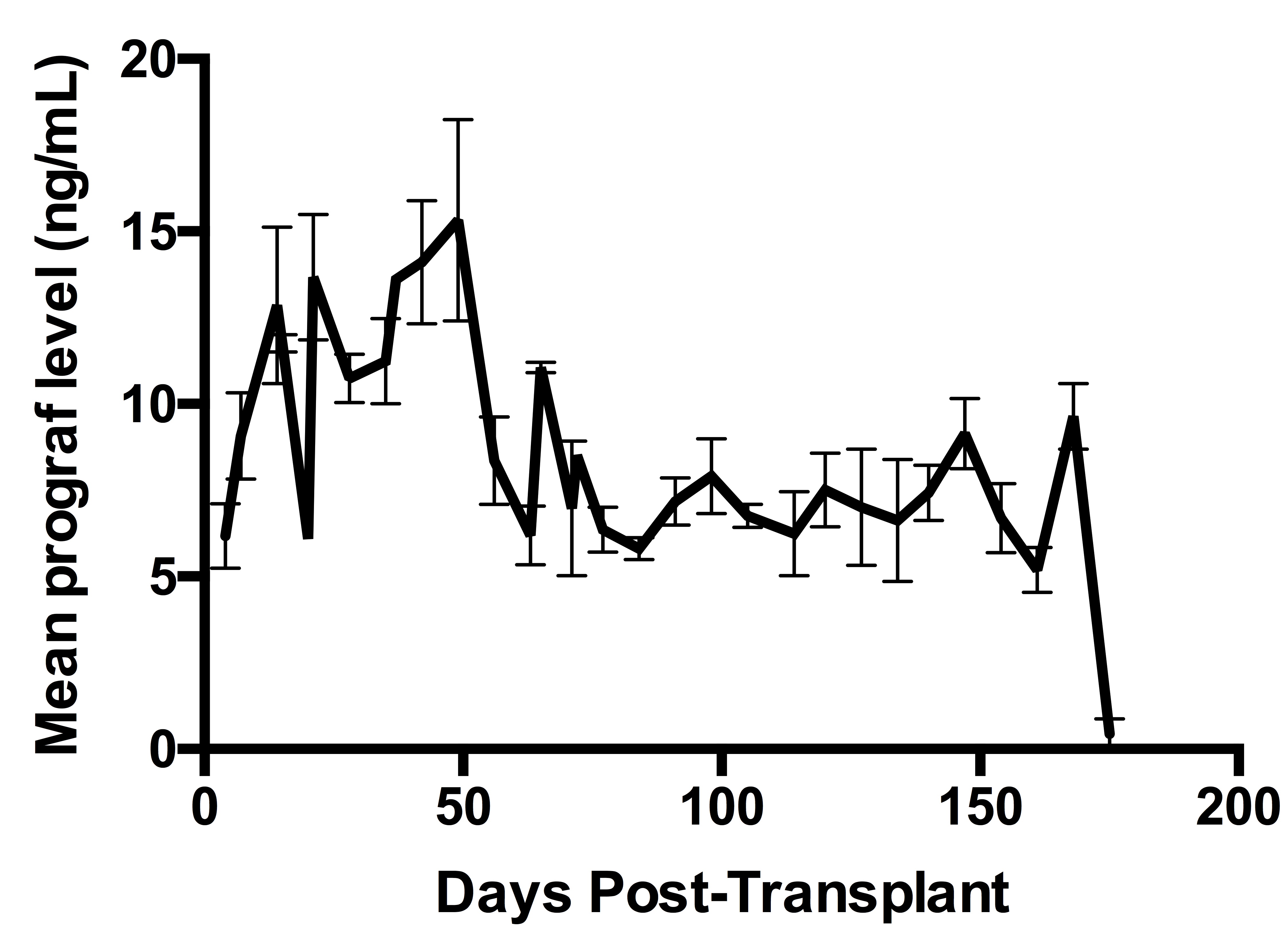
Tacrolimus treated animals were monitored weekly for prograf levels, and doses were adjusted in order to maintain blood levels from (8-12ng/ml up to day 56, and 5-8ng/ml until day 140).
{kind=link}
Animals in all treatment groups (a) belatacept, (b) belatacept + basiliximab, (c) tacrolimus + basiliximab experienced infrequent (n=1/group) and equivalent rates of CMV reactivation, defined as greater than 10,000 copies of viral DNA/ul of blood, as measured by quantitative-PCR.
Gating strategy involved gating first on lymphocytes, then CD3+ cells, then CD8+ (vs. CD4+). Gates were then set on CD28+CD95+ cells as pictured. CD28+CD95+ cells were then further analyzed by CD45RA and CCR7, as pictured. Sample plots show pre-transplant flow cytometry from one therapy resistant animal (top) and therapy susceptible animal (bottom).
Acknowledgments
This work was supported by funding from Bristol-Myers Squibb, Non-Human Primate Transplant Tolerance Cooperative Study Group,- NIH grant U19AI051731 and NIH grant P51OD11132 in support of Yerkes. DM was supported by NIH F30DK109665. WCW was supported by ASTS Fellowship. The authors would like to thank all collaborators at the Emory Transplant Center and at the Yerkes Primate Research Center, especially all veterinary technicians and pathology lab staff, as well as all collaborators at Bristol Myers-Squibb.
Abbreviations
- CNI
Calcineurin Inhibitor
- TCM
central memory T cells
- TEM
effector memory T cells
- TEMRA
T Effector Memory CD45RA+ cells
- NHP
Non-Human Primate
- TCR
T Cell Receptor
Footnotes
Disclosure
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. ABA and MLF have received research funding and consulting fees from BMS. The other authors have no conflicts of interest to disclose.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- 1.Lamb KE, Lodhi S, Meier-Kriesche H-U. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant. 2011 Mar;11(3):450–62. doi: 10.1111/j.1600-6143.2010.03283.x. [DOI] [PubMed] [Google Scholar]
- 2.Gaston RS. Chronic Calcineurin Inhibitor Nephrotoxicity: Reflections on an Evolving Paradigm. Clin J Am Soc Nephrol. American Society of Nephrology. 2009 Dec 1;4(12):2029–34. doi: 10.2215/CJN.03820609. [DOI] [PubMed] [Google Scholar]
- 3.Kasiske BL, Snyder JJ, Gilbertson D, Matas AJ. Diabetes mellitus after kidney transplantation in the United States. Am J Transplant. 2003;3(2):178–85. doi: 10.1034/j.1600-6143.2003.00010.x. [DOI] [PubMed] [Google Scholar]
- 4.Ojo A, Held P, Port F, Wolfe R, Leichtman A, Young E, et al. Chronic Renal Failure after Transplantation of a Nonrenal Organ. N Engl J Med. 2003;349:931–40. doi: 10.1056/NEJMoa021744. [DOI] [PubMed] [Google Scholar]
- 5.Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, et al. Costimulation blockade with belatacept in renal transplantation. The New England journal of medicine. 2005 doi: 10.1056/NEJMoa050085. [DOI] [PubMed] [Google Scholar]
- 6.Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005 Mar;5(3):443–53. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 7.Vanrenterghem Y, Bresnahan B, Campistol J, Durrbach A, Grinyó J, Neumayer H-H, et al. Belatacept-based regimens are associated with improved cardiovascular and metabolic risk factors compared with cyclosporine in kidney transplant recipients (BENEFIT and BENEFIT-EXT studies) Transplantation. 2011 May 15;91(9):976–83. doi: 10.1097/TP.0b013e31820c10eb. [DOI] [PubMed] [Google Scholar]
- 8.Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and Long-Term Outcomes in Kidney Transplantation. N Engl J Med. 2016;374(4):333–43. doi: 10.1056/NEJMoa1506027. [DOI] [PubMed] [Google Scholar]
- 9.Vincenti F, Larsen CP, Alberu J, Bresnahan B, Garcia VD, Kothari J, et al. Three-year outcomes from BENEFIT, a randomized, active-controlled, parallel-group study in adult kidney transplant recipients. Am J Transplant. 2012;12(1):210–7. doi: 10.1111/j.1600-6143.2011.03785.x. [DOI] [PubMed] [Google Scholar]
- 10.Rostaing L, Vincenti F, Grinyó J, Rice KM, Bresnahan B, Steinberg S, et al. Long-term belatacept exposure maintains efficacy and safety at 5 years: results from the long-term extension of the BENEFIT study. Am J Transplant. 2013 Nov;13(11):2875–83. doi: 10.1111/ajt.12460. [DOI] [PubMed] [Google Scholar]
- 11.Vincenti F, Charpentier B, Vanrenterghem Y, Rostaing L, Bresnahan B, Darji P, et al. A phase III study of belatacept-based immunosuppression regimens versus cyclosporine in renal transplant recipients (BENEFIT study) Am J Transplant. 2010 Mar;10(3):535–46. doi: 10.1111/j.1600-6143.2009.03005.x. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan B. Editorial: Belatacept: The promises and challenges of belatacept and costimulatory blockade. American Journal of Transplantation. 2010;10(3):441–2. doi: 10.1111/j.1600-6143.2010.03026.x. [DOI] [PubMed] [Google Scholar]
- 13.Weng N, Akbar AN, Goronzy J. CD28− T cells: their role in the age-associated decline of immune function. Trends Immunol. 2009 Jul;30(7):306–12. doi: 10.1016/j.it.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev. 2005;205(1):158–69. doi: 10.1111/j.0105-2896.2005.00256.x. [DOI] [PubMed] [Google Scholar]
- 15.Bryl E, Vallejo aN, Weyand CM, Goronzy JJ. Down-regulation of CD28 expression by TNF-alpha. J Immunol. 2001;167(6):3231–8. doi: 10.4049/jimmunol.167.6.3231. [DOI] [PubMed] [Google Scholar]
- 16.Borthwick NJ, Lowdell M, Salmon M, Akbar AN. Loss of CD28 expression on CD8(+) T cells is induced by IL-2 receptor gamma chain signalling cytokines and type I IFN, and increases susceptibility to activation-induced apoptosis. Int Immunol. 2000 Jul;12(7):1005–13. doi: 10.1093/intimm/12.7.1005. [DOI] [PubMed] [Google Scholar]
- 17.Chiu WK, Fann M, Weng N. Generation and growth of CD28nullCD8+ memory T cells mediated by IL-15 and its induced cytokines. J Immunol. 2006 Dec 1;177(11):7802–10. doi: 10.4049/jimmunol.177.11.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valenzuela HF, Effros RB. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin Immunol. 2002;105(2):117–25. doi: 10.1006/clim.2002.5271. [DOI] [PubMed] [Google Scholar]
- 19.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GMa, Papagno L, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8(4):379–85. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 20.Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia a. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401(6754):708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 21.Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, et al. Development and homeostasis of T cell memory in rhesus macaque. J Immunol. 2002;168(1):29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- 22.Vaccari M, Trindade CJ, Venzon D, Zanetti M, Franchini G. Vaccine-induced CD8+ central memory T cells in protection from simian AIDS. J Immunol. 2005;175(6):3502–7. doi: 10.4049/jimmunol.175.6.3502. [DOI] [PubMed] [Google Scholar]
- 23.Appay V, van Lier RAW, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73(11):975–83. doi: 10.1002/cyto.a.20643. [DOI] [PubMed] [Google Scholar]
- 24.De Rosa SC, Herzenberg LA, Roederer M. 11-color, 13-parameter flow cytometry: identification of human naive T cells by phenotype, function, and T-cell receptor diversity. Nat Med. Nature Publishing Group. 2001 Feb 1;7(2):245–8. doi: 10.1038/84701. [DOI] [PubMed] [Google Scholar]
- 25.Vaccari M, Franchini G. Memory T cells in Rhesus macaques. Adv Exp Med Biol. 2010;684:126–44. doi: 10.1007/978-1-4419-6451-9_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur J Immunol. 2013;43(11):2797–809. doi: 10.1002/eji.201343751. [DOI] [PubMed] [Google Scholar]
- 27.Yap M, Tilly G, Giral M, Brouard S, Degauque N. Benefits of Using CD45RA and CD28 to Investigate CD8 Subsets in Kidney Transplant Recipients. Am J Transplant. 2016 Mar;16(3):999–1006. doi: 10.1111/ajt.13581. [DOI] [PubMed] [Google Scholar]
- 28.Chan KS, Kaur A. Flow cytometric detection of degranulation reveals phenotypic heterogeneity of degranulating CMV-specific CD8+ T lymphocytes in rhesus macaques. J Immunol Methods. 2007 Aug 31;325(1–2):20–34. doi: 10.1016/j.jim.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azuma M, Phillips JH, Lanier LL. CD28- T lymphocytes. Antigenic and functional properties. J Immunol. 1993;150(4):1147–59. [PubMed] [Google Scholar]
- 30.Judge AD, Zhang X, Fujii H, Surh CD, Sprent J. Interleukin 15 controls both proliferation and survival of a subset of memory-phenotype CD8(+) T cells. J Exp Med. 2002;196(7):935–46. doi: 10.1084/jem.20020772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villinger F, Miller R, Mori K, Mayne AE, Bostik P, Sundstrom JB, et al. IL-15 is superior to IL-2 in the generation of long-lived antigen specific memory CD4 and CD8 T cells in rhesus macaques. Vaccine. 2004 Sep 3;22(25–26):3510–21. doi: 10.1016/j.vaccine.2003.07.022. [DOI] [PubMed] [Google Scholar]
- 32.Sneller MC, Kopp WC, Engelke KJ, Yovandich JL, Creekmore SP, Waldmann TA, et al. IL-15 administered by continuous infusion to rhesus macaques induces massive expansion of CD8 + T effector memory population in peripheral blood. Blood. 2011;118(26):6845–8. doi: 10.1182/blood-2011-09-377804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Espinosa J, Herr F, Tharp G, Bosinger S, Song M, Farris AB, et al. CD57(+) CD4 T cells underlie belatacept-resistant allograft rejection. Am J Transplant. 2015 Nov 25;16(4):1102–12. doi: 10.1111/ajt.13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cortes M, Laurie SJ, Sayed B, Ford ML. Rapid Emergence of CD28null Cells Following Ex Vivo Restimulation Is Associated with Belatacept Resistant Rejection. Am J Transplant. 2016;16(suppl 3) [Google Scholar]
- 35.Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci. 1997 Aug 5;94(16):8789–94. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higginbotham L, Mathews D, Breeden C, Wakwe W, Larsen C, Nadler S, et al. CD28-Directed Therapy Prolongs Survival in a Non-Human Primate Kidney Transplant. Model. Am J Transplant. 2015;15(suppl 3) [Google Scholar]
- 37.Kim SC, Wakwe W, Higginbotham LB, Mathews DV, Breeden CP, Stephenson AC, et al. Fc-Silent Anti-CD154 Domain Antibody Effectively Prevents Nonhuman Primate Renal Allograft Rejection. Am J Transplant. 2017 doi: 10.1111/ajt.14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang DW, Lempicki Ra, Sherman BT. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 39.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nashan B, Moore R, Amlot P, Schmidt A-G, Abeywickrama K, Soulillou J-P. Randomised trial of basiliximab versus placebo for control of acute cellular rejection in renal allograft recipients. Lancet. 1997 Oct;350(9086):1193–8. doi: 10.1016/s0140-6736(97)09278-7. [DOI] [PubMed] [Google Scholar]
- 41.Bluestone JA, Liu W, Yabu JM, Laszik ZG, Putnam A, Belingheri M, et al. The Effect of Costimulatory and Interleukin 2 Receptor Blockade on Regulatory T Cells in Renal Transplantation. Am J Transplant. Blackwell Publishing Inc. 2008 Oct;8(10):2086–96. doi: 10.1111/j.1600-6143.2008.02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams AB, Williams MA, Jones TR, Shirasugi N, Durham MM, Kaech SM, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. American Society for Clinical Investigation. 2003 Jun 15;111(12):1887–95. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nadazdin O, Boskovic S, Murakami T, Tocco G, Smith R-N, Colvin RB, et al. Host alloreactive memory T cells influence tolerance to kidney allografts in nonhuman primates. Sci Transl Med. 2011 Jun 8;3(86):86ra51. doi: 10.1126/scitranslmed.3002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ford ML, Kirk AD, Larsen CP. Donor-reactive T-cell stimulation history and precursor frequency: barriers to tolerance induction. Transplantation. 2009;87(9 Suppl):S69–74. doi: 10.1097/TP.0b013e3181a2a701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen W, Ghobrial RM, Li XC. The Evolving Roles of Memory Immune Cells in Transplantation. Transplantation. 2015;99(10):2029–37. doi: 10.1097/TP.0000000000000802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mou D, Espinosa J, Lo DJ, Kirk AD. CD28 negative T cells: Is their loss our gain? American Journal of Transplantation. 2014:2460–6. doi: 10.1111/ajt.12937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turgeon NA, Avila JG, Cano JA, Hutchinson JJ, Badell IR, Page AJ, et al. Experience with a Novel Efalizumab-Based Immunosuppressive Regimen to Facilitate Single Donor Islet Cell Transplantation. Am J Transplant. 2010 Sep 25;10(9):2082–91. doi: 10.1111/j.1600-6143.2010.03212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Badell IR, Russell MC, Thompson PW, Turner AP, Weaver TA, Robertson JM, et al. LFA-1-specific therapy prolongs allograft survival in rhesus macaques. J Clin Invest. 2010 Dec;120(12):4520–31. doi: 10.1172/JCI43895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitchens WH, Haridas D, Wagener ME, Song M, Kirk AD, Larsen CP, et al. Integrin antagonists prevent costimulatory blockade-resistant transplant rejection by CD8(+) memory T cells. Am J Transplant. 2012 Jan;12(1):69–80. doi: 10.1111/j.1600-6143.2011.03762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kitchens WH, Haridas D, Wagener ME, Song M, Ford ML. Combined costimulatory and leukocyte functional antigen-1 blockade prevents transplant rejection mediated by heterologous immune memory alloresponses. Transplantation. 2012 May 27;93(10):997–1005. doi: 10.1097/TP.0b013e31824e75d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vallejo aN, Brandes JC, Weyand CM, Goronzy JJ. Modulation of CD28 expression: distinct regulatory pathways during activation and replicative senescence. J Immunol. 1999;162(11):6572–9. [PubMed] [Google Scholar]
- 52.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2(4):251–62. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 53.Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. NIH Public Access. 2009 Dec 15;183(12):7919–30. doi: 10.4049/jimmunol.0803903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Animals were assessed weekly for renal function utilizing serum creatinine. Depressed renal function pursuant to allograft rejection was determined by two consecutive Cr > 5. Resistant animals (black lines) and susceptible animals (grey lines). Weekly serum creatinine and corresponding survival curves of belatacept treated animals (a-b), belatacept treated animals who received basiliximab induction (c-d) and tacrolimus treated animals (e-f). Survival analysis revealed no statistically significant differences between treatment groups, belatacept vs. basiliximab+belatacept (b, P1 =0 .1122) or belatacept vs. basiliximab+tacrolimus (b, P2 = 0.1175), or basiliximab+belatacept vs. basiliximab+tacrolimus (P = 0.6678). Belatacept and basiliximab+belatacept treated animals demonstrated an identical rate of therapeutic resistance (n=3/grp).
Pre-transplant immunophenotyping of therapy resistant (black circles) versus therapy susceptible animals (grey circles), with corresponding survival curves. Belatacept and basiliximab+belatacept treated animals demonstrate similarly elevated pre-transplant CD28+CD95+CD45RA+CCR7 cells as a frequency of total CD8+ T cells (c and e), as well as identical rates of therapy resistance (d and f). These differences were not observed in tacrolimus resistance (a and b)
Kinetics of CD8+ TEMRA in Belatacept Therapy overall percentage of CD45RA+CCR7 (CD8+ TEMRA, all CD95+) as a subset of CD8 T cells remained stable over time in (a) belatacept and (c) belatacept+basiliximab treated animals while the fraction of CD8+ TEMRA which were CD28+ decreased rapidly post-transplantation and remained low in belatacept resistant animals (black lines), compared to animals susceptible to therapy (grey lines), in both (b) belatacept and (d) basiliximab+belatacept treated animals. Belatacept and basiliximab+belatacept treated animals demonstrate similar kinetics of CD28+CD8+ TEMRA.
Tacrolimus treated animals were monitored weekly for prograf levels, and doses were adjusted in order to maintain blood levels from (8-12ng/ml up to day 56, and 5-8ng/ml until day 140).
Animals in all treatment groups (a) belatacept, (b) belatacept + basiliximab, (c) tacrolimus + basiliximab experienced infrequent (n=1/group) and equivalent rates of CMV reactivation, defined as greater than 10,000 copies of viral DNA/ul of blood, as measured by quantitative-PCR.
Gating strategy involved gating first on lymphocytes, then CD3+ cells, then CD8+ (vs. CD4+). Gates were then set on CD28+CD95+ cells as pictured. CD28+CD95+ cells were then further analyzed by CD45RA and CCR7, as pictured. Sample plots show pre-transplant flow cytometry from one therapy resistant animal (top) and therapy susceptible animal (bottom).